
Promoted Ru/PrOx Catalysts for Mild Ammonia Synthesis

 , , , and
, , , and
Abstract
:
1. Introduction
2. Results and Discussion
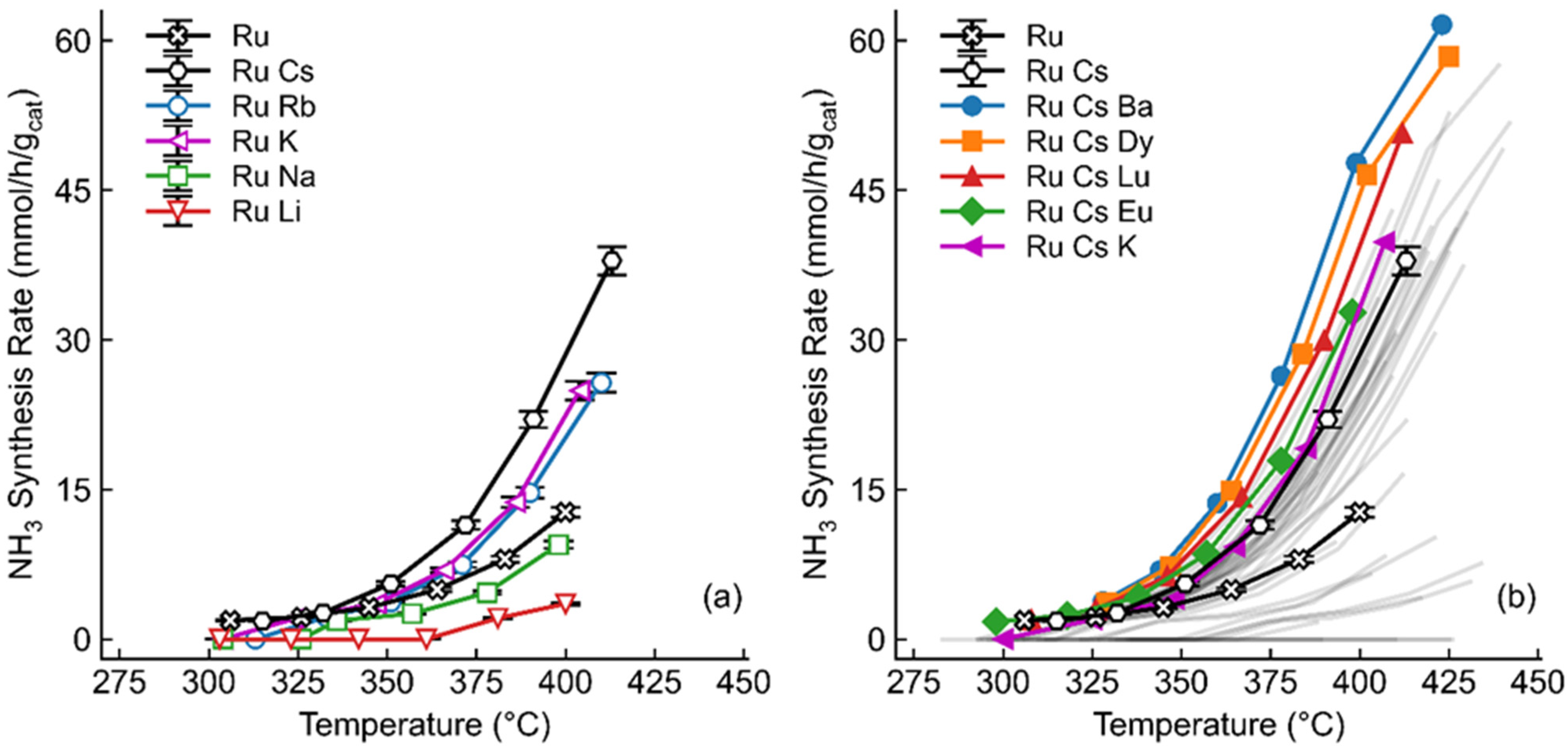
2.1. Screening of Promoters
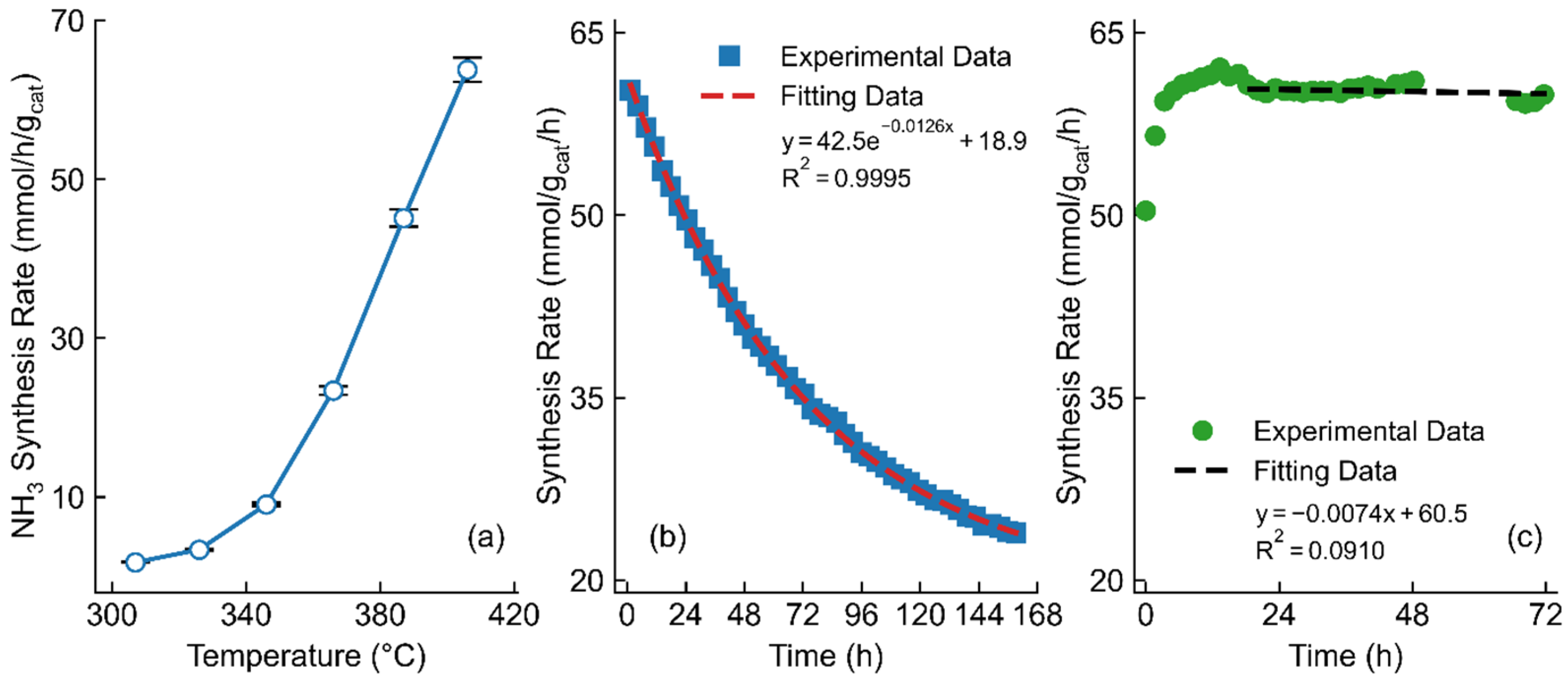
2.2. Optimization of Catalysts
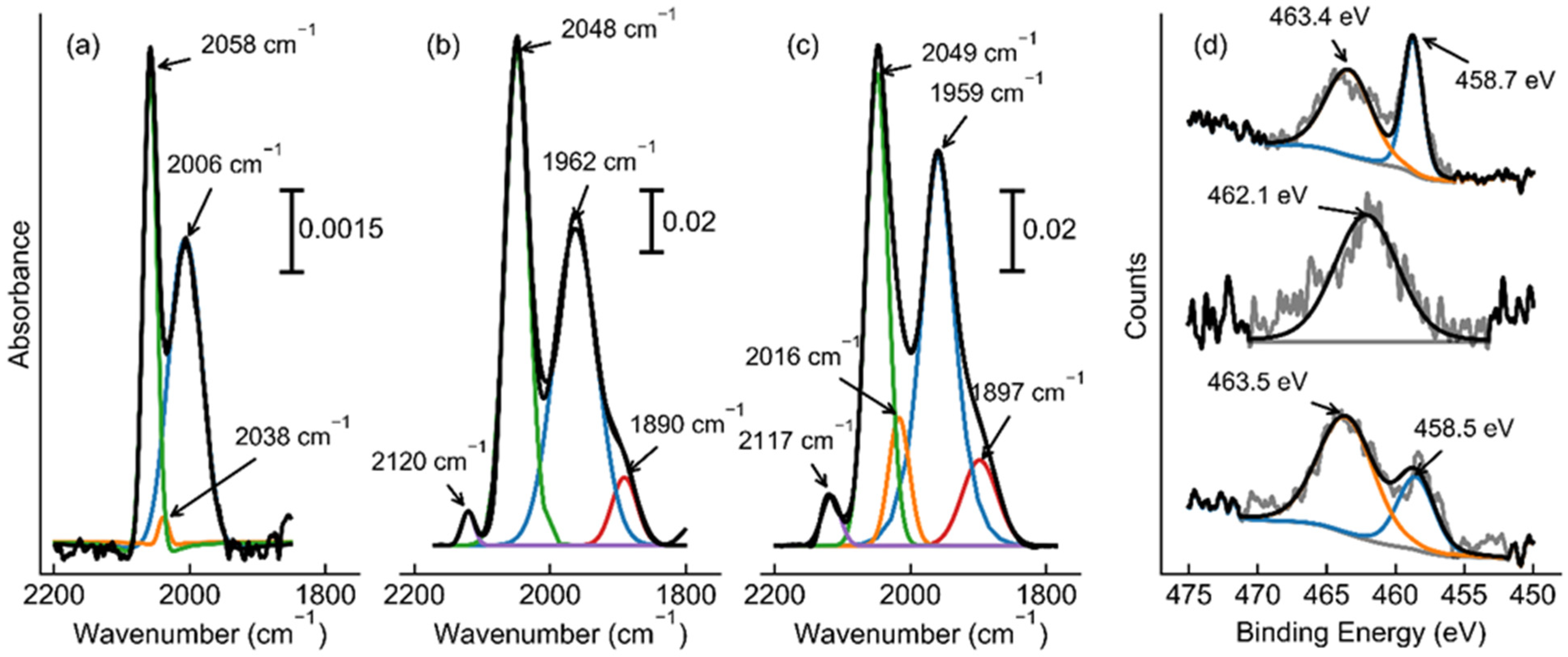
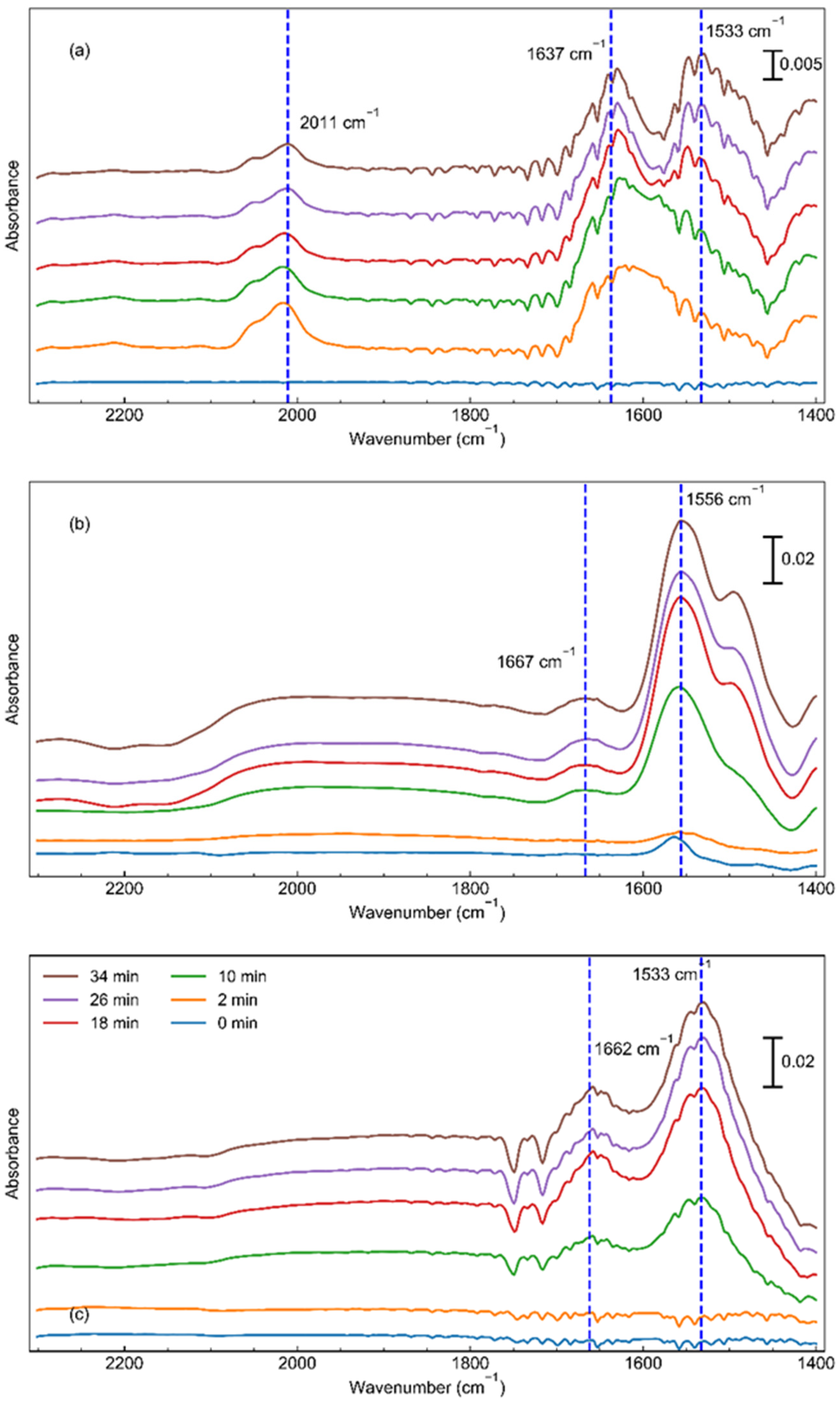
2.3. The Role of the Promoters and the Mechanism
3. Materials and Methods
3.1. Catalyst Preparation
3.2. Catalyst Evaluation
3.3. Catalyst Characterization
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Daisley, A.; Hargreaves, J.S.J.; Hermann, R.; Poya, Y.; Wang, Y. A Comparison of the activities of various supported catalysts for ammonia synthesis. Catal. Today 2020, 357, 534–540. [Google Scholar] [CrossRef]
- Ogawa, T.; Yoshida, M.; Ishihara, K. Techno-economic analysis on recent heterogeneous catalysts for ammonia synthesis. ChemRxiv 2022. [Google Scholar] [CrossRef]
- Knaebel, K.S. Pressure Swing Adsorption System for Ammonia Synthesis. U.S. Patent 5711926A, 14 May 1996. [Google Scholar]
- Bhadra, S.J. Purification of Ammonia by Pressure Swing Adsorption. Ph.D. Thesis, University of South Carolina, Columbia, SC, USA, 2012. [Google Scholar]
- Padinjarekutt, S.; Sengupta, B.; Li, H.; Friedman, K.; Behera, D.; Lecaros, R.; Yu, M. Synthesis of Na+-gated nanochannel membranes for the ammonia (NH3) separation. J. Memb. Sci. 2023, 674, 121512. [Google Scholar] [CrossRef]
- Saadatjou, N.; Jafari, A.; Sahebdelfar, S. Ruthenium nanocatalysts for ammonia synthesis: A review. Chem. Eng. Commun. 2015, 202, 420–448. [Google Scholar] [CrossRef]
- Rosowski, F.; Hornung, A.; Hinrichsen, O.; Herein, D.; Muhler, M.; Ertl, G. Ruthenium Catalysts for ammonia synthesis at high pressures: Preparation, characterization, and power-law kinetics. Appl. Catal. A Gen. 1997, 151, 443–460. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, L.; Wang, J.; Ju, X.; Si, R.; Feng, J.; Guo, J.; Chen, P. The role of lanthanum hydride species in La2O3 supported Ru cluster catalyst for ammonia synthesis. J. Catal. 2023, 417, 382–395. [Google Scholar] [CrossRef]
- Li, C.; Zhang, Z.; Zheng, Y.; Fang, B.; Ni, J.; Lin, J.; Lin, B.; Wang, X.; Jiang, L. Titanium modified Ru/CeO2 catalysts for ammonia synthesis. Chem. Eng. Sci. 2022, 251, 117434. [Google Scholar] [CrossRef]
- Imamura, K.; Miyahara, S.; Kawano, Y.; Sato, K.; Nakasaka, Y.; Nagaoka, K. Kinetics of ammonia synthesis over Ru/Pr2O3. J Taiwan Inst. Chem. Eng. 2019, 105, 50–56. [Google Scholar] [CrossRef]
- Sato, K.; Imamura, K.; Kawano, Y.; Miyahara, S.; Yamamoto, T.; Matsumura, S.; Nagaoka, K. A low-crystalline ruthenium nano-layer supported on praseodymium oxide as an active catalyst for ammonia synthesis. Chem. Sci. 2017, 8, 674–679. [Google Scholar] [CrossRef]
- Li, W.; Wang, S.; Li, J. Highly effective Ru/BaCeO3 catalysts on supports with strong basic sites for ammonia synthesis. Chem. Asian J. 2019, 14, 2815–2821. [Google Scholar] [CrossRef]
- Truszkiewicz, E.; Raróg-Pilecka, W.; Schmidt-Szałowski, K.; Jodzis, S.; Wilczkowska, E.; Łomot, D.; Kaszkur, Z.; Karpiński, Z.; Kowalczyk, Z. Barium-promoted Ru/carbon catalyst for ammonia synthesis: State of the system when operating. J. Catal. 2009, 265, 181–190. [Google Scholar] [CrossRef]
- Osozawa, M.; Hori, A.; Fukai, K.; Honma, T.; Oshima, K.; Satokawa, S. Improvement in ammonia synthesis activity on ruthenium catalyst using ceria support modified a large amount of cesium promoter. Int. J. Hydrogen Energy 2022, 47, 2433–2441. [Google Scholar] [CrossRef]
- Chen, S.Y.; Chang, C.L.; Nishi, M.; Hsiao, W.C.; Reyes, Y.I.A.; Tateno, H.; Chou, H.H.; Yang, C.M.; Chen, H.Y.T.; Mochizuki, T.; et al. Unraveling the active sites of cs-promoted Ru/γ-Al2O3 catalysts for ammonia synthesis. Appl. Catal. B 2022, 310, 121269. [Google Scholar] [CrossRef]
- Aika, K. Role of alkali promoter in ammonia synthesis over ruthenium catalysts—effect on reaction mechanism. Catal. Today 2017, 286, 14–20. [Google Scholar] [CrossRef]
- Wang, Q.; Guo, J.; Chen, P. The impact of alkali and alkaline earth metals on green ammonia synthesis. Chem 2021, 7, 3203–3220. [Google Scholar] [CrossRef]
- Zeinalipour-Yazdi, C.D.; Hargreaves, J.S.J.; Laassiri, S.; Catlow, C.R.A. A Comparative analysis of the mechanisms of ammonia synthesis on various catalysts using density functional theory. R. Soc. Open Sci. 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, L.; Fang, Z.; Zhang, Y.; Ni, J.; Lin, B.; Zheng, L.; Au, C.; Jiang, L. Atomically dispersed Ru catalyst for low-temperature nitrogen activation to ammonia via an associative mechanism. ACS Catal. 2020, 10, 9504–9514. [Google Scholar] [CrossRef]
- Ghoreishian, S.M.; Shariati, K.; Huh, Y.S.; Lauterbach, J. Recent advances in ammonia synthesis over ruthenium single-atom-embedded catalysts: A focused review. Chem. Eng. J. 2023, 467, 143533. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, J.; Zhou, Y.; Au, C.; Wang, X.; Jiang, L. Recent progress of thermocatalytic ammonia synthesis via an associative mechanism. Fundam. Res. 2024; in press. [Google Scholar] [CrossRef]
- Vieri, H.M.; Badakhsh, A.; Choi, S.H. Comparative study of Ba, Cs, K, and Li as promoters for Ru/La2Ce2O7-based catalyst for ammonia synthesis. Int. J. Energy Res. 2023, 2023, 2072245. [Google Scholar] [CrossRef]
- Humphreys, J.; Lan, R.; Tao, S. Development and recent progress on ammonia synthesis catalysts for haber-bosch process. Adv. Energy Sustain. Res. 2021, 2, 2000043. [Google Scholar] [CrossRef]
- Forni, L.; Molinari, D.; Rossetti, I.; Pernicone, N. Carbon-supported promoted Ru catalyst for ammonia synthesis. Appl. Catal. A Gen. 1999, 185, 269–275. [Google Scholar] [CrossRef]
- Aika, K.; Takano, T.; Murato, S. Preparation and characterization of chlorine-free ruthenium catalysts and the promoter effect in ammonia synthesis 3. A magnesia-supported ruthenium catalyst. J. Catal. 1992, 136, 126–140. [Google Scholar] [CrossRef]
- Zheng, J.; Liao, F.; Wu, S.; Jones, G.; Chen, T.; Fellowes, J.; Sudmeier, T.; McPherson, I.J.; Wilkinson, I.; Tsang, S.C.E. Efficient non-dissociative activation of dinitrogen to ammonia over lithium-promoted ruthenium nanoparticles at low pressure. Angew. Chem. Int. Ed. 2019, 58, 17335–17341. [Google Scholar] [CrossRef]
- Lin, B.; Wang, R.; Lin, J.; Ni, J.; Wei, K. Effect of chlorine on the chemisorptive properties and ammonia synthesis activity of alumina-supported Ru catalysts. Catal. Lett. 2011, 141, 1557–1568. [Google Scholar] [CrossRef]
- Zeng, H.S.; Inazu, K.; Aika, K. The working state of the barium promoter in ammonia synthesis over an active-carbon-supported ruthenium catalyst using barium nitrate as the promoter precursor. J. Catal. 2002, 211, 33–41. [Google Scholar] [CrossRef]
- Ren, Y.; Sun, X.; Huang, J.; Zhang, L.; Zhang, B.; Haruta, M.; Lu, A.-H. dual-component sodium and cesium promoters for Au/TS-1: Enhancement of propene epoxidation with hydrogen and oxygen. Ind. Eng. Chem. Res. 2020, 59, 8155–8163. [Google Scholar] [CrossRef]
- Tikhonov, P.A.; Nakusov, A.T.; Popova, I.O.; Konyukhov, G.S. Investigation of the surface structure of dysprosium and praseodymium oxide-based polycrystalline films sensitive to ozone and vapors of ethanol and methanol. Glass Phys. Chem. 2005, 31, 252–258. [Google Scholar] [CrossRef]
- Ogura, Y.; Sato, K.; Miyahara, S.; Kawano, Y.; Toriyama, T.; Yamamoto, T.; Matsumura, S.; Hosokawa, S.; Nagaoka, K. Efficient ammonia synthesis over a Ru/La0.5Ce0.5O1.75 catalyst pre-reduced at high temperature. Chem. Sci. 2018, 9, 2230–2237. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Miyahara, S.; Ogura, Y.; Tsujimaru, K.; Wada, Y.; Toriyama, T.; Yamamoto, T.; Matsumura, S.; Nagaoka, K. Surface dynamics for creating highly active Ru sites for ammonia synthesis: Accumulation of a low-crystalline, oxygen-deficient nanofraction. ACS Sustain. Chem. Eng. 2020, 8, 2726–2734. [Google Scholar] [CrossRef]
- Bardwell, C.J.; Bickley, R.I.; Poulston, S.; Twigg, M.V. Thermal decomposition of bulk and supported barium nitrate. Thermochim. Acta 2015, 613, 94–99. [Google Scholar] [CrossRef]
- Miyajima, K.; Mafuné, F. Thermal decomposition of triruthenium dodecacarbonyl investigated by variable-temperature mass spectrometry in the gas phase. Chem. Phys. Lett. 2022, 786, 139191. [Google Scholar] [CrossRef]
- Zhao, X.; Hrbek, J.; Rodriguez, J.A. The decomposition and chemistry of Ru3(CO)12 on TiO2(110) studied with X-ray photoelectron spectroscopy and temperature programmed desorption. Surf. Sci. 2005, 575, 115–124. [Google Scholar] [CrossRef]
- Meier, D.C.; Rizzi, G.A.; Granozzi, G.; Lai, X.; Goodman, D.W. Ru3(CO)12 adsorption and decomposition on TiO2. Langmuir. 2002, 18, 698–705. [Google Scholar] [CrossRef]
- Ferro, S. Physicochemical and electrical properties of praseodymium oxides. Int. J. Electrochem. 2011, 2011, 561204. [Google Scholar] [CrossRef]
- Javaid, R.; Nanba, T. Stability of Cs/Ru/MgO catalyst for ammonia synthesis as a hydrogen and energy carrier. Energies 2022, 15, 3506. [Google Scholar] [CrossRef]
- Schwegmann, S.; Seitsonen, A.P.; Dietrich, H.; Bludau, H.; Over, H.; Jacobi, K.; Ertl, G. The adsorption of atomic nitrogen. on Ru(0001): Geometry and energetics. Chem. Phys. Lett. 1997, 264, 680–686. [Google Scholar] [CrossRef]
- Peng, X.; Chen, X.; Zhou, Y.; Sun, F.; Zhang, T.; Zheng, L.; Jiang, L.; Wang, X. Size-dependent activity of supported Ru catalysts for ammonia synthesis at mild conditions. J. Catal. 2022, 408, 98–108. [Google Scholar] [CrossRef]
- Baik, Y.; Kwen, M.; Lee, K.; Chi, S.; Lee, S.; Cho, K.; Kim, H.; Choi, M. Splitting of hydrogen atoms into proton–electron pairs at BaO-Ru interfaces for promoting ammonia synthesis under mild conditions. J. Am. Chem. Soc. 2023, 145, 11364–11374. [Google Scholar] [CrossRef]
- Chin, S.Y.; Williams, C.T.; Amiridis, M.D. FTIR Studies of CO adsorption on Al2O3- and SiO2-supported Ru catalysts. J. Phys. Chem. B 2006, 110, 871–882. [Google Scholar] [CrossRef]
- Rini, A.S.; Radiman, S.; Yarmo, M.A. XPS and TPR studies of 2%(Ru 1/2 -Sn 1/2)/Al2O3 catalyst synthesized by microwave techniques. J. Phys. Conf. Ser. 2018, 1120, 012036. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, L.; Feng, J.; Ju, X.; Wang, J.; He, T.; Chen, P. Ru nanoparticles on Pr2O3 as an efficient catalyst for hydrogen production from ammonia decomposition. Catal. Letters 2022, 152, 1170–1181. [Google Scholar] [CrossRef]
- Deng, Y.; Yang, L.; Wang, Y.; Zeng, L.; Yu, J.; Chen, B.; Zhang, X.; Zhou, W. Ruthenium nanoclusters anchored on cobalt phosphide hollow microspheres by green phosphating process for full water splitting in acidic electrolyte. Chin. Chem. Lett. 2021, 32, 511–515. [Google Scholar] [CrossRef]
- Kubota, J.; Aika, K. Infrared studies of adsorbed dinitrogen on supported ruthenium catalysts for ammonia synthesis: Effects of the alumina and magnesia supports and the cesium compound promoter. J. Phys. Chem. 1994, 98, 11293–11300. [Google Scholar] [CrossRef]
- Zimmermann, F.; Lippert, T.; Beyer, C.; Stebani, J.; Nuyken, O.; Wokaun, A. N=N vibrational frequencies and fragmentation patterns of substituted 1-Aryl-3,3-dialkyl-triazenes: Comparison with other high-nitrogen compounds. Appl. Spectrosc. 1993, 47, 986–993. [Google Scholar] [CrossRef]
- Larkin, P.J. General outline for IR and raman spectral interpretation. In Infrared and Raman Spectroscopy; Elsevier Publishing: Stamford, CT, USA, 2018; pp. 135–151. [Google Scholar]
- Rayner-Canham, G.W.; Sutton, D. Identification of ν(N=N) in metal arylazo complexes: The infrared and raman spectra of some arylazo complexes of rhodium(III). Can. J. Chem. 1971, 49, 3994–3996. [Google Scholar] [CrossRef]
- Sun, K.; Zou, X.; Sun, X.; Pang, W.; Hao, X.; Xu, Y.; Su, H.-Y. Structure and reaction condition dependent mechanism for ammonia synthesis on Ru-based catalyst. Appl. Surf. Sci. 2023, 613, 156060. [Google Scholar] [CrossRef]
- Jacobsen, C.J.H.; Dahl, S.; Hansen, P.L.; Törnqvist, E.; Jensen, L.; Topsøe, H.; Prip, D.V.; Møenshaug, P.B.; Chorkendorff, I. Structure sensitivity of supported ruthenium catalysts for ammonia synthesis. J. Mol. Catal. A Chem. 2000, 163, 19–26. [Google Scholar] [CrossRef]
- Logadóttir, Á.; Nørskov, J.K. Ammonia Synthesis over a Ru(0001) surface studied by density functional calculations. J. Catal. 2003, 220, 273–279. [Google Scholar] [CrossRef]
- Li, Q.; Chen, S.; Wang, Y.; Li, K.; Li, M.; Liu, L.; Wang, F. Elucidating the dissociative and associative mechanisms on the surface-anchored Fe3 cluster under the effect of external electric field. Appl. Surf. Sci. 2023, 623, 157021. [Google Scholar] [CrossRef]
- Lee, K.; Woo, R.; Woo, H.C.; Ko, G.; Cho, K.; Park, Y.; Choi, M.; Yoon, H.C. Unraveling the role of MgO in the Ru-Ba/MgO catalyst for boosting ammonia synthesis: Comparative study of MgO and MgAlO supports. J. Catal. 2024, 434, 115530. [Google Scholar] [CrossRef]
- Han, W.; Li, Z.; Liu, H. La2Ce2O7 Supported Ruthenium as a Robust Catalyst for Ammonia Synthesis. J. Rare Earth. 2019, 37, 492–499. [Google Scholar] [CrossRef]
- Lin, B.; Liu, Y.; Heng, L.; Wang, X.; Ni, J.; Lin, J.; Jiang, L. Morphology Effect of Ceria on the Catalytic Performances of Ru/CeO2 Catalysts for Ammonia Synthesis. Ind. Eng. Chem. Res. 2018, 57, 9127–9135. [Google Scholar] [CrossRef]
- Lin, B.; Guo, Y.; Cao, C.; Ni, J.; Lin, J.; Jiang, L. Carbon Support Surface Effects in the Catalytic Performance of Ba-Promoted Ru Catalyst for Ammonia Synthesis. Catal. Today 2018, 316, 230–236. [Google Scholar] [CrossRef]
- Ma, Y.; Lan, G.; Fu, W.; Lai, Y.; Han, W.; Tang, H.; Liu, H.; Li, Y. Role of Surface Defects of Carbon Nanotubes on Catalytic Performance of Barium Promoted Ruthenium Catalyst for Ammonia Synthesis. J. Energy Chem. 2020, 41, 79–86. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Dispersion (%) | Chemisorption Particle Size (nm) | BET Surface Area (m2/g) | Pore Volume (cc/g) | TOFRu (s−1) | TOFSurface Ru (s−1) | Apparent Activation Energy (kJ/mol) |
|---|---|---|---|---|---|---|---|
| 1 wt.% Ru/PrOx | ~100 | <0.9 | 27.1 | 0.745 | 5.95 × 10−2 | 5.95 × 10−2 | 73.9 ± 0.7 |
| 1 wt.% Ru, 2 wt.% Cs/PrOx | 44.2 | 3.0 | 17.6 | 0.199 | 1.63 × 10−1 | 3.69 × 10−1 | 102.3 ± 1.0 |
| 1 wt.% Ru, 4.12 wt.% Cs, 3.86 wt.% Ba/PrOx | 29.3 | 4.6 | 18.1 | 0.361 | 3.06 × 10−1 | 1.04 × 100 | 133.8 ± 1.3 |
| Catalyst | Temperature (°C) | Pressure (bar) | Nitrogen Reaction Order | Hydrogen Reaction Order | Ammonia Reaction Order |
|---|---|---|---|---|---|
| 1 wt.% Ru/PrOx | 400 | 30 | 1.05 | 0.01 | −0.38 |
| 1 wt.% Ru, 2 wt.% Cs/PrOx | 400 | 30 | 1.04 | −0.25 | −0.13 |
| 1 wt.% Ru, 4.12 wt.% Cs, 3.86 wt.% Ba/PrOx | 400 | 30 | 0.82 | 0.25 | −0.59 |
| 1 wt.% Ru/PrOx | 360 | 30 | 0.39 | −0.14 | −0.49 |
| 1 wt.% Ru, 2 wt.% Cs/PrOx | 360 | 30 | 0.66 | −0.63 | −0.28 |
| 1 wt.% Ru, 4.12 wt.% Cs, 3.86 wt.% Ba/PrOx | 360 | 30 | 0.72 | −1.08 | −0.17 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drummond, S.M.; Naglic, J.; Onsree, T.; Balijepalli, S.K.; Allegro, A.; Orraca Albino, S.N.; O’Connell, K.M.; Lauterbach, J. Promoted Ru/PrOx Catalysts for Mild Ammonia Synthesis. Catalysts 2024, 14, 572. https://doi.org/10.3390/catal14090572
Drummond SM, Naglic J, Onsree T, Balijepalli SK, Allegro A, Orraca Albino SN, O’Connell KM, Lauterbach J. Promoted Ru/PrOx Catalysts for Mild Ammonia Synthesis. Catalysts. 2024; 14(9):572. https://doi.org/10.3390/catal14090572
Chicago/Turabian StyleDrummond, Samuel M., Jennifer Naglic, Thossaporn Onsree, Santosh K. Balijepalli, Alexis Allegro, Stephanie N. Orraca Albino, Katherine M. O’Connell, and Jochen Lauterbach. 2024. "Promoted Ru/PrOx Catalysts for Mild Ammonia Synthesis" Catalysts 14, no. 9: 572. https://doi.org/10.3390/catal14090572