

Controlled Synthesis of Nickel Phosphides in Hollow N, P Co-Doped Carbon: In Situ Transition to (Oxy)hydroxide Phases During Oxygen Evolution Reaction

 , ,
, ,  , and
, and 
Abstract

1. Introduction
2. Results and Discussion
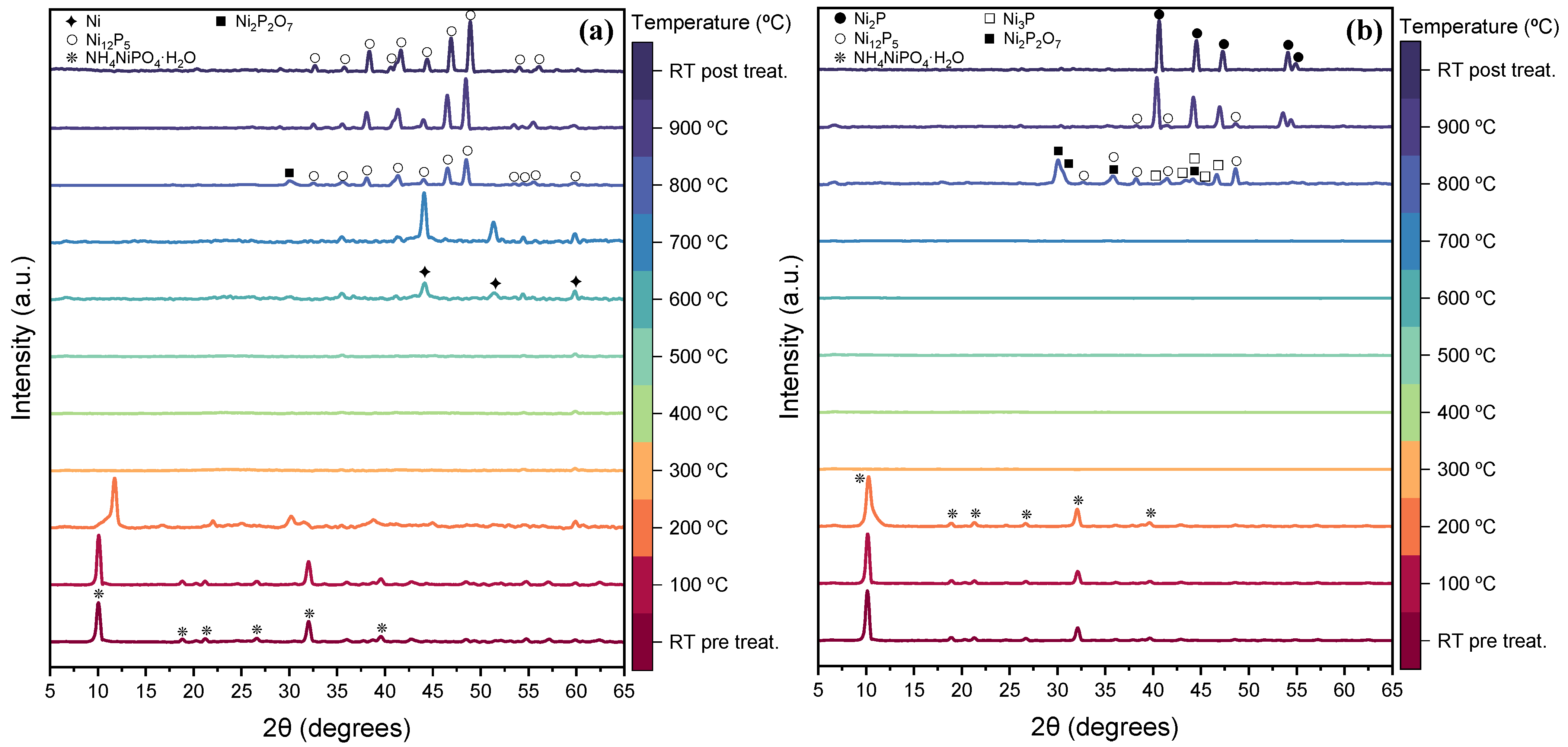
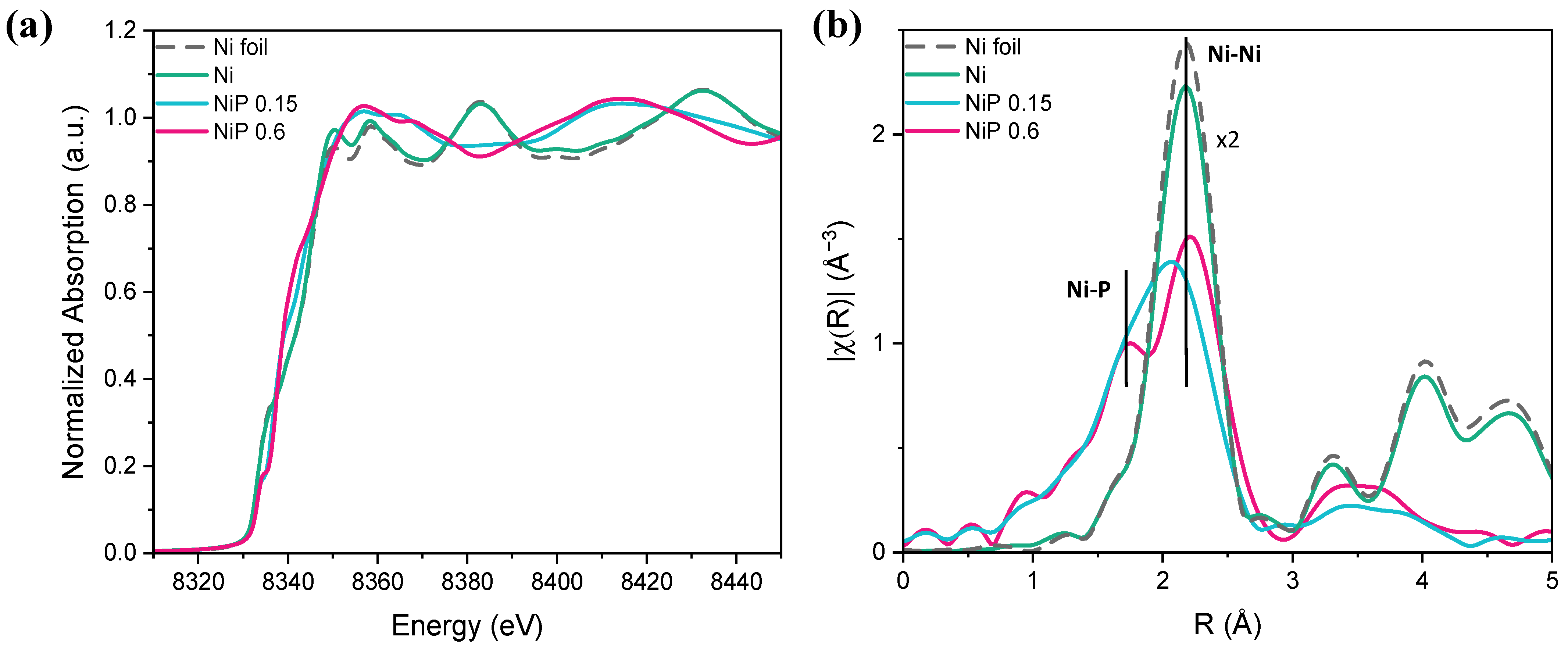
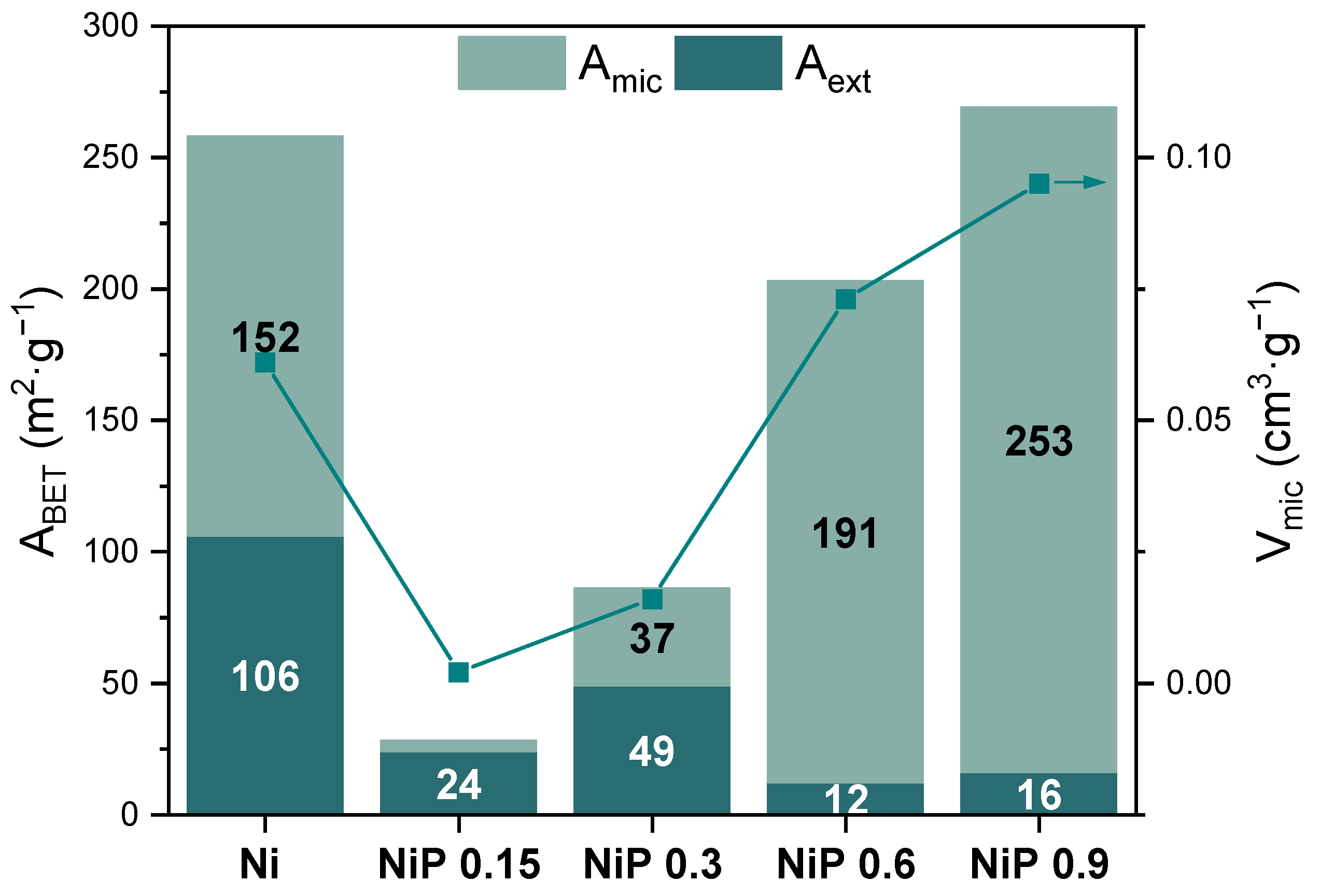
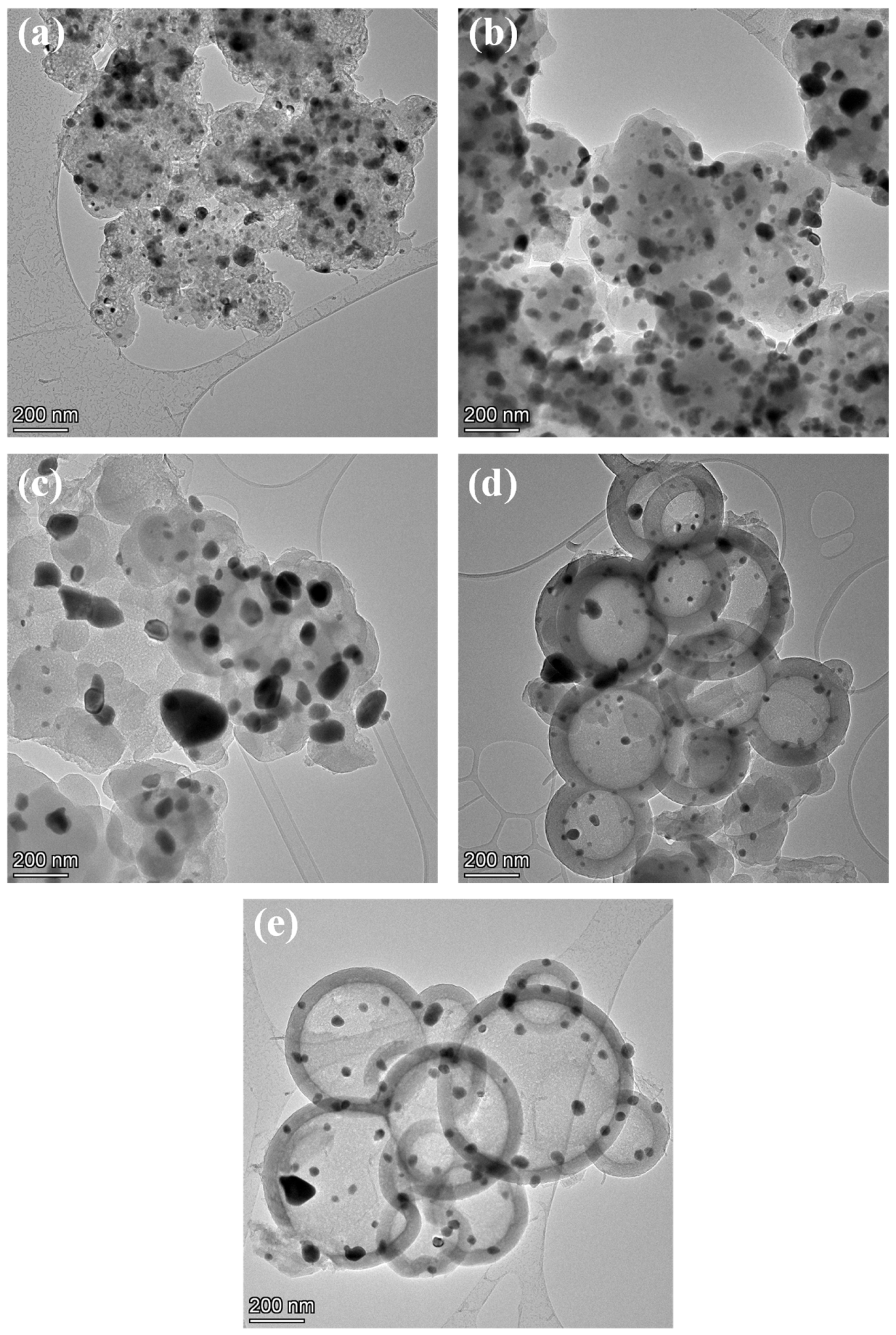
2.1. Preparation and Physico-Chemical Characterization
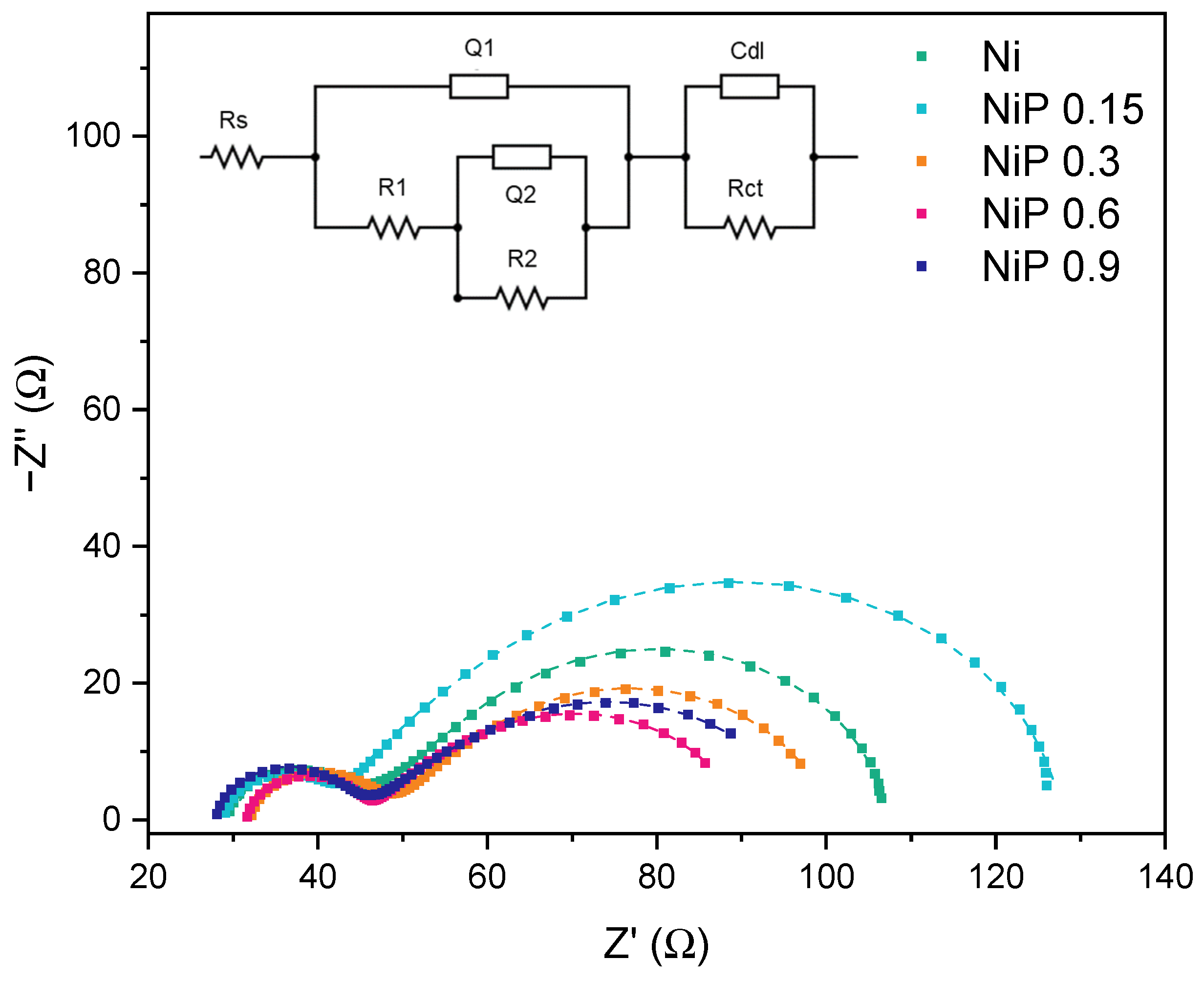
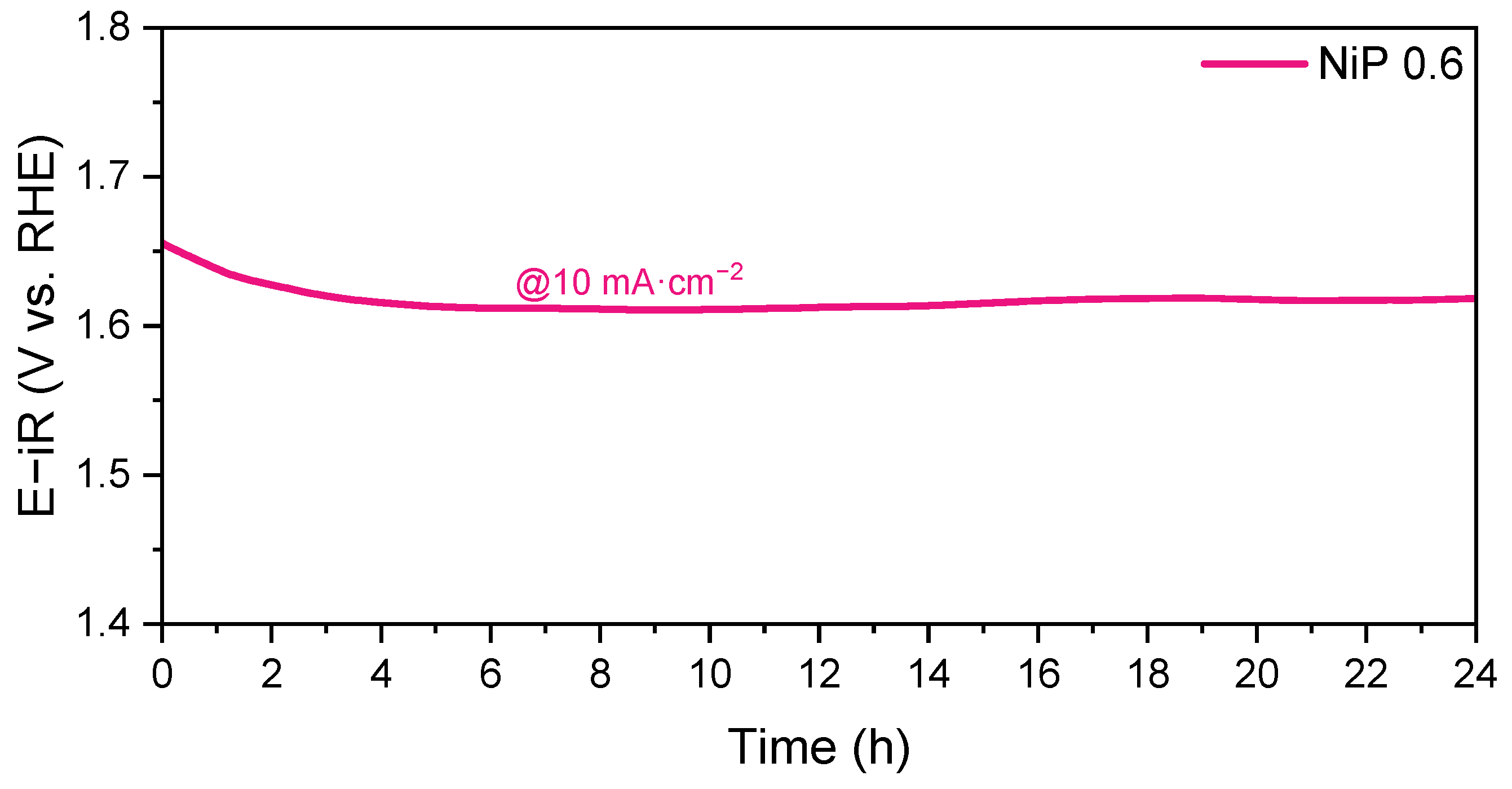
2.2. Electrochemical Characterization: OER Studies
3. Materials and Methods
3.1. Materials
3.2. Synthesis of Nickel Phosphides in N, P Co-Doped Carbon
3.3. Physico-Chemical Characterization
3.4. Electrochemical Characterization
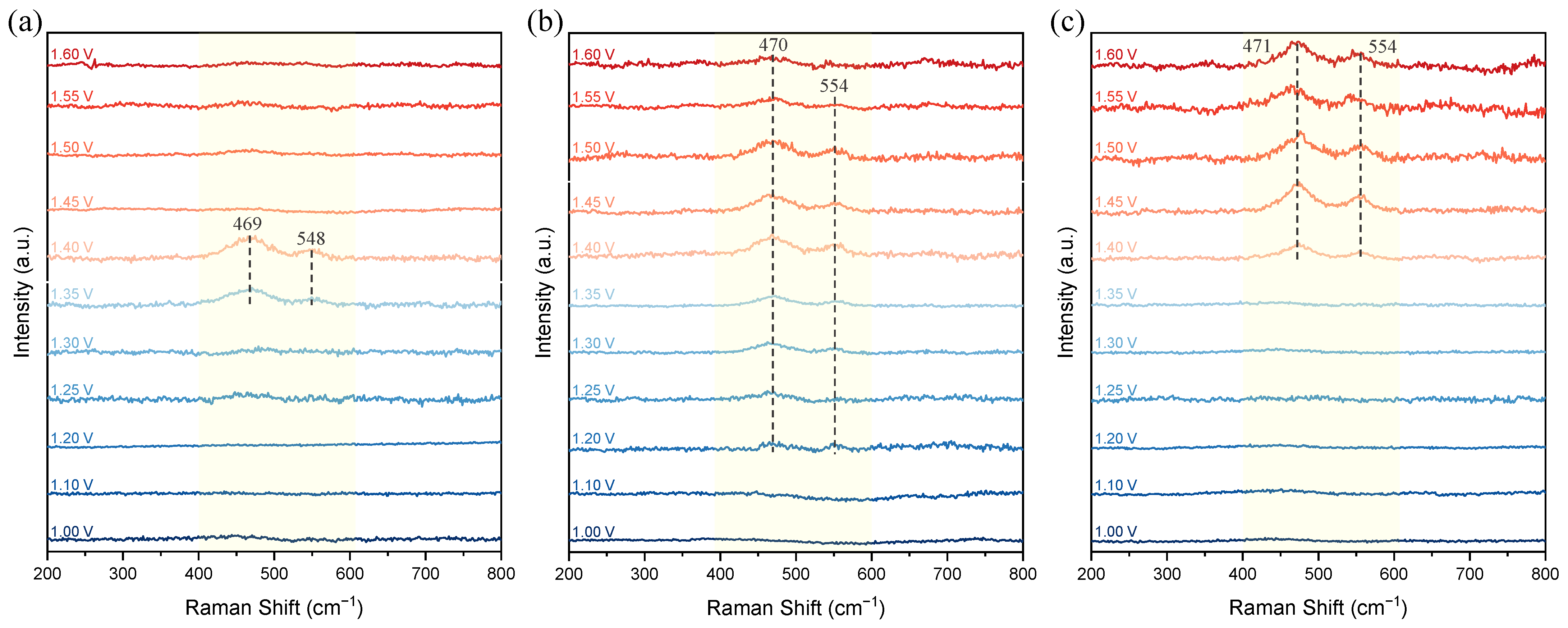
3.5. In Situ/Operando Raman Spectroscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vass, Á.; Kormányos, A.; Kószó, Z.; Endrődi, B.; Janáky, C. Anode Catalysts in CO2 Electrolysis: Challenges and Untapped Opportunities. ACS Catal. 2022, 12, 1037–1051. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Du, L.; Yan, L.; Park, S.; Qiu, Y.; Sokolowski, J.; Wang, W.; Shao, Y. Oxygen Evolution Reaction in Alkaline Environment: Material Challenges and Solutions. Adv. Funct. Mater. 2022, 32, 2110036. [Google Scholar] [CrossRef]
- Chatenet, M.; Pollet, B.G.; Dekel, D.R.; Dionigi, F.; Deseure, J.; Millet, P.; Braatz, R.D.; Bazant, M.Z.; Eikerling, M.; Staffell, I.; et al. Water electrolysis: From textbook knowledge to the latest scientific strategies and industrial developments. Chem. Soc. Rev. 2022, 51, 4583–4762. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhou, D.; Deng, T.; He, G.; Chen, A.; Sun, X.; Yang, Y.; Miao, P. Research Progress of Oxygen Evolution Reaction Catalysts for Electrochemical Water Splitting. ChemSusChem 2021, 14, 5359–5383. [Google Scholar] [CrossRef]
- Dionigi, F.; Strasser, P. NiFe-Based (Oxy)hydroxide Catalysts for Oxygen Evolution Reaction in Non-Acidic Electrolytes. Adv. Energy Mater. 2016, 6, 1600621. [Google Scholar] [CrossRef]
- Gebreslase, G.A.; Martínez-Huerta, M.V.; Lázaro, M.J. Recent progress on bimetallic NiCo and CoFe based electrocatalysts for alkaline oxygen evolution reaction: A review. J. Energy Chem. 2022, 67, 101–137. [Google Scholar] [CrossRef]
- Jamadar, A.S.; Sutar, R.; Patil, S.; Khandekar, R.; Yadav, J.B. Progress in metal oxide-based electrocatalysts for sustainable water splitting. Mater. Rep. Energy 2024, 4, 100283. [Google Scholar] [CrossRef]
- Li, Y.; Dong, Z.; Jiao, L. Multifunctional Transition Metal-Based Phosphides in Energy-Related. Adv. Energy Mater. 2020, 10, 1902104. [Google Scholar]
- Hu, C.; Lv, C.; Liu, S.; Shi, Y.; Song, J.; Zhang, Z.; Cai, J.; Watanabe, A. Nickel phosphide electrocatalysts for hydrogen evolution reaction. Catalysts 2020, 10, 188. [Google Scholar] [CrossRef]
- Sun, M.; Liu, H.; Qu, J.; Li, J. Earth-Rich Transition Metal Phosphide for Energy Conversion and Storage. Adv. Energy Mater. 2016, 6, 1600087. [Google Scholar] [CrossRef]
- Read, C.G.; Callejas, J.F.; Holder, C.F.; Schaak, R.E. General Strategy for the Synthesis of Transition Metal Phosphide Films for Electrocatalytic Hydrogen and Oxygen Evolution. ACS Appl. Mater. Interfaces 2016, 8, 12798–12803. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, H.; Niu, S.; Zhou, Y.; Ni, Z.; Wei, Q.; Chen, A.; Zhang, S.; Sun, T.; Dai, R.; et al. Bifunctional heterostructured nitrogen and phosphorus co-doped carbon-layer-encapsulated Co2P electrocatalyst for efficient water splitting. Cell Rep. Phys. Sci. 2021, 2, 100586. [Google Scholar] [CrossRef]
- Dutta, A.; Pradhan, N. Developments of Metal Phosphides as Efficient OER Precatalysts. J. Phys. Chem. Lett. 2017, 8, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Sultana, S.; Paramanik, L.; Parida, K.M. Recent advances in phase, size, and morphology-oriented nanostructured nickel phosphide for overall water splitting. J. Mater. Chem. A 2020, 8, 19196–19245. [Google Scholar] [CrossRef]
- Popczun, E.J.; McKone, J.R.; Read, C.G.; Biacchi, A.J.; Wiltrout, A.M.; Lewis, N.S.; Schaak, R.E. Nanostructured Nickel Phosphide as an Electrocatalyst for the Hydrogen Evolution Reaction. J. Am. Chem. Soc. 2013, 135, 9267–9270. [Google Scholar] [CrossRef]
- Pan, Y.; Liu, Y.; Zhao, J.; Yang, K.; Liang, J.; Liu, D.; Hu, W.; Liu, D.; Liu, Y.; Liu, C. Monodispersed nickel phosphide nanocrystals with different phases: Synthesis, characterization and electrocatalytic properties for hydrogen evolution. J. Mater. Chem. A 2015, 3, 1656–1665. [Google Scholar] [CrossRef]
- Lin, Y.; Pan, Y.; Zhang, J. In Situ Construction of Nickel Phosphosulfide (Ni5P4|S) Active Species on 3D Ni Foam through Chemical Vapor Deposition for Electrochemical Hydrogen Evolution. ChemElectroChem 2017, 4, 1108–1116. [Google Scholar] [CrossRef]
- Ramesh, S.K.; Son, J.; Ganesan, V.; Kim, J. Carbon-incorporated Ni2P-Fe2P hollow nanorods as superior electrocatalysts for the oxygen evolution reaction. Nanoscale 2022, 136, 16262–16269. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Huang, L. Improving the oxygen evolution performance of nickel phosphide nanoparticles with satellite nitrogen-doped carbon quantum dots. Mater. Lett. 2017, 209, 106–110. [Google Scholar] [CrossRef]
- Yuan, M.; Sun, Y.; Yang, Y.; Zhang, J.; Dipazir, S.; Zhao, T.; Li, S.; Xie, Y.; Zhao, H.; Liu, Z.; et al. Boosting oxygen evolution reactivity by modulating electronic structure and honeycomb-like architecture in Ni2P/N,P-codoped carbon hybrids. Green Energy Environ. 2021, 6, 866–874. [Google Scholar] [CrossRef]
- Kandel, M.R.; Pan, U.N.; Dhakal, P.P.; Ghising, R.B.; Sidra, S.; Kim, D.H.; Kim, N.H.; Lee, J.H. Manganese-Doped Bimetallic (Co,Ni)2P Integrated CoP in N,S Co−Doped Carbon: Unveiling a Compatible Hybrid Electrocatalyst for Overall Water Splitting. Small 2024, 20, 2307241. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Liu, B.; Dai, L. Non-N-Doped Carbons as Metal-Free Electrocatalysts. Adv. Sustain. Syst. 2021, 5, 2000134. [Google Scholar] [CrossRef]
- Zhan, X.; Tong, X.; Gu, M.; Tian, J.; Gao, Z.; Ma, L.; Xie, Y.; Chen, Z.; Ranganathan, H.; Zhang, G.; et al. Phosphorus-Doped Graphene Electrocatalysts for Oxygen Reduction Reaction. Nanomaterials 2022, 12, 1141. [Google Scholar] [CrossRef]
- Choi, C.H.; Chung, M.W.; Kwon, H.C.; Park, S.H.; Woo, S.I. B, N- and P, N-doped graphene as highly active catalysts for oxygen reduction reactions in acidic media. J. Mater. Chem. A 2013, 1, 3694. [Google Scholar] [CrossRef]
- Li, J.-C.; Hou, P.-X.; Cheng, M.; Liu, C.; Cheng, H.-M.; Shao, M. Carbon nanotube encapsulated in nitrogen and phosphorus co-doped carbon as a bifunctional electrocatalyst for oxygen reduction and evolution reactions. Carbon 2018, 139, 156–163. [Google Scholar] [CrossRef]
- Choi, C.H.; Park, S.H.; Woo, S.I. Phosphorus–nitrogen dual doped carbon as an effective catalyst for oxygen reduction reaction in acidic media: Effects of the amount of P-doping on the physical and electrochemical properties of carbon. J. Mater. Chem. 2012, 22, 12107. [Google Scholar] [CrossRef]
- Luo, S.; Wang, R.; Hei, P.; Gao, L.; Yang, J.; Jiao, T. Self-assembled Ni2P nanosheet-implanted reduced graphene oxide composite as highly efficient electrocatalyst for oxygen evolution reaction. Colloids Surf. A Physicochem. Eng. Asp. 2021, 612, 125992. [Google Scholar] [CrossRef]
- Ghadge, S.D.; Velikokhatnyi, O.I.; Datta, M.K.; Damodaran, K.; Shanthi, P.M.; Kumta, P.N. Highly Efficient Fluorine Doped Ni2P Electrocatalysts for Alkaline Mediated Oxygen Evolution Reaction. J. Electrochem. Soc. 2021, 168, 064512. [Google Scholar] [CrossRef]
- Sun, H.; Xu, X.; Yan, Z.; Chen, X.; Cheng, F.; Weiss, P.S.; Chen, J. Porous Multishelled Ni2P Hollow Microspheres as an Active Electrocatalyst for Hydrogen and Oxygen Evolution. Chem. Mater. 2017, 29, 8539–8547. [Google Scholar] [CrossRef]
- Berhault, G.; Afanasiev, P.; Loboué, H.; Geantet, C.; Cseri, T.; Pichon, C.; Guillot-Deudon, C.; Lafond, A. In Situ XRD, XAS, and Magnetic Susceptibility Study of the Reduction of Ammonium Nickel Phosphate NiNH4PO4·H2O into Nickel Phosphide. Inorg. Chem. 2009, 48, 2985–2992. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Kim, J.Y.; Hanson, J.C.; Sawhill, S.J.; Bussell, M.E. Physical and chemical properties of MoP, Ni2P, and MoNiP hydrodesulfurization catalysts: Time-resolved X-ray diffraction, density functional, and hydrodesulfurization activity studies. J. Phys. Chem. B 2003, 107, 6276–6285. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiao, L.; Yang, W.; Xie, C.; Jiang, H.L. Rational Fabrication of Low-Coordinate Single-Atom Ni Electrocatalysts by MOFs for Highly Selective CO2 Reduction. Angew. Chem. Int. Ed. 2021, 60, 7607–7611. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Wang, H.; Xia, X.; Zhang, X.; Dong, Z. Integration of CoFe Alloys and Fe/Fe3C Nanoparticles into N-Doped Carbon Nanosheets as Dual Catalytic Active Sites To Promote the Oxygen Electrocatalysis of Zn–Air Batteries. ACS Sustain. Chem. Eng. 2020, 8, 9009–9016. [Google Scholar] [CrossRef]
- Artyushkova, K. Misconceptions in interpretation of nitrogen chemistry from x-ray photoelectron spectra. J. Vac. Sci. Technol. A 2020, 38, 031002. [Google Scholar] [CrossRef]
- Luque-Centeno, J.M.; Martínez-Huerta, M.V.; Sebastián, D.; Pérez-Rodríguez, S.; Lázaro, M.J. Titanium Dioxide/N-Doped Graphene Composites as Non-Noble Bifunctional Oxygen Electrocatalysts. Ind. Eng. Chem. Res. 2021, 60, 18817–18830. [Google Scholar] [CrossRef]
- Zhou, Z.; Wei, L.; Wang, Y.; Karahan, H.E.; Chen, Z.; Lei, Y.; Chen, X.; Zhai, S.; Liao, X.; Chen, Y. Hydrogen evolution reaction activity of nickel phosphide is highly sensitive to electrolyte pH. J. Mater. Chem. A 2017, 5, 20390–20397. [Google Scholar] [CrossRef]
- Tiwari, A.P.; Lee, K.; Kim, K.; Kim, J.; Novak, T.G.; Jeon, S. Conformally Coated Nickel Phosphide on 3D, Ordered Nanoporous Nickel for Highly Active and Durable Hydrogen Evolution. ACS Sustain. Chem. Eng. 2020, 8, 17116–17123. [Google Scholar] [CrossRef]
- Bernasconi, R.; Khalil, M.I.; Cakmakci, D.S.; Bektas, Y.; Nobili, L.; Magagnin, L.; Lenardi, C. Electrocatalytic layers for hydrogen evolution reaction based on nickel phosphides: Cost-effective fabrication and XPS characterization. J. Mater. Sci. 2022, 57, 9370–9388. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Payne, B.P.; Grosvenor, A.P.; Lau, L.W.M.; Gerson, A.R.; Smart, R.S.C. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni. Appl. Surf. Sci. 2011, 257, 2717–2730. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, T.; Cao, H.; Cui, S.; Du, P. Self-supported Ni2P nanosheets on low-cost three-dimensional Fe foam as a novel electrocatalyst for efficient water oxidation. J. Energy Chem. 2020, 42, 71–76. [Google Scholar] [CrossRef]
- Xiong, K.; Huang, L.; Gao, Y.; Zhang, H.; Zhuo, Y.; Shen, H.; Wang, Y.; Peng, L.; Wei, Z. Formation of a thin-layer of nickel hydroxide on nickel phosphide nanopillars for hydrogen evolution. Electrochem. Commun. 2018, 92, 9–13. [Google Scholar] [CrossRef]
- Kumar, A.; Bui, V.Q.; Lee, J.; Jadhav, A.R.; Hwang, Y.; Kim, M.G.; Kawazoe, Y.; Lee, H. Modulating Interfacial Charge Density of NiP2-FeP2 via Coupling with Metallic Cu for Accelerating Alkaline Hydrogen Evolution. ACS Energy Lett. 2021, 6, 354–363. [Google Scholar] [CrossRef]
- Kalaiyarasan, G.; Joseph, J.; Kumar, P. Phosphorus-Doped Carbon Quantum Dots as Fluorometric Probes for Iron Detection. ACS Omega 2020, 5, 22278–22288. [Google Scholar] [CrossRef]
- Yu, Y.; Ma, J.; Chen, C.; Fu, Y.; Wang, Y.; Li, K.; Liao, Y.; Zheng, L.; Zuo, X. General Method for Synthesis Transition-Metal Phosphide/Nitrogen and Phosphide Doped Carbon Materials with Yolk-Shell Structure for Oxygen Reduction Reaction. ChemCatChem 2019, 11, 1722–1731. [Google Scholar] [CrossRef]
- Wu, J.; Yang, Z.; Sun, Q.; Li, X.; Strasser, P.; Yang, R. Synthesis and electrocatalytic activity of phosphorus-doped carbon xerogel for oxygen reduction. Electrochim. Acta 2014, 127, 53–60. [Google Scholar] [CrossRef]
- Sun, H.; Zhu, Y.; Yang, B.; Wang, Y.; Wu, Y.; Du, J. Template-free fabrication of nitrogen-doped hollow carbon spheres for high-performance supercapacitors based on a scalable homopolymer vesicle. J. Mater. Chem. A 2016, 4, 12088–12097. [Google Scholar] [CrossRef]
- Wang, S.; Sun, W.; Yang, D.-S.; Yang, F. Conversion of soybean waste to sub-micron porous-hollow carbon spheres for supercapacitor via a reagent and template-free route. Mater. Today Energy 2019, 13, 50–55. [Google Scholar] [CrossRef]
- Cui, C.M.; Guo, X.H.; Geng, Y.M.; Dang, T.T.; Xie, G.; Chen, S.P.; Zhao, F.Q. Facile one-pot synthesis of multi-yolk–shell Bi@C nanostructures by the nanoscale Kirkendall effect. Chem. Commun. 2015, 51, 9276–9279. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, S.; Shi, F.; Chen, K.; Nie, Y.; Wang, Y.; Wu, R.; Li, J.; Zhang, Y.; Ding, W.; et al. Structural Evolution of Solid Pt Nanoparticles to a Hollow PtFe Alloy with a Pt-Skin Surface via Space-Confined Pyrolysis and the Nanoscale Kirkendall Effect. Adv. Mater. 2016, 28, 10673–10678. [Google Scholar] [CrossRef]
- Hussain, I.; Sahoo, S.; Sayed, M.S.; Ahmad, M.; Javed, M.S.; Lamiel, C.; Li, Y.; Shim, J.J.; Ma, X.; Zhang, K. Hollow nano- and microstructures: Mechanism, composition, applications, and factors affecting morphology and performance. Coord. Chem. Rev. 2022, 458, 214429. [Google Scholar] [CrossRef]
- Cho, J.S.; Won, J.M.; Lee, J.-H.; Kang, Y.C. Synthesis and electrochemical properties of spherical and hollow-structured NiO aggregates created by combining the Kirkendall effect and Ostwald ripening. Nanoscale 2015, 7, 19620–19626. [Google Scholar] [CrossRef]
- Xie, X.; Shang, L.; Shi, R.; Waterhouse, G.I.N.; Zhao, J.; Zhang, T. Tubular assemblies of N-doped carbon nanotubes loaded with NiFe alloy nanoparticles as efficient bifunctional catalysts for rechargeable zinc-air batteries. Nanoscale 2020, 12, 13129–13136. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Morales, O.; Ferrus-Suspedra, D.; Koper, M.T.M. The importance of nickel oxyhydroxide deprotonation on its activity towards electrochemical water oxidation. Chem. Sci. 2016, 7, 2639–2645. [Google Scholar] [CrossRef]
- Stern, L.A.; Feng, L.; Song, F.; Hu, X. Ni2P as a Janus catalyst for water splitting: The oxygen evolution activity of Ni2P nanoparticles. Energy Environ. Sci. 2015, 8, 2347–2351. [Google Scholar] [CrossRef]
- Wu, Z.; Vagin, M.; Boyd, R.; Bakhit, B.; Greczynski, G.; Odén, M.; Björk, E.M. Morphology effects on electrocatalysis of anodic water splitting on nickel (II) oxide. Microporous Mesoporous Mater. 2022, 333, 111734. [Google Scholar] [CrossRef]
- Lyons, M.E.G.; Brandon, M.P. The significance of electrochemical impedance spectra recorded during active oxygen evolution for oxide covered Ni, Co and Fe electrodes in alkaline solution. J. Electroanal. Chem. 2009, 631, 62–70. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, Z.; Zhao, H.; Huang, H.; Jiao, H.; Du, Y. MOF-derived porous Ni2P nanosheets as novel bifunctional electrocatalysts for the hydrogen and oxygen evolution reactions. J. Mater. Chem. A 2018, 6, 18720–18727. [Google Scholar] [CrossRef]
- Wang, M.; Lin, M.; Li, J.; Huang, L.; Zhuang, Z.; Lin, C.; Zhou, L.; Mai, L. Metal–organic framework derived carbon-confined Ni2P nanocrystals supported on graphene for an efficient oxygen evolution reaction. Chem. Commun. 2017, 53, 8372–8375. [Google Scholar] [CrossRef]
- Li, R.; Wei, Z.; Gou, X. Nitrogen and Phosphorus Dual-Doped Graphene/Carbon Nanosheets as Bifunctional Electrocatalysts for Oxygen Reduction and Evolution. ACS Catal. 2015, 5, 4133–4142. [Google Scholar] [CrossRef]
- Li, J.; Tian, Q.; Jiang, S.; Zhang, Y.; Wu, Y. Electrocatalytic performances of phosphorus doped carbon supported Pd towards formic acid oxidation. Electrochim. Acta 2016, 213, 21–30. [Google Scholar] [CrossRef]
- McCrory, C.C.L.; Jung, S.; Peters, J.C.; Jaramillo, T.F. Benchmarking Heterogeneous Electrocatalysts for the Oxygen Evolution Reaction. J. Am. Chem. Soc. 2013, 135, 16977–16987. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Chen, X.; Wang, Y.; Song, P.; Zhang, Y. High-efficiency Ni–P catalysts in amorphous and crystalline states for the hydrogen evolution reaction. Sustain. Energy Fuels 2020, 4, 4733–4742. [Google Scholar] [CrossRef]
- Zhang, X.; Ma, G.; Shui, L.; Zhou, G.; Wang, X. Urea electrooxidation-boosted hydrogen production on nitrogen-doped porous carbon nanorod-supported nickel phosphide nanoparticles. J. Energy Chem. 2022, 72, 88–96. [Google Scholar] [CrossRef]
- Mijowska, E.; Pietrusewicz, K.; Maślana, K. Highly Porous Carbon Flakes Derived from Cellulose and Nickel Phosphide Heterostructure towards Efficient Electrocatalysis of Oxygen Evolution Reaction. Molecules 2024, 29, 352. [Google Scholar] [CrossRef]
- Yan, L.; Jiang, H.; Wang, Y.; Li, L.; Gu, X.; Dai, P.; Liu, D.; Tang, S.F.; Zhao, G.; Zhao, X.; et al. One-step and scalable synthesis of Ni2P nanocrystals encapsulated in N,P-codoped hierarchically porous carbon matrix using a bipyridine and phosphonate linked nickel metal–organic framework as highly efficient electrocatalysts for overall water splitting. Electrochim. Acta 2019, 297, 755–766. [Google Scholar] [CrossRef]
- Cebollada, J.; Sebastián, D.; Lázaro, M.J.; Martínez-Huerta, M.V. Carbonized Polydopamine-Based Nanocomposites: The Effect of Transition Metals on the Oxygen Electrocatalytic Activity. Nanomaterials 2023, 13, 1549. [Google Scholar] [CrossRef]
- Dent, A.J.; Cibin, G.; Ramos, S.; Smith, A.D.; Scott, S.M.; Varandas, L.; Pearson, M.R.; Krumpa, N.A.; Jones, C.P.; Robbins, P.E. B18: A core XAS spectroscopy beamline for Diamond. J. Phys. Conf. Ser. 2009, 190, 012039. [Google Scholar] [CrossRef]
- Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 2005, 12, 537–541. [Google Scholar] [CrossRef]
- Li, J.; Li, J.; Zhou, X.; Xia, Z.; Gao, W.; Ma, Y.; Qu, Y. Highly Efficient and Robust Nickel Phosphides as Bifunctional Electrocatalysts for Overall Water-Splitting. ACS Appl. Mater. Interfaces 2016, 8, 10826–10834. [Google Scholar] [CrossRef]
- Ji, X.; Lin, Y.; Zeng, J.; Ren, Z.; Lin, Z.; Mu, Y.; Qiu, Y.; Yu, J. Graphene/MoS2/FeCoNi(OH)x and Graphene/MoS2/FeCoNiPx multilayer-stacked vertical nanosheets on carbon fibers for highly efficient overall water splitting. Nat. Commun. 2021, 12, 1380. [Google Scholar] [CrossRef]
- Kumar, P.; Murthy, A.P.; Bezerra, L.S.; Martini, B.K.; Maia, G.; Madhavan, J. Carbon supported nickel phosphide as efficient electrocatalyst for hydrogen and oxygen evolution reactions. Int. J. Hydrogen Energy 2021, 46, 622–632. [Google Scholar] [CrossRef]
- Bhanja, P.; Kim, Y.; Paul, B.; Kaneti, Y.V.; Alothman, A.A.; Bhaumik, A.; Yamauchi, Y. Microporous nickel phosphonate derived heteroatom doped nickel oxide and nickel phosphide: Efficient electrocatalysts for oxygen evolution reaction. Chem. Eng. J. 2021, 405, 126803. [Google Scholar] [CrossRef]
- Battiato, S.; Urso, M.; Cosentino, S.; Pellegrino, A.L.; Mirabella, S.; Terrasi, A. Optimization of Oxygen Evolution Reaction with Electroless Deposited Ni–P Catalytic Nanocoating. Nanomaterials 2021, 11, 3010. [Google Scholar] [CrossRef] [PubMed]
- Louie, M.W.; Bell, A.T. An Investigation of Thin-Film Ni–Fe Oxide Catalysts for the Electrochemical Evolution of Oxygen. J. Am. Chem. Soc. 2013, 135, 12329–12337. [Google Scholar] [CrossRef]
- Cheng, N.; Liu, Q.; Tian, J.; Sun, X.; He, Y.; Zhai, S.; Asiri, A.M. Nickel oxide nanosheets array grown on carbon cloth as a high-performance three-dimensional oxygen evolution electrode. Int. J. Hydrogen Energy 2015, 40, 9866–9871. [Google Scholar] [CrossRef]
- Li, Z.; Dou, X.; Zhao, Y.; Wu, C. Enhanced oxygen evolution reaction of metallic nickel phosphide nanosheets by surface modification. Inorg. Chem. Front. 2016, 3, 1021–1027. [Google Scholar] [CrossRef]
- Hung, K.-M.; Wu, J.-J. Bifunctional nickel phosphide nanoparticle/nickel cobalt sulfide nanosheet framework for electrocatalytic simultaneous hydrogen evolution and 2,5-Furandicaroxylic acid production. Chem. Eng. J. 2024, 484, 149772. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | C | N | Ni | P | P/Ni |
|---|---|---|---|---|---|
| Ni | 75 | 1.7 | 19 | 0.0 | - |
| NiP 0.15 | 58 | 5.1 | 18 | 4.5 | 0.25 |
| NiP 0.3 | 50 | 4.0 | 22 | 8.1 | 0.36 |
| NiP 0.6 | 51 | 3.7 | 19 | 8.2 | 0.43 |
| NiP 0.9 | 48 | 3.0 | 16 | 7.5 | 0.46 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ríos-Ruiz, D.; Arévalo-Cid, P.; Cebollada, J.; Celorrio, V.; Čeh, M.; Drev, S.; Martínez-Huerta, M.V. Controlled Synthesis of Nickel Phosphides in Hollow N, P Co-Doped Carbon: In Situ Transition to (Oxy)hydroxide Phases During Oxygen Evolution Reaction. Catalysts 2025, 15, 292. https://doi.org/10.3390/catal15030292
Ríos-Ruiz D, Arévalo-Cid P, Cebollada J, Celorrio V, Čeh M, Drev S, Martínez-Huerta MV. Controlled Synthesis of Nickel Phosphides in Hollow N, P Co-Doped Carbon: In Situ Transition to (Oxy)hydroxide Phases During Oxygen Evolution Reaction. Catalysts. 2025; 15(3):292. https://doi.org/10.3390/catal15030292
Chicago/Turabian StyleRíos-Ruiz, David, Pablo Arévalo-Cid, Jesús Cebollada, Verónica Celorrio, Miran Čeh, Sandra Drev, and María Victoria Martínez-Huerta. 2025. "Controlled Synthesis of Nickel Phosphides in Hollow N, P Co-Doped Carbon: In Situ Transition to (Oxy)hydroxide Phases During Oxygen Evolution Reaction" Catalysts 15, no. 3: 292. https://doi.org/10.3390/catal15030292
APA StyleRíos-Ruiz, D., Arévalo-Cid, P., Cebollada, J., Celorrio, V., Čeh, M., Drev, S., & Martínez-Huerta, M. V. (2025). Controlled Synthesis of Nickel Phosphides in Hollow N, P Co-Doped Carbon: In Situ Transition to (Oxy)hydroxide Phases During Oxygen Evolution Reaction. Catalysts, 15(3), 292. https://doi.org/10.3390/catal15030292