
Effect of Metal Dispersion in Rh-Based Zeolite and SiO2 Catalysts on the Hydroformylation of Olefin Mixtures from Fischer–Tropsch Synthesis

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results and Discussion
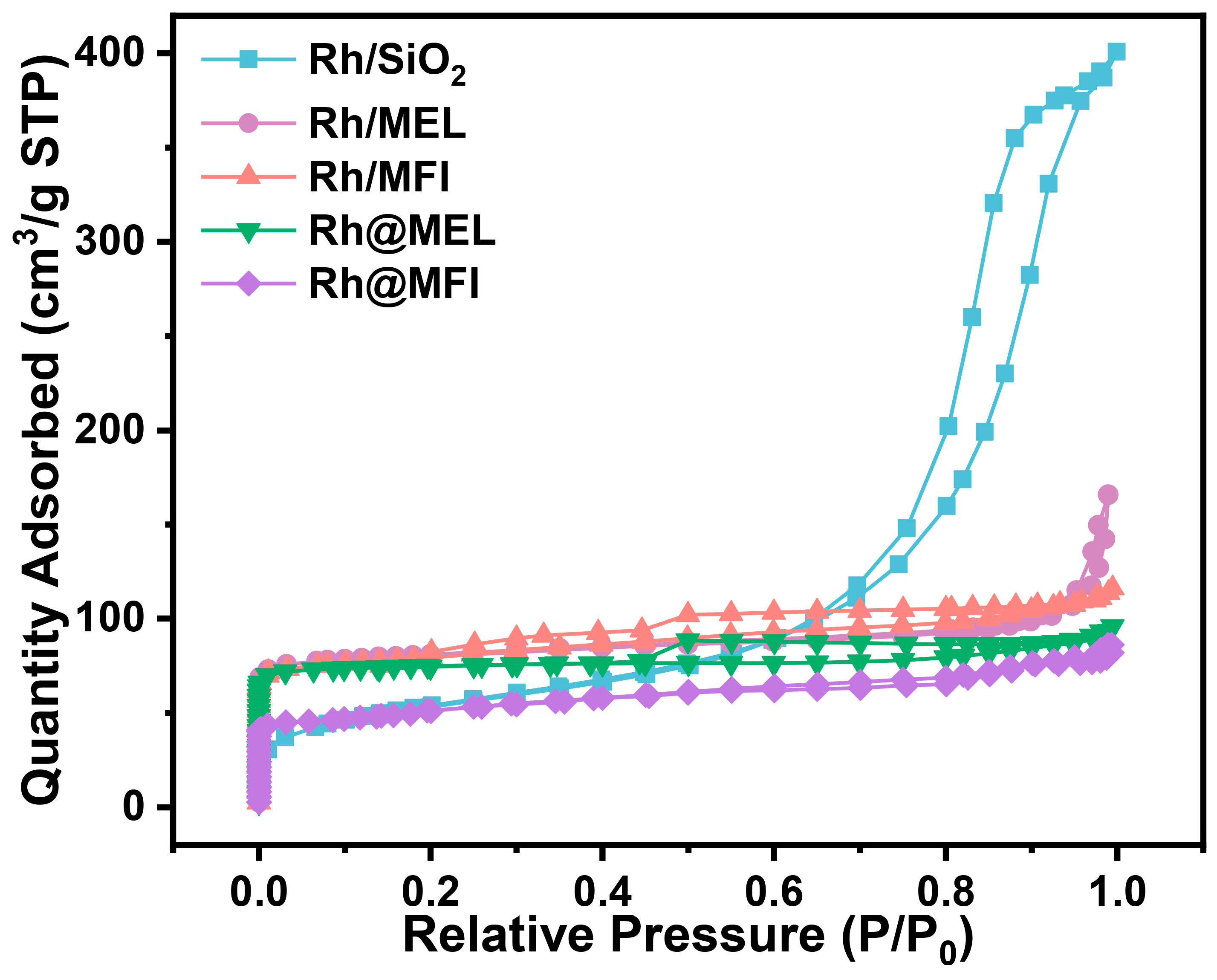
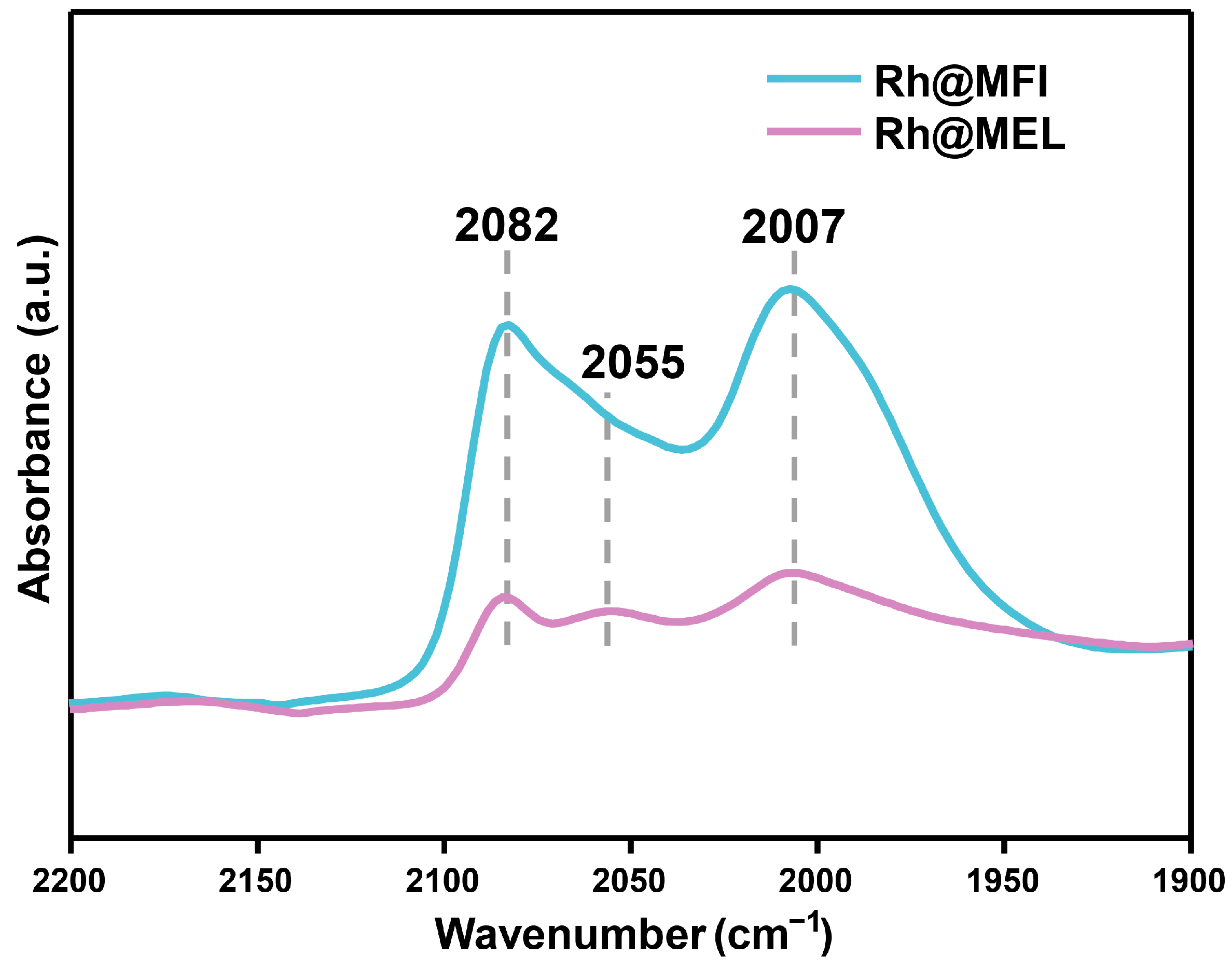
2.1. Catalyst Synthesis and Characterization
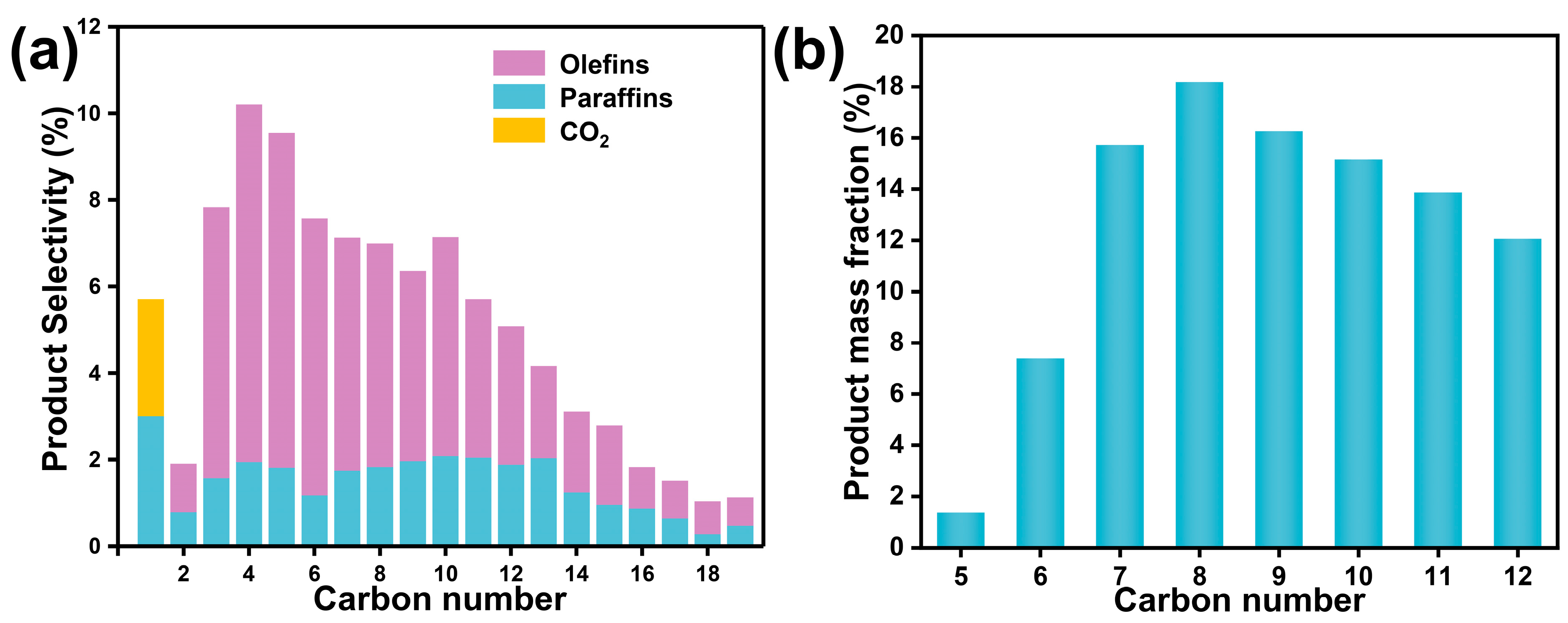
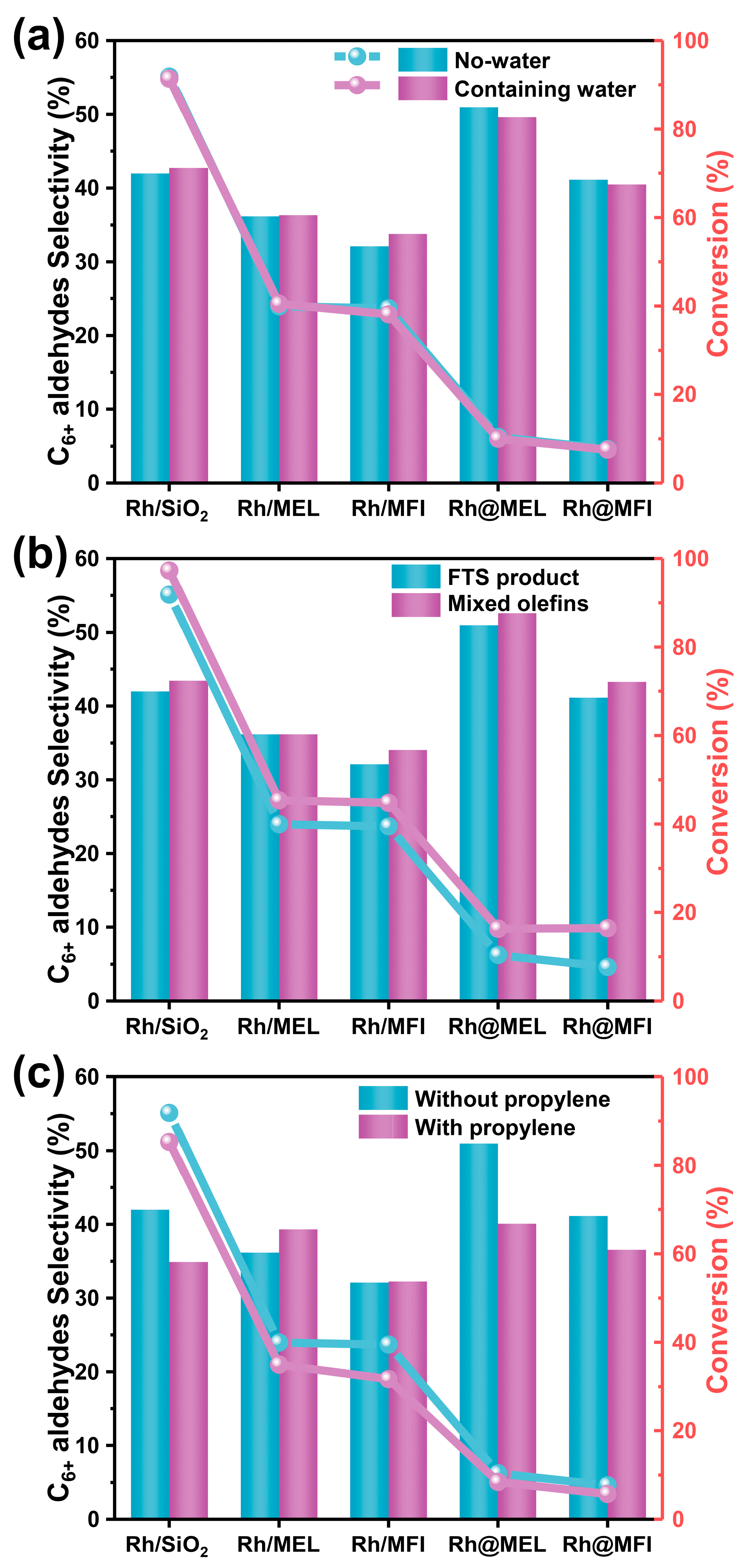
2.2. Catalytic Performance Evaluation
3. Experimental Section
3.1. Materials Synthesis
3.2. Characterization Methods
3.3. Catalytic Performance Evaluation
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kang, J.; He, S.; Zhou, W.; Shen, Z.; Li, Y.; Chen, M.; Zhang, Q.; Wang, Y. Single-Pass Transformation of Syngas into Ethanol with High Selectivity by Triple Tandem Catalysis. Nat. Commun. 2020, 11, 827. [Google Scholar] [CrossRef] [PubMed]
- Foit, S.R.; Vinke, I.C.; de Haart, L.G.J.; Eichel, R.A. Power-to-Syngas: An Enabling Technology for the Transition of the Energy System? Angew. Chem. Int. Ed. 2017, 56, 5402–5411. [Google Scholar] [CrossRef] [PubMed]
- Jiao, F.; Li, J.; Pan, X.; Xiao, J.; Li, H.; Ma, H.; Wei, M.; Pan, Y.; Zhou, Z.; Li, M.; et al. Selective Conversion of Syngas to Light Olefins. Science 2016, 351, 1065–1068. [Google Scholar] [CrossRef]
- Li, J.; He, Y.; Tan, L.; Zhang, P.; Peng, X.; Oruganti, A.; Yang, G.; Abe, H.; Wang, Y.; Tsubaki, N. Integrated Tuneable Synthesis of Liquid Fuels via Fischer–Tropsch Technology. Nat. Catal. 2018, 1, 787–793. [Google Scholar] [CrossRef]
- Duyckaerts, N.; Bartsch, M.; Trotuş, I.T.; Pfänder, N.; Lorke, A.; Schüth, F.; Prieto, G. Intermediate Product Regulation in Tandem Solid Catalysts with Multimodal Porosity for High-Yield Synthetic Fuel Production. Angew. Chem. Int. Ed. 2017, 56, 11480–11484. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Yu, F.; An, Y.; Zhao, Y.; Sun, Y.; Li, Z.; Lin, T.; Lin, Y.; Qi, X.; Dai, Y.; et al. Cobalt Carbide Nanoprisms for Direct Production of Lower Olefins from Syngas. Nature 2016, 538, 84–87. [Google Scholar] [CrossRef]
- Cheng, K.; Gu, B.; Liu, X.; Kang, J.; Zhang, Q.; Wang, Y. Direct and Highly Selective Conversion of Synthesis Gas into Lower Olefins: Design of a Bifunctional Catalyst Combining Methanol Synthesis and Carbon–Carbon Coupling. Angew. Chem. Int. Ed. 2016, 128, 4803–4806. [Google Scholar] [CrossRef]
- Torres Galvis, H.M.; Bitter, J.H.; Khare, C.B.; Ruitenbeek, M.; Dugulan, A.I.; de Jong, K.P. Supported Iron Nanoparticles as Catalysts for Sustainable Production of Lower Olefins. Science 2012, 335, 835–838. [Google Scholar] [CrossRef]
- Ao, M.; Pham, G.H.; Sunarso, J.; Tade, M.O.; Liu, S. Active Centers of Catalysts for Higher Alcohol Synthesis from Syngas: A Review. ACS Catal. 2018, 8, 7025–7050. [Google Scholar] [CrossRef]
- Luk, H.T.; Mondelli, C.; Ferre, D.C.; Stewart, J.A.; Perez-Ramirez, J. Status and Prospects in Higher Alcohols Synthesis from Syngas. Chem. Soc. Rev. 2017, 46, 1358–1426. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Kruse, N. Tuning the Catalytic CO Hydrogenation to Straight- and Long-Chain Aldehydes/Alcohols and Olefins/Paraffins. Nat. Commun. 2016, 7, 13058. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Zhang, K.; He, P.; Chang, H.; Zhang, M.; Yan, T.; Zhang, X.; Li, Y.; Cao, Z. Reactive Intermediate Confinement in Beta Zeolites for the Efficient Aerobic Epoxidation of α-Olefins. Angew. Chem. Int. Ed. 2025, 64, e202419900. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Yin, A.; Zhang, X.; Wang, J.; Qin, X.; Yang, Y.; Qin, G.; Sun, X.; He, P.; Yang, Y. Carbon-Coated Ni-Fe Nanocatalysts: Bridging the Gap in Cinnamaldehyde Hydrogenation Performance and Durability. Catalysts 2023, 13, 1474. [Google Scholar] [CrossRef]
- Liu, G.; Yang, G.; Peng, X.; Wu, J.; Tsubaki, N. Recent Advances in the Routes and Catalysts for Ethanol Synthesis from Syngas. Chem. Soc. Rev. 2022, 51, 5606–5659. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ma, L.; Zhang, R.; Ye, R.; Liu, W.; Wei, J.; Ordomsky, V.V.; Liu, J. Carbon-supported Fe Catalysts with Well-Defined Active Sites for Highly Selective Alcohol Production from Fischer-Tropsch Synthesis. Appl. Catal. B Environ. 2022, 312, 121393. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, Z.; Lu, W.; Jiang, M.; Li, C.; Zhao, M.; Gong, L.; Wang, S.; Guo, L.; Lyu, Y.; et al. Highly Selective Conversion of Syngas to Higher Oxygenates over Tandem Catalysts. ACS Catal. 2021, 11, 14791–14802. [Google Scholar] [CrossRef]
- Zhao, Z.; Lu, W.; Yang, R.; Zhu, H.; Dong, W.; Sun, F.; Jiang, Z.; Lyu, Y.; Liu, T.; Du, H.; et al. Insight into the Formation of Co@Co2C Catalysts for Direct Synthesis of Higher Alcohols and Olefins from Syngas. ACS Catal. 2017, 8, 228–241. [Google Scholar] [CrossRef]
- Martin, O.; Mondelli, C.; Cervellino, A.; Ferri, D.; Curulla-Ferré, D.; Pérez-Ramírez, J. Operando Synchrotron X-ray Powder Diffraction and Modulated-Excitation Infrared Spectroscopy Elucidate the CO2 Promotion on a Commercial Methanol Synthesis Catalyst. Angew. Chem. Int. Ed. 2016, 55, 11031–11036. [Google Scholar] [CrossRef]
- Xiang, Y.; Chitry, V.; Liddicoat, P.; Felfer, P.; Cairney, J.; Ringer, S.; Kruse, N. Long-Chain Terminal Alcohols through Catalytic CO Hydrogenation. J. Am. Chem. Soc. 2013, 135, 7114–7117. [Google Scholar] [CrossRef]
- Bezemer, G.L.; Bitter, J.H.; Kuipers, H.P.C.E.; Oosterbeek, H.; Holewijn, J.E.; Xu, X.; Kapteijn, F.; van Dillen, A.J.; de Jong, K.P. Cobalt Particle Size Effects in the Fischer−Tropsch Reaction Studied with Carbon Nanofiber Supported Catalysts. J. Am. Chem. Soc. 2006, 128, 3956–3964. [Google Scholar] [CrossRef]
- Li, Y.; Gao, W.; Peng, M.; Zhang, J.; Sun, J.; Xu, Y.; Hong, S.; Liu, X.; Liu, X.; Wei, M.; et al. Interfacial Fe5C2-Cu Catalysts toward Low-Pressure Syngas Conversion to Long-Chain Alcohols. Nat. Commun. 2020, 11, 61. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Zou, T.; Martín, A.J.; Pérez-Ramírez, J. ZrO2-Promoted Cu-Co, Cu-Fe and Co-Fe Catalysts for Higher Alcohol Synthesis. ACS Catal. 2023, 13, 9946–9959. [Google Scholar] [CrossRef]
- Lin, T.; Qi, X.; Wang, X.; Xia, L.; Wang, C.; Yu, F.; Wang, H.; Li, S.; Zhong, L.; Sun, Y. Direct Production of Higher Oxygenates by Syngas Conversion over a Multifunctional Catalyst. Angew. Chem. Int. Ed. 2019, 58, 4627–4631. [Google Scholar] [CrossRef] [PubMed]
- Jeske, K.; Rösler, T.; Belleflamme, M.; Rodenas, T.; Fischer, N.; Claeys, M.; Leitner, W.; Vorholt, A.J.; Prieto, G. Direct Conversion of Syngas to Higher Alcohols via Tandem Integration of Fischer–Tropsch Synthesis and Reductive Hydroformylation. Angew. Chem. Int. Ed. 2022, 61, e202201004. [Google Scholar] [CrossRef]
- An, Y.; Wang, X.; Yu, H.; Qi, X.; Lv, D.; Lin, T.; Zhong, L. Mn-Promoted Ru-Based Catalysts for Enhanced Fischer–Tropsch Synthesis of Olefins from Syngas. ACS Catal. 2023, 13, 13245–13256. [Google Scholar] [CrossRef]
- Yu, H.; Wang, C.; Lin, T.; An, Y.; Wang, Y.; Chang, Q.; Yu, F.; Wei, Y.; Sun, F.; Jiang, Z.; et al. Direct Production of Olefins from Syngas with Ultrahigh Carbon Efficiency. Nat. Commun. 2022, 13, 5987. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Lin, T.; An, Y.; Gong, K.; Wang, X.; Sun, Y.; Zhong, L. Recent Advances in Co2C-Based Nanocatalysts for Direct Production of Olefins from Syngas Conversion. Chem. Commun. 2022, 58, 9712–9727. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Li, X.; Gao, J.; Wang, J.; Ma, G.; Wen, X.; Yang, Y.; Li, Y.; Ding, M. A Hydrophobic FeMn@Si Catalyst Increases Olefins from Syngas by Suppressing C1 by-Products. Science 2021, 371, 610–613. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Yu, F.; An, Y.; Qin, T.; Li, L.; Gong, K.; Zhong, L.; Sun, Y. Cobalt Carbide Nanocatalysts for Efficient Syngas Conversion to Value-Added Chemicals with High Selectivity. Acc. Chem. Res. 2021, 54, 1961–1971. [Google Scholar] [CrossRef] [PubMed]
- Zhai, P.; Xu, C.; Gao, R.; Liu, X.; Li, M.; Li, W.; Fu, X.; Jia, C.; Xie, J.; Zhao, M.; et al. Highly Tunable Selectivity for Syngas-Derived Alkenes over Zinc and Sodium-Modulated Fe5C2 Catalyst. Angew. Chem. Int. Ed. 2016, 55, 9902–9907. [Google Scholar] [CrossRef]
- Zhang, X.; Yan, T.; Hou, H.; Yin, J.; Wan, H.; Sun, X.; Zhang, Q.; Sun, F.; Wei, Y.; Dong, M.; et al. Regioselective Hydroformylation of Propene Catalysed by Rhodium-Zeolite. Nature 2024, 629, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Dou, X.; Yan, T.; Qian, L.; Hou, H.; Lopez-Haro, M.; Marini, C.; Agostini, G.; Meira, D.M.; Zhang, X.; Zhang, L.; et al. Regioselective Hydroformylation with Subnanometre Rh Clusters in MFI Zeolite. Nat. Catal. 2024, 7, 666–677. [Google Scholar] [CrossRef]
- Sun, Q.; Wang, N.; Yu, J. Advances in Catalytic Applications of Zeolite-Supported Metal Catalysts. Adv. Mater. 2021, 33, 2104442. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, L.; Xiao, F.-S. Metal@Zeolite Hybrid Materials for Catalysis. ACS Cent. Sci. 2020, 6, 1685–1697. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Corma, A. Confining Isolated Atoms and Clusters in Crystalline Porous Materials for Catalysis. Nat. Rev. Mater. 2020, 6, 244–263. [Google Scholar] [CrossRef]
- Babucci, M.; Guntida, A.; Gates, B.C. Atomically Dispersed Metals on Well-Defined Supports including Zeolites and Metal–Organic Frameworks: Structure, Bonding, Reactivity, and Catalysis. Chem. Rev. 2020, 120, 11956–11985. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Lopez-Haro, M.; Lopes, C.W.; Li, C.; Concepcion, P.; Simonelli, L.; Calvino, J.J.; Corma, A. Regioselective Generation and Reactivity Control of Subnanometric Platinum Clusters in Zeolites for High-Temperature Catalysis. Nat. Mater. 2019, 18, 866–873. [Google Scholar] [CrossRef]
- Yan, T.; Hou, H.; Wu, C.; Cai, Y.; Yin, A.; Cao, Z.; Liu, Z.; He, P.; Xu, J. Unraveling the Molecular Mechanism for Enhanced Gas Adsorption in Mixed-Metal MOFs via Solid-State NMR Spectroscopy. Proc. Natl. Acad. Sci. USA 2024, 121, e2312959121. [Google Scholar] [CrossRef] [PubMed]
- Dou, X.; Li, W.; Zhang, K.; Hou, H.; He, Z.; Zhu, C.; Meira, D.M.; Lopez-Haro, M.; Xia, Z.; He, P.; et al. Size-Dependent Structural Features of Subnanometer PtSn Catalysts Encapsulated in Zeolite for Alkane Dehydrogenation. ACS Catal. 2024, 14, 2859–2871. [Google Scholar] [CrossRef]
- Gong, N.; Qin, G.; Li, P.; Zhang, X.; Chen, Y.; Yang, Y.; He, P. Enhanced Stability and Selectivity in Pt@MFI Catalysts for n-Butane Dehydrogenation: The Crucial Role of Sn Promoter. Catalysts 2024, 14, 760. [Google Scholar] [CrossRef]
- Qi, L.; Das, S.; Zhang, Y.; Nozik, D.; Gates, B.C.; Bell, A.T. Ethene Hydroformylation Catalyzed by Rhodium Dispersed with Zinc or Cobalt in Silanol Nests of Dealuminated Zeolite Beta. J. Am. Chem. Soc. 2023, 145, 2911–2929. [Google Scholar] [CrossRef] [PubMed]
- Matsubu, J.C.; Yang, V.N.; Christopher, P. Isolated Metal Active Site Concentration and Stability Control Catalytic CO2 Reduction Selectivity. J. Am. Chem. Soc. 2015, 137, 3076–3084. [Google Scholar] [CrossRef] [PubMed]
- McClure, S.M.; Lundwall, M.J.; Goodman, D.W. Planar Oxide Supported Rhodium Nanoparticles as Model Catalysts. Proc. Natl. Acad. Sci. USA 2010, 108, 931–936. [Google Scholar] [CrossRef] [PubMed]
- Yates, D. Ultradispersed Rhodium Rafts: Their Existence and Topology. J. Catal. 1979, 57, 41–63. [Google Scholar] [CrossRef]
- Chen, M.; Gupta, G.; Ordonez, C.W.; Lamkins, A.R.; Ward, C.J.; Abolafia, C.A.; Zhang, B.; Roling, L.T.; Huang, W. Intermetallic Nanocatalyst for Highly Active Heterogeneous Hydroformylation. J. Am. Chem. Soc. 2021, 143, 20907–20915. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Liu, B.; Lan, X.; Wang, T. Insights into the Bimetallic Effects of a RhCo Catalyst for Ethene Hydroformylation: Experimental and DFT Investigations. Ind. Eng. Chem. Res. 2020, 59, 18771–18780. [Google Scholar] [CrossRef]
- Li, C.; Wang, W.; Yan, L.; Ding, Y. A Mini Review on Strategies for Heterogenization of Rhodium-Based Hydroformylation Catalysts. Front. Chem. Sci. Eng. 2017, 12, 113–123. [Google Scholar] [CrossRef]
- Cnudde, P.; De Wispelaere, K.; Vanduyfhuys, L.; Demuynck, R.; Van der Mynsbrugge, J.; Waroquier, M.; Van Speybroeck, V. How Chain Length and Branching Influence the Alkene Cracking Reactivity on H-ZSM-5. ACS Catal. 2018, 8, 9579–9595. [Google Scholar] [CrossRef] [PubMed]
- Yoo, K.; Tsekov, R.; Smirniotis, P.G. Experimental Proof for Resonant Diffusion of Normal Alkanes in LTL and ZSM-12 Zeolites. J. Phys. Chem. B 2003, 107, 13593–13596. [Google Scholar] [CrossRef]
- Zhang, K.; Dou, X.; Zhou, Z.; Wang, Y.; Hou, H.; Meira, D.M.; Liu, L.; He, P. Tuning the Size and Spatial Distribution of Pt in Bifunctional Pt-zeolite Catalysts for Direct Coupling of Ethane and Benzene. Chem. Eng. J. 2024, 497, 154874. [Google Scholar] [CrossRef]
- Chen, Z.; Zhao, W.; Zhang, H.; Ren, J.; He, P. Influence of Intracrystalline Diffusion Constraints Enhanced by Channel Fluctuation on Zeolite Catalyst Performance in Isomerization and Aromatization. Fuel 2024, 362, 130797. [Google Scholar] [CrossRef]
- Zhang, K.; Dou, X.; Hou, H.; Zhou, Z.; Lopez-Haro, M.; Meira, D.M.; Liu, P.; He, P.; Liu, L. Generation of Subnanometer Metal Clusters in Silicoaluminate Zeolites as Bifunctional Catalysts. JACS Au 2023, 3, 3213–3226. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Zhao, H.; Guo, X.; He, Y.; Yang, G.; Tsubaki, N. Silicalite-1 Encapsulated Rhodium Nanoparticles for Hydroformylation of 1-hexene. Catal. Today 2023, 410, 150–156. [Google Scholar] [CrossRef]
- Dou, X.; Yan, T.; Li, W.; Zhu, C.; Chen, T.; Lo, B.T.W.; Marini, C.; Xiao, H.; Liu, L. Structure–Reactivity Relationship of Zeolite-Confined Rh Catalysts for Hydroformylation of Linear α-Olefins. J. Am. Chem. Soc. 2025, 147, 2726–2736. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | Surface Area (m2/g) | Pore Sizes (nm) | ICP Elemental Analysis | ||
|---|---|---|---|---|---|
| Total | External | Micro | Rh (wt %) | ||
| Rh@MFI | 291 | 7 | 284 | 0.53 | 0.21 |
| Rh@MEL | 303 | 7 | 296 | 0.48 | 0.20 |
| Rh/MFI | 289 | 6 | 283 | 0.54 | 0.23 |
| Rh/MEL | 299 | 8 | 291 | 0.50 | 0.20 |
| Rh/SiO2 | 223 | 182 | 40 | 5.60 | 0.21 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Cao, X.; Dai, Y.; Yan, T.; Zhang, X.; He, H.; Xie, Y.; Lin, T.; Song, C.; He, P. Effect of Metal Dispersion in Rh-Based Zeolite and SiO2 Catalysts on the Hydroformylation of Olefin Mixtures from Fischer–Tropsch Synthesis. Catalysts 2025, 15, 212. https://doi.org/10.3390/catal15030212
Wang Y, Cao X, Dai Y, Yan T, Zhang X, He H, Xie Y, Lin T, Song C, He P. Effect of Metal Dispersion in Rh-Based Zeolite and SiO2 Catalysts on the Hydroformylation of Olefin Mixtures from Fischer–Tropsch Synthesis. Catalysts. 2025; 15(3):212. https://doi.org/10.3390/catal15030212
Chicago/Turabian StyleWang, Yu, Xuemin Cao, Yuting Dai, Tao Yan, Xiangjie Zhang, Huizi He, Yujie Xie, Tiejun Lin, Chang Song, and Peng He. 2025. "Effect of Metal Dispersion in Rh-Based Zeolite and SiO2 Catalysts on the Hydroformylation of Olefin Mixtures from Fischer–Tropsch Synthesis" Catalysts 15, no. 3: 212. https://doi.org/10.3390/catal15030212
APA StyleWang, Y., Cao, X., Dai, Y., Yan, T., Zhang, X., He, H., Xie, Y., Lin, T., Song, C., & He, P. (2025). Effect of Metal Dispersion in Rh-Based Zeolite and SiO2 Catalysts on the Hydroformylation of Olefin Mixtures from Fischer–Tropsch Synthesis. Catalysts, 15(3), 212. https://doi.org/10.3390/catal15030212