

Engineering Xylose Isomerase for Industrial Applications


Abstract

1. Introduction
2. Structure
2.1. XI Structures in Protein Data Bank
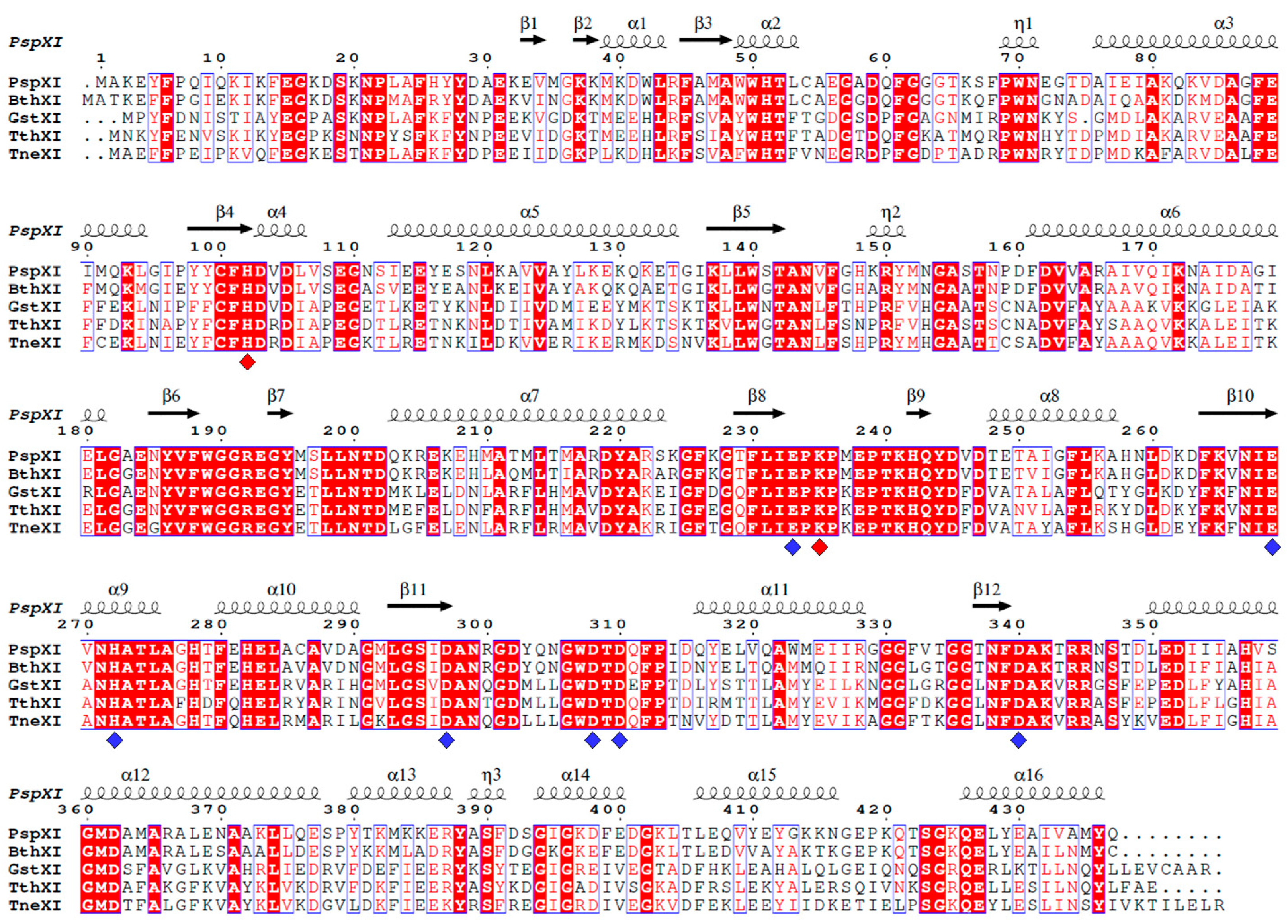
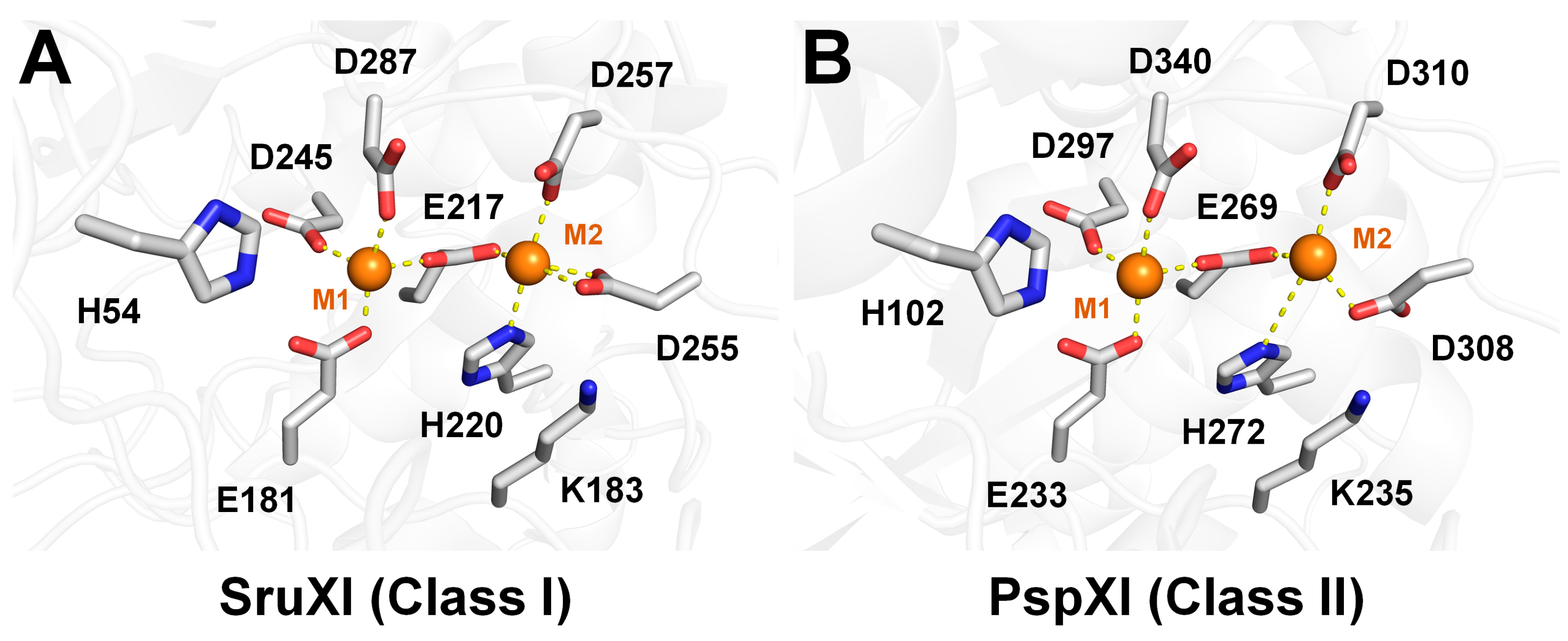
2.2. Structural Analysis of Class I and II XIs
3. Engineering of XI
3.1. GI from Thermoanaerobacterium saccharolyticum
3.2. GI from Geobacillus caldoxylosilyticus TK4
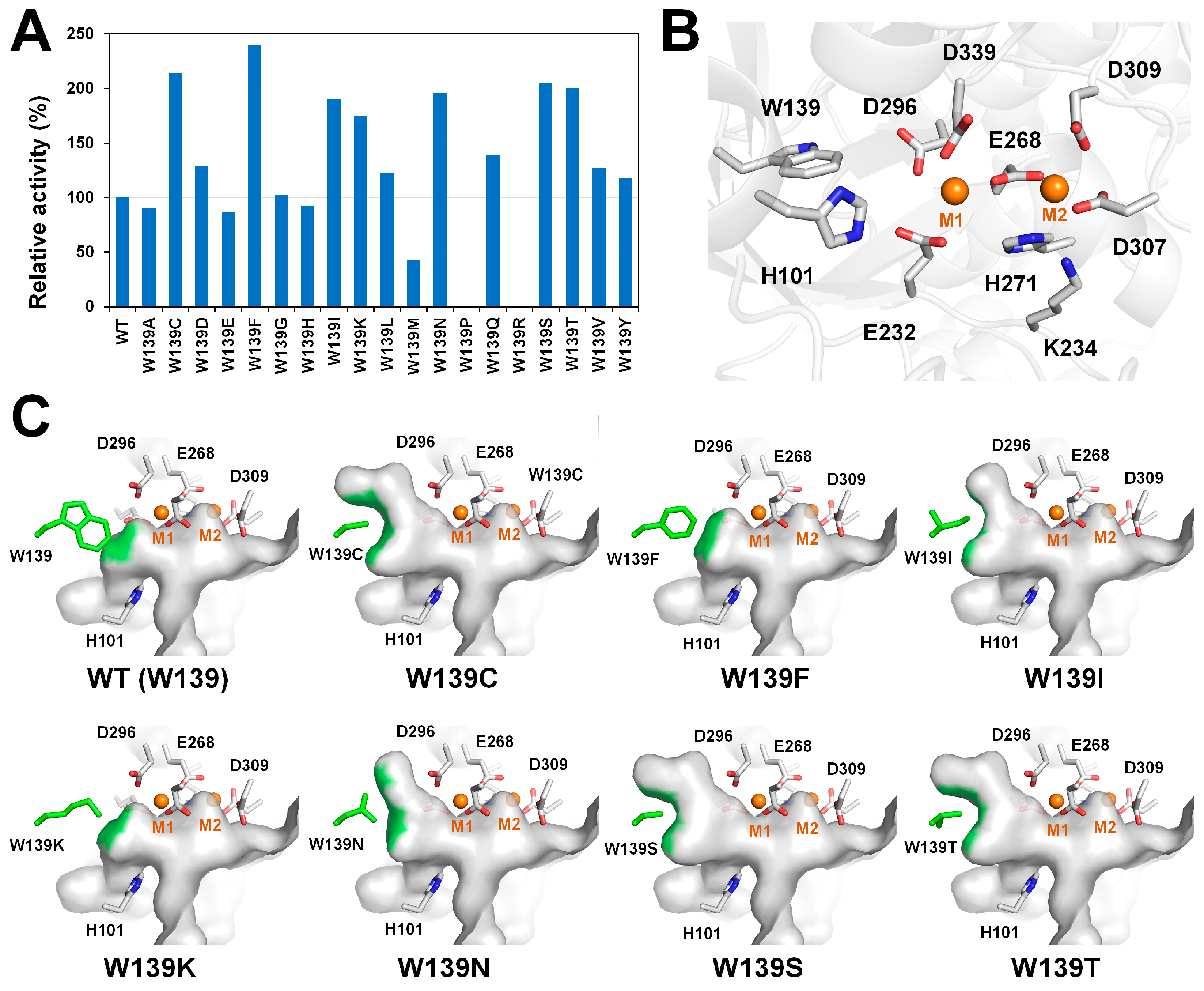
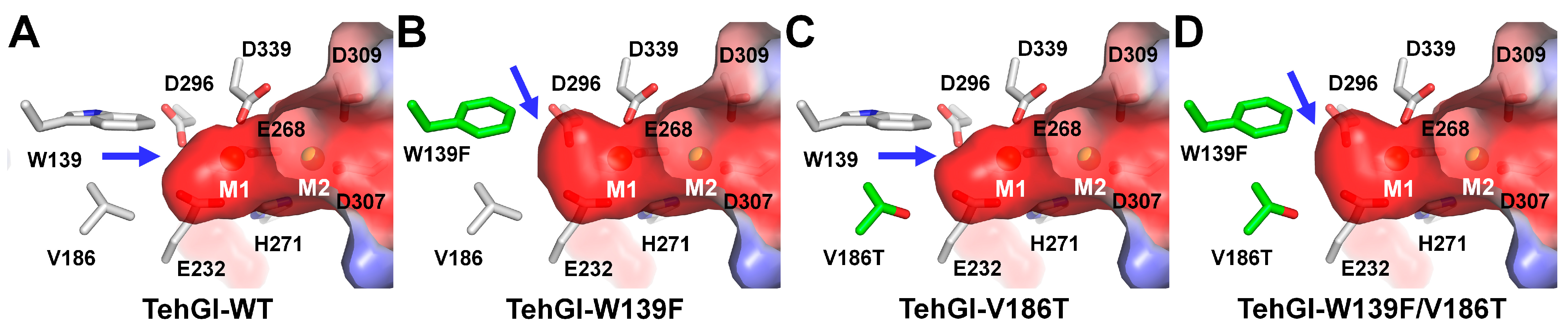
3.3. GI from Thermoanaerobacter ethanolicus
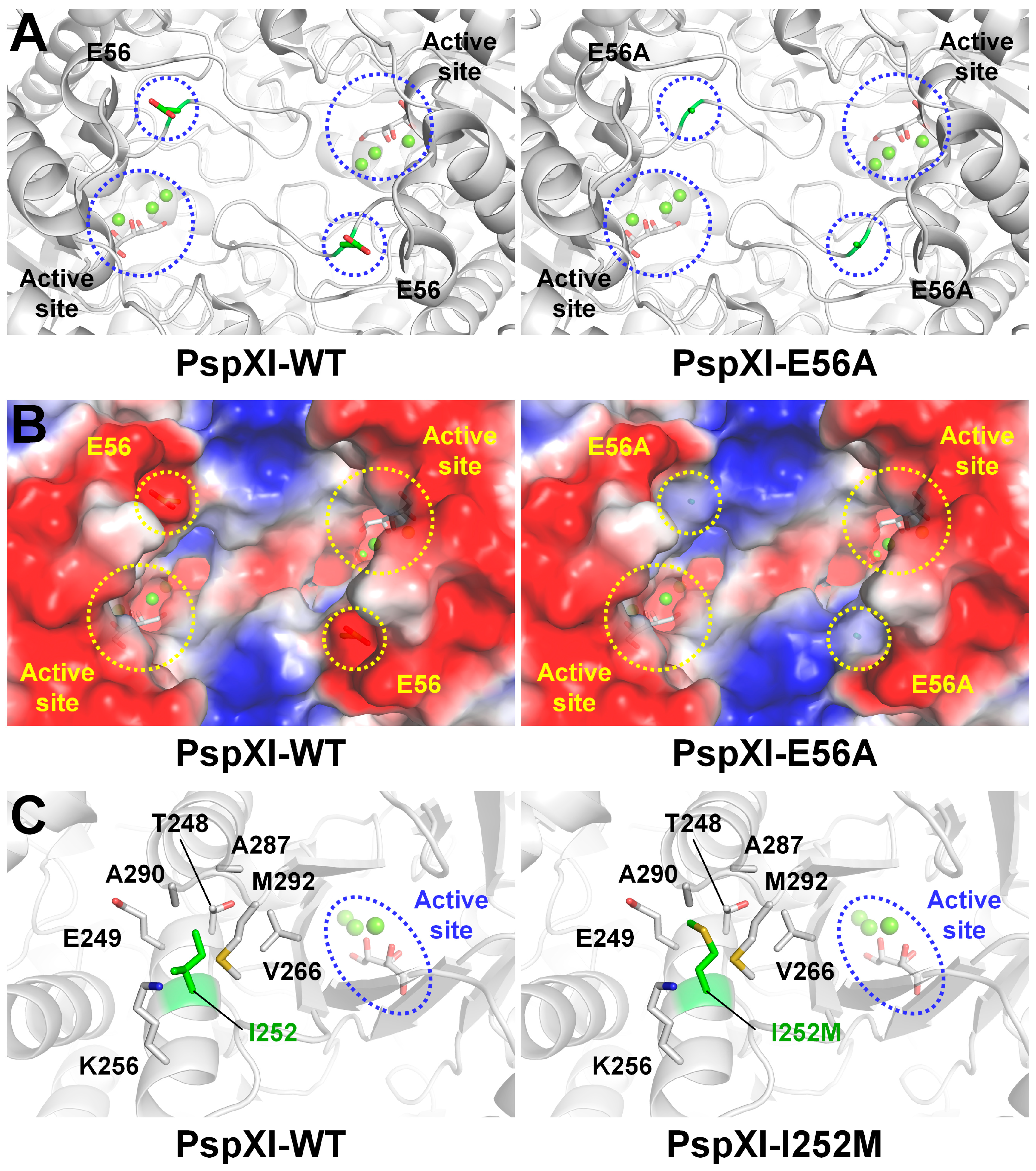
3.4. XI from Piromyces sp. E2
4. Discussion
5. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- DiCosimo, R.; McAuliffe, J.; Poulose, A.J.; Bohlmann, G. Industrial use of immobilized enzymes. Chem. Soc. Rev. 2013, 42, 6437. [Google Scholar] [CrossRef] [PubMed]
- Bhosale, S.H.; Rao, M.B.; Deshpande, V.V. Molecular and industrial aspects of glucose isomerase. Microbiol. Rev. 1996, 60, 280–300. [Google Scholar] [CrossRef] [PubMed]
- Nam, K.H. Glucose Isomerase: Functions, Structures, and Applications. Appl. Sci. 2022, 12, 428. [Google Scholar] [CrossRef]
- Barreto, M.Q.; Garbelotti, C.V.; de Moura Soares, J.; Grandis, A.; Buckeridge, M.S.; Leone, F.A.; Ward, R.J. Xylose isomerase from Piromyces sp. E2 is a promiscuous enzyme with epimerase activity. Enzym. Microb. Technol. 2023, 166, 110230. [Google Scholar] [CrossRef]
- Kasumi, T.; Hayashi, K.; Tsumura, N. Purification and enzymatic properties of glucose isomerase from Streptomyces griseofuscus, S-41. Agric. Biol. Chem. 1981, 45, 619–627. [Google Scholar] [CrossRef]
- Kim, Y.-S.; Kim, D.-Y.; Park, C.-S. Production of l-rhamnulose, a rare sugar, from l-rhamnose using commercial immobilized glucose isomerase. Biocatal. Biotransform. 2017, 36, 417–421. [Google Scholar] [CrossRef]
- Langan, P.; Sangha, A.K.; Wymore, T.; Parks, J.M.; Yang, Z.K.; Hanson, B.L.; Fisher, Z.; Mason, S.A.; Blakeley, M.P.; Forsyth, V.T.; et al. L-Arabinose Binding, Isomerization, and Epimerization by D-Xylose Isomerase: X-Ray/Neutron Crystallographic and Molecular Simulation Study. Structure 2014, 22, 1287–1300. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, J.; Zhang, W.; Zhang, T.; Guang, C.; Mu, W. Recent research on the physiological functions, applications, and biotechnological production of d-allose. Appl. Microbiol. Biotechnol. 2018, 102, 4269–4278. [Google Scholar] [CrossRef]
- Kovalevsky, A.Y.; Hanson, L.; Fisher, S.Z.; Mustyakimov, M.; Mason, S.A.; Trevor Forsyth, V.; Blakeley, M.P.; Keen, D.A.; Wagner, T.; Carrell, H.L.; et al. Metal Ion Roles and the Movement of Hydrogen during Reaction Catalyzed by D-Xylose Isomerase: A Joint X-ray and Neutron Diffraction Study. Structure 2010, 18, 688–699. [Google Scholar] [CrossRef]
- Lee, M.; Rozeboom, H.J.; de Waal, P.P.; de Jong, R.M.; Dudek, H.M.; Janssen, D.B. Metal Dependence of the Xylose Isomerase from Piromyces sp. E2 Explored by Activity Profiling and Protein Crystallography. Biochemistry 2017, 56, 5991–6005. [Google Scholar] [CrossRef]
- Allen, K.N.; Lavie, A.; Glasfeld, A.; Tanada, T.N.; Gerrity, D.P.; Carlson, S.C.; Farber, G.K.; Petsko, G.A.; Ringe, D. Role of the Divalent Metal Ion in Sugar Binding, Ring Opening, and Isomerization by D-Xylose Isomerase: Replacement of a Catalytic Metal by an Amino Acid. Biochemistry 2002, 33, 1488–1494. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.; Bagdasarian, M.; Meng, M.H.; Zeikus, J.G. Catalytic mechanism of xylose (glucose) isomerase from Clostridium thermosulfurogenes. Characterization of the structural gene and function of active site histidine. J. Biol. Chem. 1990, 265, 19082–19090. [Google Scholar] [CrossRef]
- Collyer, C.A.; Henrick, K.; Blow, D.M. Mechanism for aldose-ketose interconversion by D-xylose isomerase involving ring opening followed by a 1,2-hydride shift. J. Mol. Biol. 1990, 212, 211–235. [Google Scholar] [CrossRef] [PubMed]
- Collyer, C.A.; Blow, D.M. Observations of reaction intermediates and the mechanism of aldose-ketose interconversion by D-xylose isomerase. Proc. Natl. Acad. Sci. USA 1990, 87, 1362–1366. [Google Scholar] [CrossRef] [PubMed]
- Asboth, B.; Naray-Szabo, G. Mechanism of Action of D-Xylose Isomerase. Curr. Protein Pept. Sci. 2000, 1, 237–254. [Google Scholar] [CrossRef]
- van Maris, A.J.A.; Winkler, A.A.; Kuyper, M.; de Laat, W.T.A.M.; van Dijken, J.P.; Pronk, J.T. Development of Efficient Xylose Fermentation in Saccharomyces cerevisiae: Xylose Isomerase as a Key Component. In Biofuels; Advances in Biochemical Engineering/Biotechnology; Springer: Berlin/Heidelberg, Germany, 2007; pp. 179–204. [Google Scholar]
- Van Vleet, J.H.; Jeffries, T.W. Yeast metabolic engineering for hemicellulosic ethanol production. Curr. Opin. Biotechnol. 2009, 20, 300–306. [Google Scholar] [CrossRef]
- Kwak, S.; Jin, Y.-S. Production of fuels and chemicals from xylose by engineered Saccharomyces cerevisiae: A review and perspective. Microb. Cell Fact. 2017, 16, 82. [Google Scholar] [CrossRef]
- Hou, J.; Qiu, C.; Shen, Y.; Li, H.; Bao, X. Engineering of Saccharomyces cerevisiae for the efficient co-utilization of glucose and xylose. FEMS Yeast Res. 2017, 17, fox034. [Google Scholar] [CrossRef]
- Hassan, M.E.; Awad, G.E.A.; MohyEldin, M.S.; Haroun, B.M.; El-Diwany, A.I.; Elnashar, M.M. Carbohydrate microcapsules tailored and grafted for covalent immobilization of glucose isomerase for pharmaceutical and food industries. Biotechnol. Lett. 2022, 45, 175–189. [Google Scholar] [CrossRef]
- Visuri, K.; Klibanov, A.M. Enzymatic production of high fructose corn syrup (HFCS) containing 55% fructose in aqueous ethanol. Biotechnol. Bioeng. 2004, 30, 917–920. [Google Scholar] [CrossRef]
- Mohamed, S.A.; Janee, S.; Saha, S.; Sharmin, S.; Hasan, A.Q.F.; Zohora, U.S.; Moni, R.; Islam, M.Z.; Rahman, M.S. Construction and investigation of multi-enzyme immobilized matrix for the production of HFCS. PLoS ONE 2024, 19, e0292931. [Google Scholar] [CrossRef]
- Majore, K.; Ciprovica, I. Bioconversion of Lactose into Glucose–Galactose Syrup by Two-Stage Enzymatic Hydrolysis. Foods 2022, 11, 400. [Google Scholar] [CrossRef] [PubMed]
- Qu, C.; Chen, L.; Fu, H.; Wang, J. Engineering Thermoanaerobacterium aotearoense SCUT27 with argR knockout for enhanced ethanol production from lignocellulosic hydrolysates. Bioresour. Technol. 2020, 310, 123435. [Google Scholar] [CrossRef]
- Vieira, I.P.V.; Cordeiro, G.T.; Gomes, D.E.B.; Melani, R.D.; Vilela, L.F.; Domont, G.B.; Mesquita, R.D.; Eleutherio, E.C.A.; Neves, B.C. Understanding xylose isomerase from Burkholderia cenocepacia: Insights into structure and functionality for ethanol production. AMB Express 2019, 9, 73. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Sahara, T.; Ohgiya, S.; Kamagata, Y.; Fujimori, K.E. Systematic optimization of gene expression of pentose phosphate pathway enhances ethanol production from a glucose/xylose mixed medium in a recombinant Saccharomyces cerevisiae. AMB Express 2018, 8, 139. [Google Scholar] [CrossRef]
- Suzuki, T.; Morimoto, K. Characterization of D-xylose isomerase from Shinella zoogloeoides NN6 and its application for producing D-allulose and two D-ketopentoses in a one-pot multi-step transformation. J. Gen. Appl. Microbiol. 2022, 68, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Nam, K.H. Structural Analysis of Xylose Isomerase from Streptomyces avermitilis. Crystals 2024, 14, 446. [Google Scholar] [CrossRef]
- Silva, P.C.; Ceja-Navarro, J.A.; Azevedo, F.; Karaoz, U.; Brodie, E.L.; Johansson, B. A novel d-xylose isomerase from the gut of the wood feeding beetle Odontotaenius disjunctus efficiently expressed in Saccharomyces cerevisiae. Sci. Rep. 2021, 11, 4766. [Google Scholar] [CrossRef]
- Park, S.-H.; Kwon, S.; Lee, C.W.; Kim, C.M.; Jeong, C.S.; Kim, K.-J.; Hong, J.W.; Kim, H.J.; Park, H.H.; Lee, J.H. Crystal Structure and Functional Characterization of a Xylose Isomerase (PbXI) from the Psychrophilic Soil Microorganism, Paenibacillus sp. J. Microbiol. Biotechnol. 2019, 29, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.E.; Hwang, K.Y.; Nam, K.H. Structural analysis of substrate recognition by glucose isomerase in Mn2+ binding mode at M2 site in S. rubiginosus. Biochem. Biophys. Res. Commun. 2018, 503, 770–775. [Google Scholar] [CrossRef]
- Bae, J.E.; Kim, I.J.; Nam, K.H. Crystal structure of glucose isomerase in complex with xylitol inhibitor in one metal binding mode. Biochem. Biophys. Res. Commun. 2017, 493, 666–670. [Google Scholar] [CrossRef]
- Nam, K.H. Room-Temperature Structure of Xylitol-Bound Glucose Isomerase by Serial Crystallography: Xylitol Binding in the M1 Site Induces Release of Metal Bound in the M2 Site. Int. J. Mol. Sci. 2021, 22, 3892. [Google Scholar] [CrossRef]
- Xu, Y.; Nam, K.H. Xylitol binding to the M1 site of glucose isomerase induces a conformational change in the substrate binding channel. Biochem. Biophys. Res. Commun. 2023, 682, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.-H.; Kim, M.-J.; Sung, B.H.; Jin, Y.-S.; Sohn, J.-H. Directed evolution and secretory expression of xylose isomerase for improved utilisation of xylose in Saccharomyces cerevisiae. Biotechnol. Biofuels 2021, 14, 223. [Google Scholar] [CrossRef] [PubMed]
- Dokuzparmak, C.; Colak, A.; Kolcuoglu, Y.; Akatin, M.Y.; Ertunga, N.S.; Tuncay, F.O. Development of Some Properties of a Thermophilic Recombinant Glucose Isomerase by Mutation. Appl. Biochem. Microbiol. 2020, 56, 164–172. [Google Scholar] [CrossRef]
- Jin, L.-Q.; Jin, Y.-T.; Zhang, J.-W.; Liu, Z.-Q.; Zheng, Y.-G. Enhanced catalytic efficiency and thermostability of glucose isomerase from Thermoanaerobacter ethanolicus via site-directed mutagenesis. Enzym. Microb. Technol. 2021, 152, 109931. [Google Scholar] [CrossRef]
- Liu, Z.-Q.; Zheng, W.; Huang, J.-F.; Jin, L.-Q.; Jia, D.-X.; Zhou, H.-Y.; Xu, J.-M.; Liao, C.-J.; Cheng, X.-P.; Mao, B.-X.; et al. Improvement and characterization of a hyperthermophilic glucose isomerase from Thermoanaerobacter ethanolicus and its application in production of high fructose corn syrup. J. Ind. Microbiol. Biotechnol. 2015, 42, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Shen, D.; Wu, X.-Q.; Liu, Z.-W.; Yang, Q.-H. Characterization of a mutant glucose isomerase from Thermoanaerobacterium saccharolyticum. J. Ind. Microbiol. Biotechnol. 2014, 41, 1581–1589. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Callens, M.; Tomme, P.; Kersters-Hilderson, H.; Cornelis, R.; Vangrysperre, W.; De Bruyne, C.K. Metal ion binding to d-xylose isomerase from Streptomyces violaceoruber. Biochem. J. 1988, 250, 285–290. [Google Scholar] [CrossRef]
- Callens, M.; Kerstershilderson, H.; Vangrysperre, W.; Debruyne, C.K. D-Xylose Isomerase from Streptomyces-Violaceoruber—Structural and Catalytic Roles of Bivalent-Metal Ions. Enzym. Microb. Technol. 1988, 10, 695–700. [Google Scholar] [CrossRef]
- Epting, K.L.; Vieille, C.; Zeikus, J.G.; Kelly, R.M. Influence of divalent cations on the structural thermostability and thermal inactivation kinetics of class II xylose isomerases. FEBS J. 2005, 272, 1454–1464. [Google Scholar] [CrossRef] [PubMed]
- Van Bastelaere, P.; Vangrysperre, W.; Kersters-Hilderson, H. Kinetic studies of Mg2+-, Co2+- and Mn2+-activated D-xylose isomerases. Biochem. J. 1991, 278, 285–292. [Google Scholar] [CrossRef]
- Lee, C.Y.; Zeikus, J.G. Purification and characterization of thermostable glucose isomerase from Clostridium thermosulfurogenes and Thermoanaerobacter strain B6A. Biochem. J. 1991, 273, 565–571. [Google Scholar] [CrossRef]
- Meng, M.; Lee, C.; Bagdasarian, M.; Zeikus, J.G. Switching substrate preference of thermophilic xylose isomerase from D-xylose to D-glucose by redesigning the substrate binding pocket. Proc. Natl. Acad. Sci. USA 1991, 88, 4015–4019. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Wu, M.; Bao, H.; Liu, W.; Shen, Y. Engineering of Saccharomyces cerevisiae for co-fermentation of glucose and xylose: Current state and perspectives. Eng. Microbiol. 2023, 3, 100084. [Google Scholar] [CrossRef]
- Coimbra, L.; Malan, K.; Fagúndez, A.; Guigou, M.; Lareo, C.; Fernández, B.; Pratto, M.; Batista, S. Fermentation of D-xylose to Ethanol by Saccharomyces cerevisiae CAT-1 Recombinant Strains. Bioenerg. Res. 2022, 16, 1001–1012. [Google Scholar] [CrossRef]
- Li, F.; Bai, W.; Zhang, Y.; Zhang, Z.; Zhang, D.; Shen, N.; Yuan, J.; Zhao, G.; Wang, X. Construction of an economical Xylose-utilizing Saccharomyces cerevisiae and its ethanol fermentation. FEMS Yeast Res. 2024, 24, foae001. [Google Scholar] [CrossRef]
- Sriprapundh, D.; Vieille, C.; Zeikus, J.G. Molecular determinants of xylose isomerase thermal stability and activity: Analysis of thermozymes by site-directed mutagenesis. Protein Eng. Des. Sel. 2000, 13, 259–265. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source Organism | Accession No. | PDB Code (Ligand) |
|---|---|---|
| Type I | ||
| Streptomyces rubiginosus | P24300 | 1GW9 (β-L-xylopyranose, Ca2+), 1MNZ (Mg2+), 1O1H (Ca2+, Mg2+), 1OAD (Mn2+, Mg2+), 1XIB (Mn2+), 1XIC (Mn2+, D-xylose), 1XID (Mn2+, ascorbic acid), 1XIE (Mn2+, 1,5-anhydro-D-glucitol), 1XIF (Mn2+, α-D-glucopyranose), 1XIG (Mn2+, xylitol), 1XIH (Mn2+, sorbitol), 1XII (Mn2+, D-xyluose), 1XIJ (Mn2+, threonate ion), 1XIS (Mn2+), 2G4J (Ca2+, Mg2+, Cl−), 2GLK (Mn2+, glycerol), 2GUB (no ligand), 2GVE (Co2+), 2XIS (Mg2+, xylitol), 3CWH (Mg2+, D-xylulose), 3GNX (Mn2+, xylitol), 3KBJ (no ligand), 3KBM (Cd2+, α-D-glucopyranose), 3KBN (Ni2+, D-glucose), 3KBS (Cd2+), 3KBV (Ni2+), 3KBW (Ni2+, Mg2+), 3KCJ (no ligand), 3KCL (Cd2+, α-D-glucopyranose), 3KCO (Ni2+, D-glucose), 3N4A (Mn2+, Cl−), 3QYS (Ni2+), 3QZA (2H+), 3U3H (Mg2+, (2R)-propane-1,1,2,3-tetrol, formic acid, (4R)-2-methylpentane-2,4-diol), 3XIS (Mg2+, α-D-xylopyranose, D-xylose), 4A8I (Co2+, 1,2-ethanediol), 4A8L (Co2+, 1,2-ethanediol), 4A8N (Co2+, glycerol), 4A8R (Co2+, glycerol), 4DUO (Mg2+, xylitol), 4DVO (Ni2+, sorbitol), 4E3V (Mn2+, Mg2+, proline, sulfate), 4J4K (Zn2+, (4S)-2-methyl-2,4-pentanediol, acetate ion), 4LNC (Mn2+, Mg2+, α-D-glucopyranose), 4QDP (Cd2+, β-L-arabinopyranose),4QDW (Ni2+, L-arabinose), 4QE1 (Cd2+, α-L-ribulofuranose), 4QE4 (Ni2+, β-L-ribulofuranose), 4QE5 (Mg2+, α-L-ribulofuranose), 4QEE (Ni2+, α-L-ribulofuranose), 4QEH (Mg2+, β-L-ribofuranose), 4US6 (Mg2+, Ca2+, Na+, glycerol), 4W4Q (Ca2+), 4XIS (Mn2+, D-xylose, α-D-xylopyranose), 4ZB0 (Mn2+, α-D-glucopyranose, β-D-fructofuranose), 4ZB2 (Mn2+, α-D-glucopyranose, β-D-fructofuranose), 4ZB5 (Mn2+, α-D-glucopyranose), 4ZBC (Mn2+, α-D-glucopyranose, β-D-fructofuranose), 5AVH (no ligand), 5AVN (Mn2+, Ca2+, sulfate ion), 5I7G (no ligand), 5VR0 (Mn2+, Cl−, Ca2+, (4R)-2-methylpentane-2,4-diol), 5Y4I (Mg2+, acetate, glycerol), 5Y4J (Mg2+, xylitol), 5ZYC (Mn2+, acetate ion, 1,2-ethanediol), 5ZYD (Mg2+, acetate ion), 5ZYE (Mn2+, α-D-glucopyranose), 6IRK (Mg2+), 6KCA (Mg2+), 6KCC (Mg2+), 6KD2 (Mg2+), 6LL2 (Mg2+), 6OQZ (Mn2+, Mg2+, (4S)-2-methyl-2,4-pentanediol), 6QNC (Mg2+, Co2+, α-D-glucopyranose), 6QND (Mg2+, Co2+, D-glucose), 6QNH (Mg2+, Co2+), 6QNI (Mg2+, Co2+, α-D-glucopyranose), 6QNJ (Mg2+, Co2+, α D-glucopyranose), 6QRR (Mg2+, Mn2+, Na+, 1,2-ethanediol, 2-propanol), 6QRS (Mg2+, Mn2+, Na+, 1,2-ethanediol, 2-propanol), 6QRT (Mg2+, Mn2+, Na+, 1,2-ethanediol, 2-propanol), 6QRU (Mg2+, Mn2+, Na+, 1,2-ethanediol, 2-propanol), 6QRV (Mg2+, Mn2+, Na+, 1,2-ethanediol, 2-propanol), 6QRW (Mg2+, Mn2+, Na+, 1,2-ethanediol, 2-propanol), 6QRX (Mg2+, Mn2+, Na+, 1,2-ethanediol, 2-propanol), 6QRY (Mg2+, Mn2+, Na+, 1,2-ethanediol, 2-propanol), 6QUF (Mn2+, glycerol, sulfate ion), 6QUK (Mn2+, glycerol, sulfate ion), 6RND (Mg2+, α-D-glucopyranose), 6RNF (Mg2+, α-D-glucopyranose), 6VRS (Mn2+), 6YBO (Mg2+, Na+), 6YBR (Mg2+, Na+), 7BJZ (Mn2+), 7BVL (Mg2+), 7BVN (Mg2+), 7CJO (Mg2+, 1,2-ethanediol), 7CJP (1,2-ethanediol), 7CK0 (Mg2+), 7CVK (Mg2+), 7CVM (Mg2+), 7DFJ (Mg2+), 7DFK (Mg2+, xylitol), 7DMM (Mn2+, Ca2+, glycerol), 7E03 (Mg2+), 7NJG (Co2+), 8AW8 (Mg2+, Mn2+, glycerol), 8AW9 (Mg2+, Mn2+, glycerol), 8AWB (Mg2+, Mn2+, glycerol), 8AWC (Mg2+, Mn2+, glycerol), 8AWD (Mg2+, Mn2+, glycerol, di(hydroxyethyl)ether), 8AWE (Mg2+, Mn2+, glycerol), 8AWF (Mg2+, Mn2+, glycerol), 8AWS (Mg2+, Mn2+, β-D-glucopyranose), 8AWU (Mg2+, Mn2+, α-D-glucopyranose), 8AWV (Mg2+, Mn2+, α-D-glucopyranose), 8AWX (Mg2+, Mn2+, β-D-glucopyranose), 8AWY (Mg2+, meso-2,3-butanediol), 8XIA (Mn2+, D-xylose), 9XIA (Mn2+, 3-deoxy-3-methyl-β-D-fructofuranose) |
| Arthrobacter sp. NRRL B3728 | P12070 | 1DID (Mn2+, 2,5-dideoxy-2,5-imino-D-glucitol), 1DIE (Mg2+, 1-deoxynojirimycin), 1XLA (no ligand), 1XLB (Mg2+, xylitol), 1XLD (Mn2+, xylitol), 1XLE (Mn2+), 1XLF (Mn2+, D-gluconic acid), 1XLG (Mg2+, Al3+, xylitol), 1XLH (Al3+), 1XLI (Mn2+, 5-thio-α-D-glucopyranose), 1XLJ (Mn2+, xylitol), 1XLK (Mn2+), 1XLL (Zn2+), 1XLM (Al3+, xylitol), 4XIA (Mg2+, sorbitol), 5XIA (Mg2+, xylitol) |
| Actinoplanes missouriensis | P12851 | 1BHW (no ligand), 1XIM (Co2+, xylitol), 1XIN (Mg2+, xylitol), 2XIM (Mg2+, xylitol), 2XIN (Co2+, xylitol), 3XIM (Co2+, xylitol), 3XIN (no ligand), 4XIM (Co2+), 5XIM (Mg2+, sorbitol), 5XIN (Mg2+, D-xylose), 6XIM (Mg2+, D-xylose), 7XIM (no ligand), 8XIM (Mg2+, D-xylose), 9XIM (Mn2+, D-xylose) |
| Streptomyces olivochromogenes | P15587 | 1MUW (Mn2+, Mg2+, HO−), 1S5M (Mn2+, Na+, α-D-glucopyranose), 1S5N (Mn2+, Na+, HO−, xylitol), 1XYA (Mg2+, HO−), 1XYB (Mg2+, D-glucose), 1XYC (Mg2+, 3-O-methylfructose [linear form]), 1XYL (Mg2+, HO−), 1XYM (Mg2+, HO−, D-glucose), 2GYI (Mg2+, 2,3,4,N-tetrahydroxy-butyrimidic acid) |
| Streptomyces diastaticus | P50910 | 1CLK (Mg2+, Co2+), 1QT1 (Co2+) |
| Streptomyces sp. F-1 | A0A1K2FZ20 | 6N98 (Mg2+, sulfate ion), 6N99 (Mg2+, sulfate ion, (4S)-2-methyl-2,4-pentanediol) |
| Streptomyces sp. SK | Q9ZAI3 | 4HHL (Co2+, Mg2+,1,2-ethanediol), 4HHM (Mg2+, Co2+) |
| Bacteroides thetaiotaomicron VPI-5482 | Q8A9M2 | 4XKM (Mn2+) |
| Geobacillus stearothermophilus | P54273 | 1A0D (Mn2+) |
| Streptomyces albus | P24299 | 6XIA (no ligand) |
| Streptomyces avermitilis | A0A4D4M698 | 8YUD (Mg2+) |
| Streptomyces murinus | P37031 | 1DXI (Mg2+) |
| Thermus caldophilus | P56681 | 1BXC (no ligand) |
| Thermus thermophilus HB8 | P26997 | 1BXB (no ligand) |
| Type II | ||
| Piromyces sp. E2 | Q9P8C9 | 5NH4 (Mg2+, sulfate ion, glycerol), 5NH5 (Ca2+, Fe2+, Mg2+, sulfate ion, glycerol), 5NH6 (Mg2+, xylitol, sulfate ion), 5NH7 (Mg2+,D-xylose, β-D-xylopyranose, α-D-xylopyranose, sulfate ion), 5NH8 (Ca2+, D-xylose, β-D-xylopyranose, sulfate ion), 5NH9 (Mn2+, D-xylose, β-D-xylopyranose, sulfate ion), 5NHA (Mn2+, sorbitol, sulfate ion), 5NHB (Fe2+, sulfate ion), 5NHC (Co2+, D-xylulose, 4-hydroxyproline), 5NHD (Ni2+, D-xylose, β-D-xylopyranose, α-D-xylopyranose, sulfate ion), 5NHE (Cd2+, D-xylose, β-D-xylopyranose, α-D-xylopyranose, sulfate ion), 5NHM (glycerol, sulfate ion, acetic acid), 5YN3 (Mn2+, glycerol), 6T8E (Ca2+, D-xylose, β-D-xylopyranose, α-D-xylopyranose, sulfate ion), 6T8F (Ca2+, D-xylose, β-D-xylopyranose, α-D-xylopyranose, sulfate ion) |
| Bacteroides thetaiotaomicron VPI-5482 | Q8A9M2 | 4XKM (Mn2+) |
| Geobacillus stearothermophilus | P54273 | 1A0D (Mn2+) |
| Thermoanaerobacterium thermosulfurigenes | P19148 | 1A0C (Co2+) |
| Thermotoga neapolitana | P45687 | 1A0E (Co2+) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nam, K.H. Engineering Xylose Isomerase for Industrial Applications. Catalysts 2024, 14, 597. https://doi.org/10.3390/catal14090597
Nam KH. Engineering Xylose Isomerase for Industrial Applications. Catalysts. 2024; 14(9):597. https://doi.org/10.3390/catal14090597
Chicago/Turabian StyleNam, Ki Hyun. 2024. "Engineering Xylose Isomerase for Industrial Applications" Catalysts 14, no. 9: 597. https://doi.org/10.3390/catal14090597
APA StyleNam, K. H. (2024). Engineering Xylose Isomerase for Industrial Applications. Catalysts, 14(9), 597. https://doi.org/10.3390/catal14090597