
Recent Advances on Computational Modeling of Supported Single-Atom and Cluster Catalysts: Characterization, Catalyst–Support Interaction, and Active Site Heterogeneity



Abstract
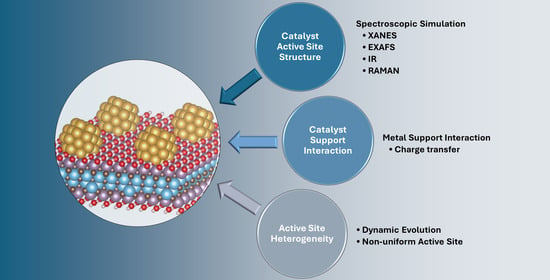
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. General Approach
2.1. Catalyst Active Site Characterization
2.2. Reaction Thermodynamics and Kinetics
2.3. Chemical and Physical Properties of the Materials
3. Best Practices
3.1. Characterization
3.2. Catalyst–Support Interaction
3.3. Active Site Heterogeneity
4. Conclusions and Outlook
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Liu, J.; Jiao, M.; Lu, L.; Barkholtz, H.M.; Li, Y.; Wang, Y.; Jiang, L.; Wu, Z.; Liu, D.-J.; Zhuang, L.; et al. High performance platinum single atom electrocatalyst for oxygen reduction reaction. Nat. Commun. 2017, 8, 15938. [Google Scholar] [CrossRef] [PubMed]
- Luengnaruemitchai, A.; Nimsuk, M.; Naknam, P.; Wongkasemjit, S.; Osuwan, S. A comparative study of synthesized and commercial A-type zeolite-supported Pt catalysts for selective CO oxidation in H2-rich stream. Int. J. Hydrogen Energy 2008, 33, 206–213. [Google Scholar] [CrossRef]
- Bhasin, M.; McCain, J.; Vora, B.; Imai, T.; Pujadó, P. Dehydrogenation and oxydehydrogenation of paraffins to olefins. Appl. Catal. A Gen. 2001, 221, 397–419. [Google Scholar] [CrossRef]
- Li, D.; Xu, F.; Tang, X.; Dai, S.; Pu, T.; Liu, X.; Tian, P.; Xuan, F.; Xu, Z.; Wachs, I.E.; et al. Induced activation of the commercial Cu/ZnO/Al2O3 catalyst for the steam reforming of methanol. Nat. Catal. 2022, 5, 99–108. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Bligaard, T.; Rossmeisl, J.; Christensen, C.H. Towards the computational design of solid catalysts. Nat. Chem. 2009, 1, 37–46. [Google Scholar] [CrossRef]
- Matsuoka, W.; Harabuchi, Y.; Maeda, S. Virtual Ligand Strategy in Transition Metal Catalysis Toward Highly Efficient Elucidation of Reaction Mechanisms and Computational Catalyst Design. ACS Catal. 2023, 13, 5697–5711. [Google Scholar] [CrossRef]
- Bligaard, T.; Bullock, R.M.; Campbell, C.T.; Chen, J.G.; Gates, B.C.; Gorte, R.J.; Jones, C.W.; Jones, W.D.; Kitchin, J.R.; Scott, S.L. Toward Benchmarking in Catalysis Science: Best Practices, Challenges, and Opportunities. ACS Catal. 2016, 6, 2590–2602. [Google Scholar] [CrossRef]
- Shan, N.; Zhou, M.; Hanchett, M.K.; Chen, J.; Liu, B. Practical principles of density functional theory for catalytic reaction simulations on metal surfaces–from theory to applications. Mol. Simul. 2017, 43, 861–885. [Google Scholar] [CrossRef]
- Durand, D.J.; Fey, N. Computational Ligand Descriptors for Catalyst Design. Chem. Rev. 2019, 119, 6561–6594. [Google Scholar] [CrossRef]
- Soyemi, A.; Szilvási, T. Trends in computational molecular catalyst design. Dalton Trans. 2021, 50, 10325–10339. [Google Scholar] [CrossRef]
- Liu, C.; Yang, B.; Tyo, E.; Seifert, S.; DeBartolo, J.; von Issendorff, B.; Zapol, P.; Vajda, S.; Curtiss, L.A. Carbon Dioxide Conversion to Methanol over Size-Selected Cu4 Clusters at Low Pressures. J. Am. Chem. Soc. 2015, 137, 8676–8679. [Google Scholar] [CrossRef]
- Qi, Z.; Xiao, C.; Liu, C.; Goh, T.W.; Zhou, L.; Maligal-Ganesh, R.V.; Pei, Y.; Li, X.; Curtiss, L.A.; Huang, W. Sub-4 nm PtZn Intermetallic Nanoparticles for Enhanced Mass and Specific Activities in Catalytic Electrooxidation Reaction. J. Am. Chem. Soc. 2017, 139, 4762–4768. [Google Scholar] [CrossRef]
- Abbasi, P.; Asadi, M.; Liu, C.; Sharifi-Asl, S.; Sayahpour, B.; Behranginia, A.; Zapol, P.; Shahbazian-Yassar, R.; Curtiss, L.A.; Salehi-Khojin, A. Tailoring the Edge Structure of Molybdenum Disulfide toward Electrocatalytic Reduction of Carbon Dioxide. ACS Nano 2017, 11, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; He, H.; Zapol, P.; Curtiss, L.A. Computational studies of electrochemical CO2 reduction on subnanometer transition metal clusters. Phys. Chem. Chem. Phys. 2014, 16, 26584–26599. [Google Scholar] [CrossRef]
- Norskov, J.K.; Abild-Pedersen, F.; Studt, F.; Bligaard, T. Density functional theory in surface chemistry and catalysis. Proc. Natl. Acad. Sci. USA 2011, 108, 937–943. [Google Scholar] [CrossRef] [PubMed]
- Hammer, B.; Nørskov, J. Electronic factors determining the reactivity of metal surfaces. Surf. Sci. 1995, 343, 211–220. [Google Scholar] [CrossRef]
- Nilsson, A.; Pettersson, L.G.M.; Hammer, B.; Bligaard, T.; Christensen, C.H.; Nørskov, J.K. The electronic structure effect in heterogeneous catalysis. Catal. Lett. 2005, 100, 111–114. [Google Scholar] [CrossRef]
- Hammer, B.; Norskov, J.K. Why gold is the noblest of all the metals. Nature 1995, 376, 238–240. [Google Scholar] [CrossRef]
- Wu, Y.A.; McNulty, I.; Liu, C.; Lau, K.C.; Liu, Q.; Paulikas, A.P.; Sun, C.-J.; Cai, Z.; Guest, J.R.; Ren, Y.; et al. Facet-dependent active sites of a single Cu2O particle photocatalyst for CO2 reduction to methanol. Nat. Energy 2019, 4, 957–968. [Google Scholar] [CrossRef]
- Stamenkovic, V.; Mun, B.S.; Mayrhofer, K.J.J.; Ross, P.N.; Markovic, N.M.; Rossmeisl, J.; Greeley, J.; Nørskov, J.K. Changing the Activity of Electrocatalysts for Oxygen Reduction by Tuning the Surface Electronic Structure. Angew. Chem. Int. Ed. 2006, 45, 2897–2901. [Google Scholar] [CrossRef]
- Yang, Y.; Lin, Z.; Gao, S.; Su, J.; Lun, Z.; Xia, G.; Chen, J.; Zhang, R.; Chen, Q. Tuning Electronic Structures of Nonprecious Ternary Alloys Encapsulated in Graphene Layers for Optimizing Overall Water Splitting Activity. ACS Catal. 2017, 7, 469–479. [Google Scholar] [CrossRef]
- Choi, C.; Yoon, S.; Jung, Y. Shifting the scaling relations of single-atom catalysts for facile methane activation by tuning the coordination number. Chem. Sci. 2021, 12, 3551–3557. [Google Scholar] [CrossRef] [PubMed]
- Aljama, H.; Nørskov, J.K.; Abild-Pedersen, F. Tuning Methane Activation Chemistry on Alkaline Earth Metal Oxides by Doping. J. Phys. Chem. C 2018, 122, 22544–22548. [Google Scholar] [CrossRef]
- He, Y.; Liu, S.; Priest, C.; Shi, Q.; Wu, G. Atomically dispersed metal-nitrogen-carbon catalysts for fuel cells: Advances in catalyst design, electrode performance, and durability improvement. Chem. Soc. Rev. 2020, 49, 1902844. [Google Scholar] [CrossRef]
- Zhu, Y.; Sokolowski, J.; Song, X.; He, Y.; Mei, Y.; Wu, G. Engineering Local Coordination Environments of Atomically Dispersed and Heteroatom-Coordinated Single Metal Site Electrocatalysts for Clean Energy-Conversion. Adv. Energy Mater. 2020, 10, 1902844. [Google Scholar] [CrossRef]
- Mun, Y.; Lee, S.; Kim, K.; Kim, S.; Lee, S.; Han, J.W.; Lee, J. Versatile Strategy for Tuning ORR Activity of a Single Fe-N4 Site by Controlling Electron-Withdrawing/Donating Properties of a Carbon Plane. J. Am. Chem. Soc. 2019, 141, 6254–6262. [Google Scholar] [CrossRef]
- Xie, Z.L.; Wang, D.; Gong, X.Q. Theoretical Insights into Nitrate Reduction to Ammonia over Pt/TiO2: Reaction Mechanism, Activity Regulation, and Catalyst Design. Acs. Catal. 2022, 12, 9887–9896. [Google Scholar] [CrossRef]
- Chen, Y.; Ji, S.; Zhao, S.; Chen, W.; Dong, J.; Cheong, W.C.; Shen, R.G.; Wen, X.D.; Zheng, L.R.; Rykov, A.I. Enhanced oxygen reduction with single-atomic-site iron catalysts for a zinc-air battery and hydrogen-air fuel cell. Nat. Commun. 2022, 13, 5422. [Google Scholar] [CrossRef]
- Schwarz, H.; González-Navarrete, P.; Li, J.; Schlangen, M.; Sun, X.; Weiske, T.; Zhou, S. Unexpected Mechanistic Variants in the Thermal Gas-Phase Activation of Methane. Organometallics 2017, 36, 8–17. [Google Scholar] [CrossRef]
- Xu, J.; Liu, B. Modeling C–H Bond Activation and Oxidations of Alkanes over Cu–MOR Using First-Principles Methods. J. Phys. Chem. C 2019, 123, 10356–10366. [Google Scholar] [CrossRef]
- Schwarz, H. Chemistry with Methane: Concepts Rather than Recipes. Angew. Chem. Int. Ed. 2011, 50, 10096–10115. [Google Scholar] [CrossRef]
- Xu, J.; Patel, P.; Liu, D.-J.; Xu, T.; Liu, C. Understanding the dynamic evolution of atomically dispersed Cu catalyst for CO2 electrochemical conversion using integrated XANES analysis and mechanistic studies. J. Catal. 2023, 425, 296–305. [Google Scholar] [CrossRef]
- Lee, S.H.; Lin, J.C.; Farmand, M.; Landers, A.T.; Feaster, J.T.; Acosta, J.E.A.; Beeman, J.W.; Ye, Y.; Yano, J.; Mehta, A.; et al. Oxidation State and Surface Reconstruction of Cu under CO2 Reduction Conditions from In Situ X-ray Characterization. J. Am. Chem. Soc. 2021, 143, 588–592. [Google Scholar] [CrossRef]
- Liu, X.; Meng, J.; Zhu, J.; Huang, M.; Wen, B.; Guo, R.; Mai, L. Comprehensive Understandings into Complete Reconstruction of Precatalysts: Synthesis, Applications, and Characterizations. Adv. Mater. 2021, 33, 2007344. [Google Scholar] [CrossRef] [PubMed]
- Lai, W.; Ma, Z.; Zhang, J.; Yuan, Y.; Qiao, Y.; Huang, H. Dynamic Evolution of Active Sites in Electrocatalytic CO2 Reduction Reaction: Fundamental Understanding and Recent Progress. Adv. Funct. Mater. 2022, 32, 2111193. [Google Scholar] [CrossRef]
- Grosse, P.; Yoon, A.; Rettenmaier, C.; Herzog, A.; Chee, S.W.; Roldan Cuenya, B. Dynamic transformation of cubic copper catalysts during CO2 electroreduction and its impact on catalytic selectivity. Nat. Commun. 2021, 12, 6736. [Google Scholar] [CrossRef] [PubMed]
- Karapinar, D.; Huan, N.T.; Sahraie, N.R.; Li, J.K.; Wakerley, D.; Touati, N.; Zanna, S.; Taverna, D.; Tizei, L.H.G.; Zitolo, A.; et al. Electroreduction of CO2 on Single-Site Copper-Nitrogen-Doped Carbon Material: Selective Formation of Ethanol and Reversible Restructuration of the Metal Sites. Angew. Chem. Int. Ed. 2019, 58, 15098–15103. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kley, C.S.; Li, Y.; Yang, P. Copper nanoparticle ensembles for selective electroreduction of CO2 to C2–C3 products. Proc. Natl. Acad. Sci. USA 2017, 114, 10560–10565. [Google Scholar] [CrossRef]
- Chapovetsky, A.; Kennedy, R.M.; Witzke, R.; Wegener, E.C.; Dogan, F.; Patel, P.; Ferrandon, M.; Niklas, J.; Poluektov, O.G.; Rui, N.; et al. Lithium-Ion Battery Materials as Tunable, “Redox Non-Innocent” Catalyst Supports. ACS Catal. 2022, 12, 7233–7242. [Google Scholar] [CrossRef]
- Gunasooriya, G.T.K.K.; Seebauer, E.G.; Saeys, M. Ethylene Hydrogenation over Pt/TiO2: A Charge-Sensitive Reaction. ACS Catal. 2017, 7, 1966–1970. [Google Scholar] [CrossRef]
- Bruix, A.; Rodriguez, J.A.; Ramírez, P.J.; Senanayake, S.D.; Evans, J.; Park, J.B.; Stacchiola, D.; Liu, P.; Hrbek, J.; Illas, F. A New Type of Strong Metal-Support Interaction and the Production of H-2 through the Transformation of Water on Pt/CeO2(111) and Pt/CeOx/TiO2(110) Catalysts. J. Am. Chem. Soc. 2012, 134, 8968–8974. [Google Scholar] [CrossRef]
- Witzke, R.J.; Chapovetsky, A.; Conley, M.P.; Kaphan, D.M.; Delferro, M. Nontraditional Catalyst Supports in Surface Organometallic Chemistry. ACS Catal. 2020, 10, 11822–11840. [Google Scholar] [CrossRef]
- Shao, Q.; Wang, P.; Huang, X. Opportunities and Challenges of Interface Engineering in Bimetallic Nanostructure for Enhanced Electrocatalysis. Adv. Funct. Mater. 2019, 29, 1806419. [Google Scholar] [CrossRef]
- Patel, P.; Lu, Z.; Jafari, M.G.; Hernández-Prieto, C.; Zatsepin, P.; Mindiola, D.J.; Kaphan, D.M.; Delferro, M.; Kropf, A.J.; Liu, C. Integrated Experimental and Computational K-Edge X-ray Absorption Near-Edge Structure Analysis of Vanadium Catalysts. J. Phys. Chem. C 2022, 126, 11949–11962. [Google Scholar] [CrossRef]
- George, S.D.; Petrenko, T.; Neese, F. Prediction of Iron K-Edge Absorption Spectra Using Time-Dependent Density Functional Theory. J. Phys. Chem. A 2008, 112, 12936–12943. [Google Scholar] [CrossRef]
- Lee, K.; Wei, H.; Blake, A.V.; Donahue, C.M.; Keith, J.M.; Daly, S.R. Ligand K-edge XAS, DFT, and TDDFT analysis of pincer linker variations in Rh(I) PNP complexes: Reactivity insights from electronic structure. Dalton Trans 2016, 45, 11198. [Google Scholar] [CrossRef]
- Krewald, V.; Lassalle-Kaiser, B.; Boron, T.T.; Pollock, C.J.; Kern, J.; Beckwith, M.A.; Yachandra, V.K.; Pecoraro, V.L.; Yano, J.; Neese, F.; et al. The Protonation States of Oxo-Bridged MnIV Dimers Resolved by Experimental and Computational Mn K Pre-Edge X-ray Absorption Spectroscopy. Inorg. Chem. 2013, 52, 12904–12914. [Google Scholar] [CrossRef] [PubMed]
- Bunău, O.; Joly, Y. Self-consistent aspects of X-ray absorption calculations. J. Phys. Condens. Matter. 2009, 21, 345501. [Google Scholar] [CrossRef]
- A Guda, A.; A Guda, S.; A Soldatov, M.; A Lomachenko, K.; Bugaev, A.L.; Lamberti, C.; Gawelda, W.; Bressler, C.; Smolentsev, G.; Soldatov, A.V.; et al. Finite difference method accelerated with sparse solvers for structural analysis of the metal-organic complexes. J. Phys. Conf. Ser. 2016, 712, 012004. [Google Scholar] [CrossRef]
- Joly, Y. Calculating X-ray absorption near-edge structure at very low energy. J. Synchrotron Radiat. 2003, 10, 58–63. [Google Scholar] [CrossRef]
- Joly, Y. X-ray absorption near-edge structure calculations beyond the muffin-tin approximation. Phys. Rev. B. 2001, 63, 125120. [Google Scholar] [CrossRef]
- Ankudinov, A.L.; Ravel, B.; Rehr, J.J.; Conradson, S.D. Real-space multiple-scattering calculation and interpretation of X-ray-absorption near-edge structure. Phys. Rev. B. 1998, 58, 7565–7576. [Google Scholar] [CrossRef]
- Blaha, P.; Schwarz, K.; Tran, F.; Laskowski, R.; Madsen, G.K.H.; Marks, L.D. WIEN2k: An APW+lo program for calculating the properties of solids. J. Chem. Phys. 2020, 152, 074101. [Google Scholar] [CrossRef]
- Te Velde, G.T.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Neese, F.; Olbrich, G. Efficient use of the resolution of the identity approximation in time-dependent density functional calculations with hybrid density functionals. Chem. Phys. Lett. 2002, 362, 170–178. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system—Version 5.0. WIREs Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Nardelli, M.B.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Gougoussis, C.; Calandra, M.; Seitsonen, A.P.; Mauri, F. First-principles calculations of X-ray absorption in a scheme based on ultrasoft pseudopotentials: From α-quartz to high-Tc compounds. Phys. Rev. B. 2009, 80, 075102. [Google Scholar] [CrossRef]
- Gougoussis, C.; Calandra, M.; Seitsonen, A.P.; Mauri, F. Projector augmented wave calculation of X-ray absorption spectra at the L2,3 edges. Phys. Rev. B. 2013, 87, 205105. [Google Scholar]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Meng, Y.; Yin, C.; Li, K.; Tang, H.; Wang, Y.; Wu, Z. Improved Oxygen Reduction Activity in Heteronuclear FeCo-Codoped Graphene: A Theoretical Study. ACS Sustain. Chem. Eng. 2019, 7, 17273–17281. [Google Scholar] [CrossRef]
- Liu, S.; Cheng, L.; Wang, W.; Li, K.; Wang, Y.; Wu, Z. Fe-porphyrin carbon matrix as a bifunctional catalyst for oxygen reduction and CO2 reduction from theoretical perspective. Mol. Phys. 2019, 117, 1805–1812. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, W.; Qu, X.; Meng, Y.; Wu, Z. M-porphyrin (M = Mn, Co) carbon materials as oxygen reduction catalysts from density functional studies. Mol. Phys. 2020, 118, e1687949. [Google Scholar] [CrossRef]
- Hunter, M.A.; Fischer, J.M.T.A.; Yuan, Q.; Hankel, M.; Searles, D.J. Evaluating the Catalytic Efficiency of Paired, Single-Atom Catalysts for the Oxygen Reduction Reaction. ACS Catal. 2019, 9, 7660–7667. [Google Scholar] [CrossRef]
- Reddy, S.N.; Krishnamurthy, C.B.; Grinberg, I. First-Principles Study of the Ligand Substituent Effect on ORR Catalysis by Metallocorroles. J. Phys. Chem. C 2020, 124, 11275–11283. [Google Scholar] [CrossRef]
- Xu, J.; Liu, B. Intrinsic properties of nitrogen-rich carbon nitride for oxygen reduction reaction. Appl. Surf. Sci. 2020, 500, 144020. [Google Scholar] [CrossRef]
- Xu, J.; Elangovan, A.; Li, J.; Liu, B. Graphene-Based Dual-Metal Sites for Oxygen Reduction Reaction: A Theoretical Study. J. Phys. Chem. C 2021, 125, 2334–2344. [Google Scholar] [CrossRef]
- Elangovan, A.; Xu, J.; Brown, E.; Liu, B.; Li, J. Fundamental Electrochemical Insights of Vertically Aligned Carbon Nanofiber Architecture as a Catalyst Support for ORR. J. Electrochem. Soc. 2020, 167, 066523. [Google Scholar] [CrossRef]
- Elangovan, A.; Xu, J.; Sekar, A.; Rajendran, S.; Liu, B.; Li, J. Platinum Deposited Nitrogen-Doped Vertically Aligned Carbon Nanofibers as Methanol Tolerant Catalyst for Oxygen Reduction Reaction with Improved Durability. Appl. Nano 2021, 2, 303–318. [Google Scholar] [CrossRef]
- Abidi, N.; Lim, K.R.G.; Seh, Z.W.; Steinmann, S.N. Atomistic modeling of electrocatalysis: Are we there yet? WIREs Comput. Mol. Sci. 2020, 11, e1499. [Google Scholar] [CrossRef]
- Hutchison, P.; Rice, P.S.; Warburton, R.E.; Raugei, S.; Hammes-Schiffer, S. Multilevel Computational Studies Reveal the Importance of Axial Ligand for Oxygen Reduction Reaction on Fe–N–C Materials. J. Am. Chem. Soc. 2022, 144, 16524–16534. [Google Scholar] [CrossRef] [PubMed]
- Jónsson, H.; Mills, G.; Jacobsen, K.W. Nudged elastic band method for finding minimum energy paths of transitions. In Classical and Quantum Dynamics in Condensed Phase Simulations; WORLD SCIENTIFIC: Singapore; River Edge, NJ, USA, 1998; pp. 385–404. [Google Scholar]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Henkelman, G.; Jónsson, H. A dimer method for finding saddle points on high dimensional potential surfaces using only first derivatives. J. Chem. Phys. 1999, 111, 7010–7022. [Google Scholar] [CrossRef]
- Zeng, F.; Tenn, W.J., III; Aki, S.N.; Xu, J.; Liu, B.; Hohn, K.L. Influence of basicity on 1,3-butadiene formation from catalytic 2,3-butanediol dehydration over gamma-alumina. J. Catal. 2016, 344, 77–89. [Google Scholar] [CrossRef]
- Manavi, N.; Liu, B. Mitigating Coke Formations for Dry Reforming of Methane on Dual-Site Catalysts: A Microkinetic Modeling Study. J. Phys. Chem. C 2023, 127, 2274–2284. [Google Scholar] [CrossRef]
- Vera, C.Y.R.; Manavi, N.; Zhou, Z.; Wang, L.-C.; Diao, W.; Karakalos, S.; Liu, B.; Stowers, K.J.; Zhou, M.; Luo, H.; et al. Mechanistic understanding of support effect on the activity and selectivity of indium oxide catalysts for CO2 hydrogenation. Chem. Eng. J. 2021, 426, 131767. [Google Scholar] [CrossRef]
- Liu, B.; Manavi, N.; Deng, H.; Huang, C.; Shan, N.; Chikan, V.; Pfromm, P. Activation of N2 on Manganese Nitride-Supported Ni3 and Fe3 Clusters and Relevance to Ammonia Formation. J. Phys. Chem. Lett. 2021, 12, 6535–6542. [Google Scholar] [CrossRef]
- Cheng, J.; Hu, P.; Ellis, P.; French, S.; Kelly, G.; Lok, C.M. Bronsted-Evans-Polanyi relation of multistep reactions and volcano curve in heterogeneous catalysis. J. Phys. Chem. C 2008, 112, 1308–1311. [Google Scholar] [CrossRef]
- Bligaard, T.; Nørskov, J.K.; Dahl, S.; Matthiesen, J.; Christensen, C.H.; Sehested, J. The Bronsted-Evans-Polanyi relation and the volcano curve in heterogeneous catalysis. J. Catal. 2004, 224, 206–217. [Google Scholar] [CrossRef]
- Nørskov, J.; Bligaard, T.; Logadottir, A.; Bahn, S.; Hansen, L.; Bollinger, M.; Bengaard, H.; Hammer, B.; Sljivancanin, Z.; Mavrikakis, M.; et al. Universality in Heterogeneous Catalysis. J. Catal. 2002, 209, 275–278. [Google Scholar] [CrossRef]
- Liu, C.; Cundari, T.R.; Wilson, A.K. CO2 Reduction on Transition Metal (Fe, Co, Ni, and Cu) Surfaces: In Comparison with Homogeneous Catalysis. J. Phys. Chem. C 2012, 116, 5681–5688. [Google Scholar] [CrossRef]
- Medford, A.J.; Shi, C.; Hoffmann, M.J.; Lausche, A.C.; Fitzgibbon, S.R.; Bligaard, T.; Nørskov, J.K. CatMAP: A Software Package for Descriptor-Based Microkinetic Mapping of Catalytic Trends. Catal. Lett. 2015, 145, 794–807. [Google Scholar] [CrossRef]
- Medasani, B.; Kasiraju, S.; Vlachos, D.G. OpenMKM: An Open-Source C plus plus Multiscale Modeling Simulator for Homogeneous and Heterogeneous Catalytic Reactions. J. Chem. Inf. Model. 2023, 63, 3227–3628. [Google Scholar] [CrossRef] [PubMed]
- Takigawa, I.; Shimizu, K.-I.; Tsuda, K.; Takakusagi, S. Machine-learning prediction of the d-band center for metals and bimetals. RSC Adv. 2016, 6, 52587–52595. [Google Scholar] [CrossRef]
- Ando, F.; Tanabe, T.; Gunji, T.; Kaneko, S.; Takeda, T.; Ohsaka, T.; Matsumoto, F. Effect of the d-Band Center on the Oxygen Reduction Reaction Activity of Electrochemically Dealloyed Ordered Intermetallic Platinum–Lead (PtPb) Nanoparticles Supported on TiO2-Deposited Cup-Stacked Carbon Nanotubes. ACS Appl. Nano Mater. 2018, 1, 2844–2850. [Google Scholar] [CrossRef]
- Ando, F.; Gunji, T.; Tanabe, T.; Fukano, I.; Abruña, H.D.; Wu, J.; Ohsaka, T.; Matsumoto, F. Enhancement of the Oxygen Reduction Reaction Activity of Pt by Tuning Its d-Band Center via Transition Metal Oxide Support Interactions. ACS Catal. 2021, 11, 9317–9332. [Google Scholar] [CrossRef]
- Tang, F.; Wang, L.; Walle, M.D.; Mustapha, A.; Liu, Y.-N. An alloy chemistry strategy to tailoring the d-band center of Ni by Cu for efficient and selective catalytic hydrogenation of furfural. J. Catal. 2020, 383, 172–180. [Google Scholar] [CrossRef]
- Ma, X.; Li, Z.; Achenie, L.E.K.; Xin, H. Machine-Learning-Augmented Chemisorption Model for CO2 Electroreduction Catalyst Screening. J. Phys. Chem. Lett. 2015, 6, 3528–3533. [Google Scholar] [CrossRef]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Sanville, E.; Kenny, S.D.; Smith, R.; Henkelman, G. Improved grid-based algorithm for Bader charge allocation. J. Comput. Chem. 2007, 28, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Sanville, E.; Henkelman, G. A grid-based Bader analysis algorithm without lattice bias. J. Phys. Condens. Matter 2009, 21, 084204. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R. Natural Bond Orbitals and Extensions of localized Bonding Concepts. Chem. Educ. Res. Pract. 2001, 2, 91–104. [Google Scholar] [CrossRef]
- Landis, C.R.; Weinhold, F. The NBO View of Chemical Bonding. In The Chemical Bond; Wiley: Weinheim, Germany, 2014; pp. 91–120. [Google Scholar]
- Weinhold, F.; Landis, C.; Glendening, E. What is NBO analysis and how is it useful? Int. Rev. Phys. Chem. 2016, 35, 399–440. [Google Scholar] [CrossRef]
- Deringer, V.L.; Tchougréeff, A.L.; Dronskowski, R. Crystal Orbital Hamilton Population (COHP) Analysis as Projected from Plane-Wave Basis Sets. J. Phys. Chem. A. 2011, 115, 5461–5466. [Google Scholar] [CrossRef]
- Steinberg, S.; Dronskowski, R. The Crystal Orbital Hamilton Population (COHP) Method as a Tool to Visualize and Analyze Chemical Bonding in Intermetallic Compounds. Crystals 2018, 8, 225. [Google Scholar] [CrossRef]
- Abdelgaid, M.; Mpourmpakis, G. Structure–Activity Relationships in Lewis Acid–Base Heterogeneous Catalysis. Acs. Catal. 2022, 12, 4268–4289. [Google Scholar] [CrossRef]
- Larabi, C.; Norsic, S.; Khrouz, L.; Boyron, O.; Szeto, K.C.; Lucas, C.; Taoufik, M.; De Mallmann, A. Oxide-Supported Titanium Catalysts: Structure–Activity Relationship in Heterogeneous Catalysis, with the Choice of Support as a Key Step. Organometallics 2020, 39, 4608–4617. [Google Scholar] [CrossRef]
- Muylaert, I.; Van Der Voort, P. Supported vanadium oxide in heterogeneous catalysis: Elucidating the structure–activity relationship with spectroscopy. Phys. Chem. Chem. Phys. 2009, 11, 2826–2832. [Google Scholar] [CrossRef] [PubMed]
- Boronat, M.; Concepción, P.; Corma, A. Unravelling the Nature of Gold Surface Sites by Combining IR Spectroscopy and DFT Calculations. Implications in Catalysis. J. Phys. Chem. C 2009, 113, 16772–16784. [Google Scholar] [CrossRef]
- Bauer, T.; Maisel, S.; Blaumeiser, D.; Vecchietti, J.; Taccardi, N.; Wasserscheid, P.; Bonivardi, A.; Görling, A.; Libuda, J. Operando DRIFTS and DFT Study of Propane Dehydrogenation over Solid- and Liquid-Supported GaxPty Catalysts. Acs. Catal. 2019, 9, 2842–2853. [Google Scholar] [CrossRef]
- Kargar, H.; Fallah-Mehrjardi, M.; Behjatmanesh-Ardakani, R.; Munawar, K.S. Synthesis, spectra (FT-IR, NMR) investigations, DFT, FMO, MEP, NBO analysis and catalytic activity of MoO2(VI) complex with ONO tridentate hydrazone Schiff base ligand. J. Mol. Struct. 2021, 1245, 131259. [Google Scholar] [CrossRef]
- Dreimann, J.M.; Kohls, E.; Warmeling, H.F.W.; Stein, M.; Guo, L.F.; Garland, M.; Dinh, T.N.; Vorholt, A.J. In Situ Infrared Spectroscopy as a Tool for Monitoring Molecular Catalyst for Hydroformylation in Continuous Processes. ACS Catal. 2019, 9, 4308–4319. [Google Scholar] [CrossRef]
- Artyushkova, K.; Kiefer, B.; Halevi, B.; Knop-Gericke, A.; Schlogl, R.; Atanassov, P. Density functional theory calculations of XPS binding energy shift for nitrogen-containing graphene-like structures. Chem. Commun. 2013, 49, 2539–2541. [Google Scholar] [CrossRef]
- Giesbers, M.; Marcelis, A.T.M.; Zuilhof, H. Simulation of XPS C1s Spectra of Organic Monolayers by Quantum Chemical Methods. Langmuir 2013, 29, 4782–4788. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Greeley, J. Characterization of oxygenated species at water/Pt(111) interfaces from DFT energetics and XPS simulations. Nano Energy 2016, 29, 369–377. [Google Scholar] [CrossRef]
- Beale, A.M.; Weckhuysen, B.M. EXAFS as a tool to interrogate the size and shape of mono and bimetallic catalyst nanoparticles. Phys. Chem. Chem. Phys. 2010, 12, 5562–5574. [Google Scholar] [CrossRef]
- Wang, H.; Liao, J.; Zhong, J.; Liu, Y.; Yao, L.; Qian, L.; Lai, Y.; Wang, X.; Li, Y.; Song, J.; et al. Evolution of Ni coordination configuration during one-pot pyrolysis synthesis of Ni-g-C3N4 single atom catalyst. Carbon 2023, 214, 118348. [Google Scholar] [CrossRef]
- Grundner, S.; Markovits, M.A.; Li, G.; Tromp, M.; Pidko, E.A.; Hensen, E.J.; Jentys, A.; Sanchez-Sanchez, M.; Lercher, J.A. Single-site trinuclear copper oxygen clusters in mordenite for selective conversion of methane to methanol. Nat. Commun. 2015, 6, 7546. [Google Scholar] [CrossRef]
- Pankin, I.A.; Martini, A.; Lomachenko, K.A.; Soldatov, A.V.; Bordiga, S.; Borfecchia, E. Identifying Cu-oxo species in Cu-zeolites by XAS: A theoretical survey by DFT-assisted XANES simulation and EXAFS wavelet transform. Catal. Today 2020, 345, 125–135. [Google Scholar] [CrossRef]
- Timoshenko, J.; Cuenya, B.R. In Situ/Operando Electrocatalyst Characterization by X-ray Absorption Spectroscopy. Chem. Rev. 2020, 121, 882–961. [Google Scholar] [CrossRef]
- Wang, M.; Árnadóttir, L.; Xu, Z.J.; Feng, Z. In Situ X-ray Absorption Spectroscopy Studies of Nanoscale Electrocatalysts. Nano-Micro Lett. 2019, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Huang, Z.; Liu, W.; Chang, C.; Tang, H.; Li, Z.; Chen, W.; Jia, C.; Yao, T.; Wei, S.; et al. Design of N-Coordinated Dual-Metal Sites: A Stable and Active Pt-Free Catalyst for Acidic Oxygen Reduction Reaction. J. Am. Chem. Soc. 2017, 139, 17281–17284. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.N.; Li, J.; Xi, S.B.; Du, Y.H.; Hai, X.; Wang, J.Y.; Xu, H.M.; Wu, G.; Zhang, J.; Lu, J.; et al. A Graphene-Supported Single-Atom FeN5 Catalytic Site for Efficient Electrochemical CO2 Reduction. Angew. Chem. Int. Edit. 2019, 58, 14871–14876. [Google Scholar] [CrossRef]
- Zandkarimi, B.; Sun, G.; Halder, A.; Seifert, S.; Vajda, S.; Sautet, P.; Alexandrova, A.N. Interpreting the Operando XANES of Surface-Supported Subnanometer Clusters: When Fluxionality, Oxidation State, and Size Effect Fight. J. Phys. Chem. C 2020, 124, 10057–10066. [Google Scholar] [CrossRef]
- Chen, Y.; Rana, R.; Sours, T.; Vila, F.D.; Cao, S.; Blum, T.; Hong, J.; Hoffman, A.S.; Fang, C.-Y.; Huang, Z.; et al. A Theory-Guided X-ray Absorption Spectroscopy Approach for Identifying Active Sites in Atomically Dispersed Transition-Metal Catalysts. J. Am. Chem. Soc. 2021, 143, 20144–20156. [Google Scholar] [CrossRef]
- Trummer, D.; Searles, K.; Algasov, A.; Guda, S.A.; Soldatov, A.V.; Ramanantoanina, H.; Safonova, O.V.; Guda, A.A.; Coperet, C. Deciphering the Phillips Catalyst by Orbital Analysis and Supervised Machine Learning from Cr Pre-edge XANES of Molecular Libraries. J. Am. Chem. Soc. 2021, 143, 7326–7341. [Google Scholar] [CrossRef]
- Zitolo, A.; Goellner, V.; Armel, V.; Sougrati, M.-T.; Mineva, T.; Stievano, L.; Fonda, E.; Jaouen, F. Identification of catalytic sites for oxygen reduction in iron- and nitrogen-doped graphene materials. Nat. Mater. 2015, 14, 937–942. [Google Scholar] [CrossRef]
- Jia, Q.; Ramaswamy, N.; Hafiz, H.; Tylus, U.; Strickland, K.; Wu, G.; Barbiellini, B.; Bansil, A.; Holby, E.F.; Zelenay, P.; et al. Experimental Observation of Redox-Induced Fe–N Switching Behavior as a Determinant Role for Oxygen Reduction Activity. ACS Nano 2015, 9, 12496–12505. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-T.; Hsu, C.-W.; Lee, J.-F.; Pao, C.-W.; Hsu, I.-J. Theoretical Analysis of Fe K-Edge XANES on Iron Pentacarbonyl. ACS Omega 2020, 5, 4991–5000. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, S.; McCormick, L.J.; Conradie, J.; Gagnon, K.J.; Sarangi, R.; Ghosh, A. Electronic Structure of Manganese Corroles Revisited: X-ray Structures, Optical and X-ray Absorption Spectroscopies, and Electrochemistry as Probes of Ligand Noninnocence. Inorg. Chem. 2018, 57, 9656–9669. [Google Scholar] [CrossRef]
- Xu, J.; Patel, P.; Kropf, A.J.; Kaphan, D.; Delferro, M.; Liu, C. Theoretical Investigation of the Hydrogenation of Cyclohexene Catalyzed by Supported Single-Atom Sites on Redox Noninnocent LiMn2O4 and Li2Mn2O4 Surfaces. J. Phys. Chem. C 2024. [Google Scholar] [CrossRef]
- Poths, P.; Sun, G.; Sautet, P.; Alexandrova, A.N. Interpreting the Operando X-ray Absorption Near-Edge Structure of Supported Cu and CuPd Clusters in Conditions of Oxidative Dehydrogenation of Propane: Dynamic Changes in Composition and Size. J. Phys. Chem. C 2022, 126, 1972–1981. [Google Scholar] [CrossRef]
- Guda, A.A.; Guda, S.A.; Martini, A.; Kravtsova, A.N.; Algasov, A.; Bugaev, A.; Kubrin, S.P.; Guda, L.V.; Šot, P.; van Bokhoven, J.A.; et al. Understanding X-ray absorption spectra by means of descriptors and machine learning algorithms. npj Comput. Mater. 2021, 7, 203. [Google Scholar] [CrossRef]
- Liu, Y.; Marcella, N.; Timoshenko, J.; Halder, A.; Yang, B.; Kolipaka, L.; Pellin, M.J.; Seifert, S.; Vajda, S.; Liu, P.; et al. Mapping XANES spectra on structural descriptors of copper oxide clusters using supervised machine learning. J. Chem. Phys. 2019, 151, 164201. [Google Scholar] [CrossRef]
- Mizoguchi, T.; Kiyohara, S. Machine learning approaches for ELNES/XANES. Microscopy 2020, 69, 92–109. [Google Scholar] [CrossRef]
- Gong, Y.; Cao, C.; Shi, W.; Zhang, J.; Deng, J.; Lu, T.; Zhong, D. Modulating the Electronic Structures of Dual-Atom Catalysts via Coordination Environment Engineering for Boosting CO2 Electroreduction. Angew. Chem. Int. Ed. 2022, 61, e202215187. [Google Scholar] [CrossRef]
- Qi, J.; Yang, X.; Pan, P.-Y.; Huang, T.; Yang, X.; Wang, C.-C.; Liu, W. Interface Engineering of Co(OH)2 Nanosheets Growing on the KNbO3 Perovskite Based on Electronic Structure Modulation for Enhanced Peroxymonosulfate Activation. Environ. Sci. Technol. 2022, 56, 5200–5212. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Zhong, L.; Du, C.; Luo, Y.; Zhao, J.; Zheng, Y.; Xu, J.; Ma, J.; Liu, C.; Li, S.; et al. Interfacing Epitaxial Dinickel Phosphide to 2D Nickel Thiophosphate Nanosheets for Boosting Electrocatalytic Water Splitting. ACS Nano 2019, 13, 7975–7984. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wang, J.A.; Klysubun, W.; Wang, G.G.; Sattayaporn, S.; Li, F.; Cai, Y.-W.; Zhang, F.; Yu, J.; Yang, Y. Interfacial electronic structure engineering on molybdenum sulfide for robust dual-pH hydrogen evolution. Nat. Commun. 2021, 12, 5260. [Google Scholar] [CrossRef]
- Jiang, Y.; Yang, L.; Sun, T.; Zhao, J.; Lyu, Z.; Zhuo, O.; Wang, X.; Wu, Q.; Ma, J.; Hu, Z. Significant Contribution of Intrinsic Carbon Defects to Oxygen Reduction Activity. ACS Catal. 2015, 5, 6707–6712. [Google Scholar] [CrossRef]
- Deng, J.; Li, H.; Xiao, J.; Tu, Y.; Deng, D.; Yang, H.; Tian, H.; Li, J.; Ren, P.; Bao, X. Triggering the electrocatalytic hydrogen evolution activity of the inert two-dimensional MoS2 surface via single-atom metal doping. Energy Environ. Sci. 2015, 8, 1594–1601. [Google Scholar] [CrossRef]
- Yang, N.; Li, L.; Li, J.; Ding, W.; Wei, Z. Modulating the oxygen reduction activity of heteroatom-doped carbon catalysts via the triple effect: Charge, spin density and ligand effect. Chem. Sci. 2018, 9, 5795–5804. [Google Scholar] [CrossRef]
- Han, Q.; Ding, J.; Bai, Y.; Li, T.; Ma, J.-Y.; Chen, Y.-X.; Zhou, Y.; Liu, J.; Ge, Q.-Q.; Chen, J.; et al. Carrier Dynamics Engineering for High-Performance Electron-Transport-Layer-free Perovskite Photovoltaics. Chem 2018, 4, 2405–2417. [Google Scholar] [CrossRef]
- Yang, C.; Lu, Y.; Zhang, L.; Kong, Z.; Yang, T.; Tao, L.; Zou, Y.; Wang, S. Defect Engineering on CeO2-Based Catalysts for Heterogeneous Catalytic Applications. Small Struct. 2021, 2, 2100058. [Google Scholar] [CrossRef]
- Li, L.; Chang, X.; Lin, X.; Zhao, Z.-J.; Gong, J. Theoretical insights into single-atom catalysts. Chem. Soc. Rev. 2020, 49, 8156–8178. [Google Scholar] [CrossRef]
- Yang, X.-F.; Wang, A.; Qiao, B.; Li, J.; Liu, J.; Zhang, T. Single-Atom Catalysts: A New Frontier in Heterogeneous Catalysis. Acc. Chem. Res. 2013, 46, 1740–1748. [Google Scholar] [CrossRef]
- Liang, Z.; Guo, W.; Zhao, R.; Qiu, T.; Tabassum, H.; Zou, R. Engineering atomically dispersed metal sites for electrocatalytic energy conversion. Nano Energy 2019, 64, 103917. [Google Scholar] [CrossRef]
- Syed, Z.H.; Kaphan, D.M.; Perras, F.A.; Pruski, M.; Ferrandon, M.S.; Wegener, E.C.; Celik, G.; Wen, J.; Liu, C.; Dogan, F.; et al. Electrophilic Organoiridium(III) Pincer Complexes on Sulfated Zirconia for Hydrocarbon Activation and Functionalization. J. Am. Chem. Soc. 2019, 141, 6325–6337. [Google Scholar] [CrossRef]
- Klet, R.C.; Kaphan, D.M.; Liu, C.; Yang, C.; Kropf, A.J.; Perras, F.A.; Pruski, M.; Hock, A.S.; Delferro, M. Evidence for Redox Mechanisms in Organometallic Chemisorption and Reactivity on Sulfated Metal Oxides. J. Am. Chem. Soc. 2018, 140, 6308–6316. [Google Scholar] [CrossRef]
- Ahmadi, M.; Mistry, H.; Roldan Cuenya, B. Tailoring the Catalytic Properties of Metal Nanoparticles via Support Interactions. J. Phys. Chem. Lett. 2016, 7, 3519–3533. [Google Scholar] [CrossRef]
- Ro, I.; Resasco, J.; Christopher, P. Approaches for Understanding and Controlling Interfacial Effects in Oxide-Supported Metal Catalysts. ACS Catal. 2018, 8, 7368–7387. [Google Scholar] [CrossRef]
- Pan, C.-J.; Tsai, M.-C.; Su, W.-N.; Rick, J.; Akalework, N.G.; Agegnehu, A.K.; Cheng, S.-Y.; Hwang, B.-J. Tuning/exploiting Strong Metal-Support Interaction (SMSI) in Heterogeneous Catalysis. J. Taiwan Inst. Chem. Eng. 2017, 74, 154–186. [Google Scholar] [CrossRef]
- Fujiwara, K.; Okuyama, K.; Pratsinis, S.E. Metal–support interactions in catalysts for environmental remediation. Environ. Sci. Nano 2017, 4, 2076–2092. [Google Scholar] [CrossRef]
- van Deelen, T.W.; Hernández Mejía, C.; de Jong, K.P. Control of metal-support interactions in heterogeneous catalysts to enhance activity and selectivity. Nat. Catal. 2019, 2, 955–970. [Google Scholar] [CrossRef]
- Wan, W.; Nie, X.; Janik, M.J.; Song, C.; Guo, X. Adsorption, Dissociation, and Spillover of Hydrogen over Au/TiO2 Catalysts: The Effects of Cluster Size and Metal-Support Interaction from DFT. J. Phys. Chem. C 2018, 122, 17895–17916. [Google Scholar] [CrossRef]
- Prats, H.; Stamatakis, M. Atomistic and electronic structure of metal clusters supported on transition metal carbides: Implications for catalysis. J. Mater. Chem. A 2021, 10, 1522–1534. [Google Scholar] [CrossRef]
- Han, B.; Yu, B.; Wang, J.; Liu, M.; Gao, G.; Xia, K.; Gao, Q.; Zhou, C. Understanding the electronic metal-support interactions of the supported Ni cluster for the catalytic hydrogenation of ethylene. Mol. Catal. 2021, 511, 111731. [Google Scholar] [CrossRef]
- Liu, J.; Fan, X.; Sun, C.Q.; Zhu, W. DFT Study on Intermetallic Pd–Cu Alloy with Cover Layer Pd as Efficient Catalyst for Oxygen Reduction Reaction. Materials 2017, 11, 33. [Google Scholar] [CrossRef]
- Qian, S.; Chen, Y.; Wang, Y.; Yan, B.; Cheng, Y. Identification of the Intrinsic Active Site in Phase-Pure M1 Catalysts for Oxidation Dehydrogenation of Ethane by Density Functional Theory Calculations. J. Phys. Chem. C 2022, 126, 17536–17543. [Google Scholar] [CrossRef]
- Comer, B.M.; Li, J.; Abild-Pedersen, F.; Bajdich, M.; Winther, K.T. Unraveling Electronic Trends in O* and OH* Surface Adsorption in the MO2 Transition-Metal Oxide Series. J. Phys. Chem. C 2022, 126, 7903–7909. [Google Scholar] [CrossRef]
- Shan, N.; Huang, C.; Lee, R.T.; Manavi, N.; Xu, L.; Chikan, V.; Pfromm, P.H.; Liu, B. Manipulating the Geometric and Electronic Structures of Manganese Nitrides for Ammonia Synthesis. ChemCatChem 2020, 12, 2233–2244. [Google Scholar] [CrossRef]
- Nie, L.; Mei, D.; Xiong, H.; Peng, B.; Ren, Z.; Hernandez, X.I.P.; DeLaRiva, A.; Wang, M.; Engelhard, M.H.; Kovarik, L.; et al. Activation of surface lattice oxygen in single-atom Pt/CeO2 for low-temperature CO oxidation. Science 2019, 363, 1419–1423. [Google Scholar]
- Tang, Y.; Li, Y.; Fung, V.; Jiang, D.-E.; Huang, W.; Zhang, S.; Iwasawa, Y.; Sakata, T.; Nguyen, L.; Zhang, X.; et al. Single rhodium atoms anchored in micropores for efficient transformation of methane under mild conditions. Nat. Commun. 2018, 9, 1231. [Google Scholar] [CrossRef]
- Shan, J.; Li, M.; Allard, L.F.; Lee, S.; Flytzani-Stephanopoulos, M. Mild oxidation of methane to methanol or acetic acid on supported isolated rhodium catalysts. Nature 2017, 551, 605–608. [Google Scholar] [CrossRef]
- Chen, Z.; Zhao, J.; Cabrera, C.R.; Chen, Z. Computational Screening of Efficient Single-Atom Catalysts Based on Graphitic Carbon Nitride (g-C3N4) for Nitrogen Electroreduction. Small Methods 2019, 3, 1800368. [Google Scholar] [CrossRef]
- Choi, C.; Back, S.; Kim, N.-Y.; Lim, J.; Kim, Y.-H.; Jung, Y. Suppression of Hydrogen Evolution Reaction in Electrochemical N2 Reduction Using Single-Atom Catalysts: A Computational Guideline. ACS Catal. 2018, 8, 7517–7525. [Google Scholar] [CrossRef]
- Patel, P.; Wells, R.H.; Kaphan, D.M.; Delferro, M.; Skodje, R.T.; Liu, C. Computational Investigation of the Role of Active Site Heterogeneity for a Supported Organovanadium(III) Hydrogenation Catalyst. ACS Catal. 2021, 11, 7257–7269. [Google Scholar] [CrossRef]
- Yang, Y.; Li, J.; Zhang, C.; Yang, Z.; Sun, P.; Liu, S.; Cao, Q. Theoretical Insights into Nitrogen-Doped Graphene-Supported Fe, Co, and Ni as Single-Atom Catalysts for CO2 Reduction Reaction. J. Phys. Chem. C 2022, 126, 4338–4346. [Google Scholar] [CrossRef]
- Khan, S.A.; Vandervelden, C.A.; Scott, S.L.; Peters, B. Grafting metal complexes onto amorphous supports: From elementary steps to catalyst site populations via kernel regression. React. Chem. Eng. 2020, 5, 66–76. [Google Scholar] [CrossRef]
- Vandervelden, C.A.; Khan, S.A.; Scott, S.L.; Peters, B. Site-averaged kinetics for catalysts on amorphous supports: An importance learning algorithm. React. Chem. Eng. 2019, 5, 77–86. [Google Scholar] [CrossRef]
- Khan, S.A.; Godahewa, S.M.; Wimalasiri, P.N.; Thompson, W.H.; Scott, S.L.; Peters, B. Modeling the Structural Heterogeneity of Vicinal Silanols and Its Effects on TiCl4 Grafting onto Amorphous Silica. Chem. Mater. 2022, 34, 3920–3930. [Google Scholar] [CrossRef]
- Zadeh, A.S.; Khan, S.A.; Vandervelden, C.; Peters, B. Site-Averaged Ab Initio Kinetics: Importance Learning for Multistep Reactions on Amorphous Supports. J. Chem. Theory Comput. 2023, 19, 2873–2886. [Google Scholar] [CrossRef]
- An, S.; Patel, P.; Liu, C.; Skodje, R.T. Computational Aspects of Single-Molecule Kinetics for Coupled Catalytic Cycles: A Spectral Analysis. J. Phys. Chem. A. 2022, 126, 3783–3796. [Google Scholar] [CrossRef]
- An, S.; Patel, P.; Liu, C.; Skodje, R.T. Active Site Engineering via Optimizing the Heterogeneous Support Structure for Single-Atom Catalysis. J. Phys. Chem. C 2023, 127, 16901–16913. [Google Scholar] [CrossRef]
- Wells, R.H.; An, S.; Patel, P.; Liu, C.; Skodje, R.T. Single-Molecule Kinetics of Styrene Hydrogenation on Silica-Supported Vanadium: The Role of Disorder for Single-Atom Catalysts. J. Phys. Chem. C 2021, 125, 20286–20300. [Google Scholar] [CrossRef]
- Sohn, H.; Camacho-Bunquin, J.; Langeslay, R.R.; Leon, P.A.I.-D.; Niklas, J.; Poluektov, O.G.; Liu, C.; Connell, J.G.; Yang, D.; Kropf, J.; et al. Isolated, well-defined organovanadium(iii) on silica: Single-site catalyst for hydrogenation of alkenes and alkynes. Chem. Commun. 2017, 53, 7325–7328. [Google Scholar] [CrossRef]
- Feher, F.J.; Newman, D.A.; Walzer, J.F. Silsesquioxanes as models for silica surfaces. J. Am. Chem. Soc. 1989, 111, 1741–1748. [Google Scholar] [CrossRef]
- Liu, C.; Camacho-Bunquin, J.; Ferrandon, M.; Savara, A.; Sohn, H.; Yang, D.; Kaphan, D.M.; Langeslay, R.R.; Leon, P.A.I.-D.; Liu, S.; et al. Development of activity–descriptor relationships for supported metal ion hydrogenation catalysts on silica. Polyhedron 2018, 152, 73–83. [Google Scholar] [CrossRef]
- Quadrelli, E.A.; Basset, J.-M. On silsesquioxanes’ accuracy as molecular models for silica-grafted complexes in heterogeneous catalysis. Coord. Chem. Rev. 2010, 254, 707–728. [Google Scholar] [CrossRef]
- Das, U.; Zhang, G.; Hu, B.; Hock, A.S.; Redfern, P.C.; Miller, J.T.; Curtiss, L.A. Effect of Siloxane Ring Strain and Cation Charge Density on the Formation of Coordinately Unsaturated Metal Sites on Silica: Insights from Density Functional Theory (DFT) Studies. ACS Catal. 2015, 5, 7177–7185. [Google Scholar] [CrossRef]
- Plascencia, C.; Curtiss, L.A.; Liu, C. Hydrogen Activation by Silica-Supported Metal Ion Catalysts: Catalytic Properties of Metals and Performance of DFT Functionals. J. Phys. Chem. A. 2019, 123, 171–186. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, J.; Lund, C.; Patel, P.; Kim, Y.L.; Liu, C. Recent Advances on Computational Modeling of Supported Single-Atom and Cluster Catalysts: Characterization, Catalyst–Support Interaction, and Active Site Heterogeneity. Catalysts 2024, 14, 224. https://doi.org/10.3390/catal14040224
Xu J, Lund C, Patel P, Kim YL, Liu C. Recent Advances on Computational Modeling of Supported Single-Atom and Cluster Catalysts: Characterization, Catalyst–Support Interaction, and Active Site Heterogeneity. Catalysts. 2024; 14(4):224. https://doi.org/10.3390/catal14040224
Chicago/Turabian StyleXu, Jiayi, Colton Lund, Prajay Patel, Yu Lim Kim, and Cong Liu. 2024. "Recent Advances on Computational Modeling of Supported Single-Atom and Cluster Catalysts: Characterization, Catalyst–Support Interaction, and Active Site Heterogeneity" Catalysts 14, no. 4: 224. https://doi.org/10.3390/catal14040224
APA StyleXu, J., Lund, C., Patel, P., Kim, Y. L., & Liu, C. (2024). Recent Advances on Computational Modeling of Supported Single-Atom and Cluster Catalysts: Characterization, Catalyst–Support Interaction, and Active Site Heterogeneity. Catalysts, 14(4), 224. https://doi.org/10.3390/catal14040224