Abstract
Glycoside hydrolase family 11 (GH11) xylanases are used in various industries, such as biorefining, animal feed production, and baking, making them key industrial enzymes. Operating bioprocesses at elevated temperatures enhances the reaction rate and product yield and thus requires thermostable enzymes to sustain catalytic performance. The limited availability of naturally occurring thermostable GH11 xylanases necessitates targeted modifications via protein engineering to enhance their thermal stability. In this review, we present the key drivers of thermostability, an overview of engineering strategies, and the underlying mechanisms of action. Finally, we investigated state-of-the-art technologies involving artificial intelligence (AI)- and ancestral sequence reconstruction-guided approaches.
1. Introduction
Xylanase plays a crucial role in xylan degradation, a major component of hemicellulose in plant cell walls. Among the glycoside hydrolase (GH) family members in the Carbohydrate-Active Enzymes (CAZy) database, xylanase catalyzes the hydrolysis of β-1,4-xylosidic linkages in xylan, generating xylooligosaccharides and/or xylose. Xylan degradation is an important bioprocess in various industries, including biofuel production, paper/pulp production, animal feed production, and food processing/production.
GH11 xylanase exhibits a high specificity towards xylan and is thus considered “true xylanase”. This property is beneficial for limiting byproduct generation from non-xylan substrates in lignocellulosic biomass, such as cellulose. Operating enzyme reactions at elevated temperatures, such as in pulp and feed/food bioprocessing, can enhance the reaction rate and yield by increasing substrate/product solubility and reducing microbial contamination [1]. Heat-resistant xylanases must maintain their activity and structural integrity under high-temperature conditions for sustained periods. However, most naturally derived GH11 xylanases are heat-susceptible. Therefore, native enzymes require artificial tailoring, for example, using protein engineering, to achieve the desired thermal stability.
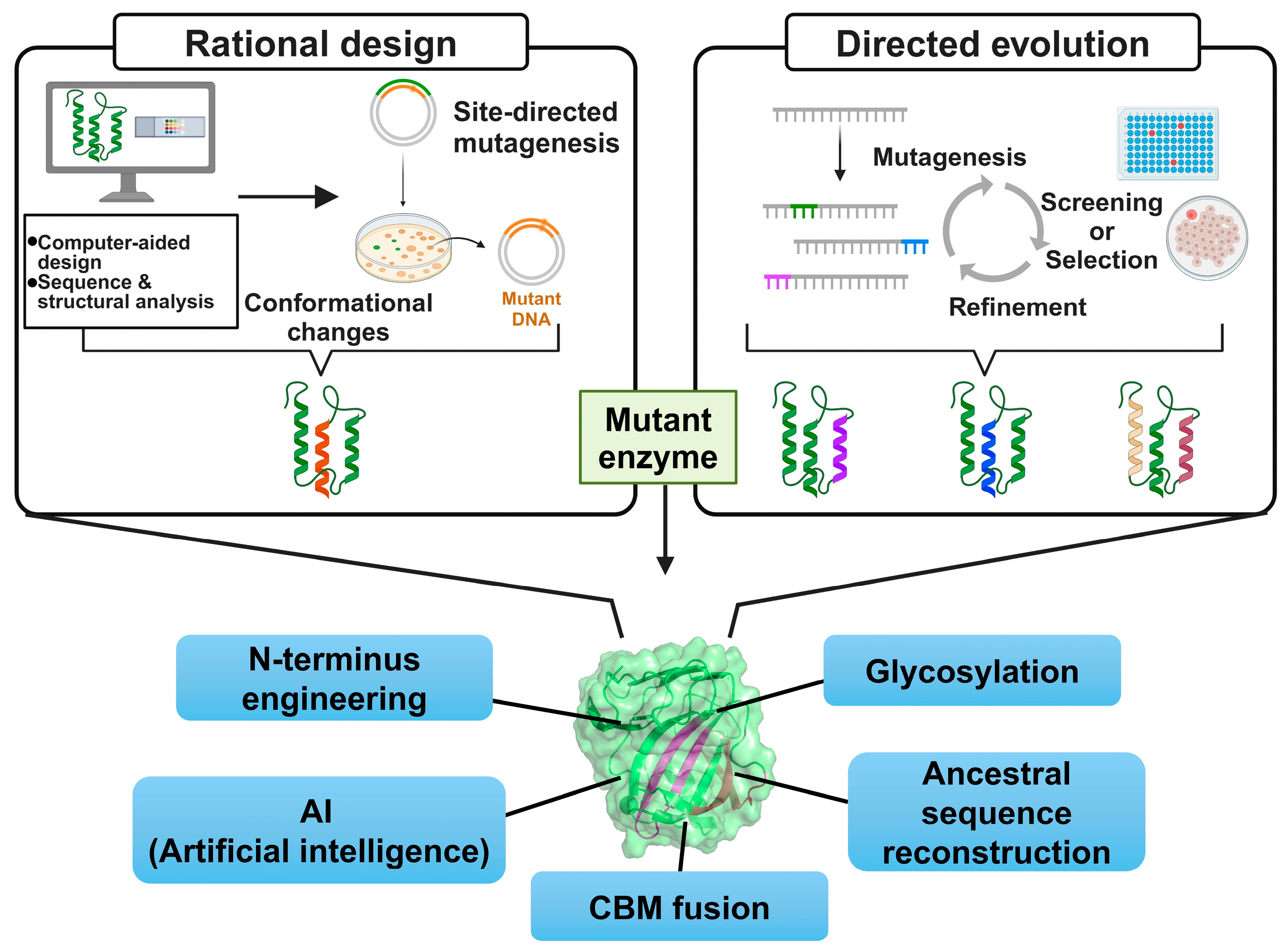
Protein engineering involves amino acid sequence design and modification to enhance or introduce specific properties and has been applied to generate various xylanase genes conferring improved thermostability. Rational design (RD) and directed evolution (DE) are well-established conventional protein engineering approaches (Figure 1). RD is a structure-, sequence-, and computation-based method that allows for targeted modifications [2]. This approach reduces the number of variants required for experimental examination by incorporating detailed structural information and simulations, lowering experimental costs and duration. However, this approach is limited by its dependence on accurate 3D structural data and sparse information regarding the effects of amino acid changes. DE mimics Darwin’s natural selection theory and, unlike RD, does not require structural information but rather functions by introducing random mutations [3]. DE can reveal unexpected protein functions and enables the creation of novel proteins with new properties.
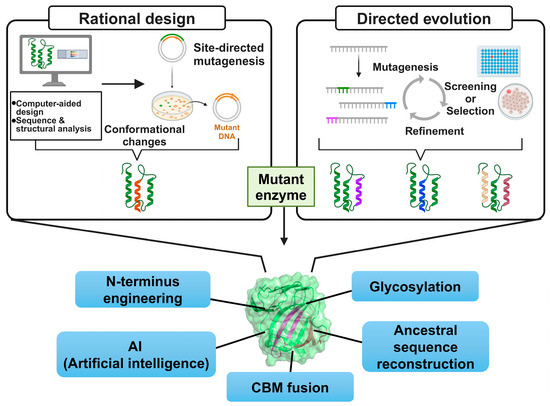
Figure 1.
Overall scheme of protein engineering approaches to generate thermostable GH11 xylanases.
Conventional protein engineering methods are limited by the limited availability of structural data, the need for large-scale screening, and the inability to explore the full sequence space of protein variants [4]. Emerging artificial intelligence (AI) technologies present promising strategies for overcoming these limitations and advancing the protein engineering field. Data-driven machine learning (ML) can approximate protein fitness landscapes and identify catalytic patterns using limited experimental data [5], promoting efficient protein mutation design and catalytic performance prediction. AI systems can be applied to predict new protein structures with high accuracy and speed, producing vast protein structure databases, such as AlphaFold (https://alphafoldserver.com/), without needing to experimentally elucidate the crystal structure, which is especially useful in protein engineering [6].
This review investigates the key factors associated with GH11 xylanase thermostability, recent trends and advances in engineering strategies and technologies, and potential underlying mechanisms of action. Finally, we discuss the prospects of emerging ML- and ancestral sequence reconstruction (ASR)-guided engineering strategies for GH11 xylanases.
2. Structure and Reaction Mechanism of GH11 Xylanases
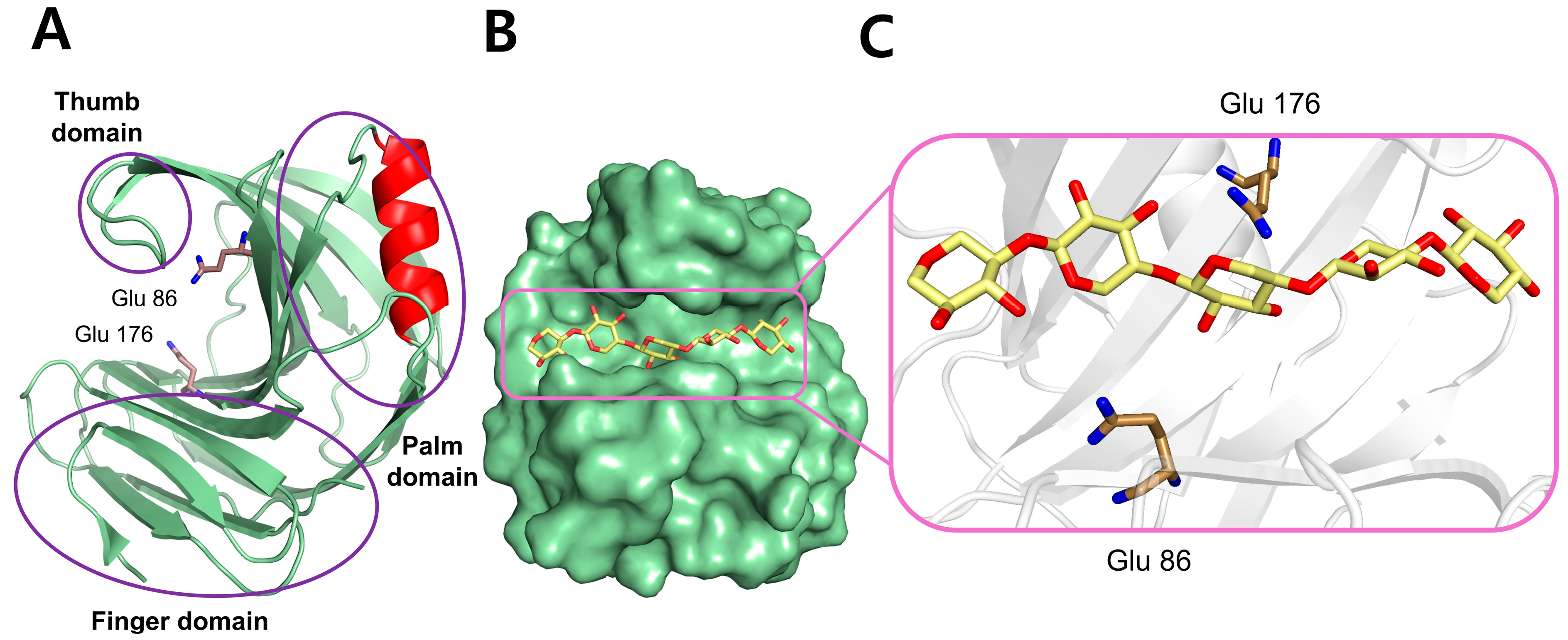
GH11 xylanase has a β-jelly roll fold. Because of its resemblance to the human hand, the three domains of GH11 xylanase are called the palm, fingers, and thumb (Figure 2A) [7]. The GH11 xylanase consists of a single α-helix and two antiparallel twisted β-sheets, stably formed by hydrogen bonds between β-strands. The active site exhibits an open cleft, consistent with the endo-mode of the enzyme, which can accommodate the linear chain of the xylan substrate (Figure 2B,C); two conserved catalytic Glu residues are positioned in the middle of the substrate-binding cleft. The substrate-binding cleft has six subunits, of which glycosidic linkages within the xylan backbone are cleaved at the positions between −1 and +1 by the two Glu catalytic residues. The substrate-binding cleft harbors aromatic amino acids, such as Trp and Tyr, which form π–π stacking interactions with the aromatic rings present in xylan substrates for specific recognition [8].
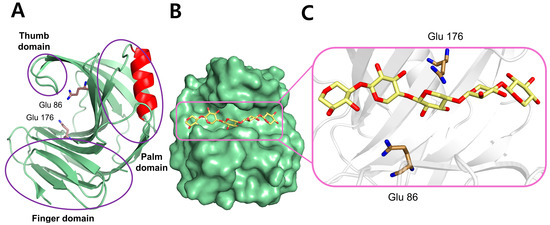
Figure 2.
The structure of a representative GH11 xylanase (PDB: 1XYO) from Trichoderma reesei is displayed, including the domain organization and substrate binding. (A) Cartoon representation of the overall structure emphasizing the domain composition. (B) Surface representation of the enzyme complexed with a xylan substrate (yellow stick), clearly showing the open binding cleft. (C) Active site enlargement showing the interaction between the xylan substrate and the catalytic residues (Glu86 and Glu176).
GH11 xylanases are categorized as retaining enzymes because they retain the anomeric configuration of the substrate during hydrolysis [9]. The retention mechanism involves a two-step reaction, glycosylation and deglycosylation, during which two glutamates in the active sites of GH11 xylanases act respectively as the acid/base catalyst and nucleophile. The glycosylation step involves general acid–base catalysis, where proton transfer initiates the cleavage of the glycosidic bond in the xylan substrate. Here, the acid/base Glu residue donates a proton to the glycosidic oxygen, facilitated by the nucleophilic attack of the anomeric carbon by the other Glu residue. This leads to the formation of the α-glycosyl enzyme intermediate—a covalent intermediate between the enzyme and xylan substrate. During deglycosylation, the glycosyl moiety of the intermediate is released from the active site of the enzyme. The acid/base Glu residue deprotonated in the previous step serves as the proton acceptor in this step, abstracting a proton from the water molecule. This leads to the formation of GluH (protonated glutamic acid) and a hydroxide ion (OH−). The OH− attacks the anomeric carbon of the sugar substrate, separating the remaining sugar product from the nucleophilic Glu of the enzyme. Notably, this process occurs with inversion of stereochemistry, thus retaining the β configuration of the anomeric carbon for hydrolysis products.
3. Protein Engineering Strategies for GH11 Xylanases
Various engineering strategies have been used to generate gene diversity in GH11 xylanases for improved thermal stability by rigidifying the enzyme structure (Table 1). A compact protein structure with few exposed hydrophobic patches confers little flexibility and enhances thermostability [10]. Therefore, conventional engineering technologies focus on reducing flexibility and increasing surface entropy [11]. In contrast, there are also cases in which the stabilizing mutation is located in the rigid β-sheet area rather than a flexible loop [12]. A computationally designed mutant (XynCDBFV-m) was thermally stabilized with the Tm increasing by 14 °C by substituting residues in the rigid β-sheet, thereby strengthening hydrophobic interaction. The mutant exhibited an approximately 10-fold higher xylooligosaccharide production than the wild-type, a product that is usable as a sweetener and prebiotic in the food and feed industries.

Table 1.
Various engineering strategies and underlying mechanisms for improving thermostability of GH11 xylanases.
Here, we investigate up-to-date engineering approaches and their underlying mechanisms using representative example enzymes (Figure 1).
3.1. N-Terminal Engineering
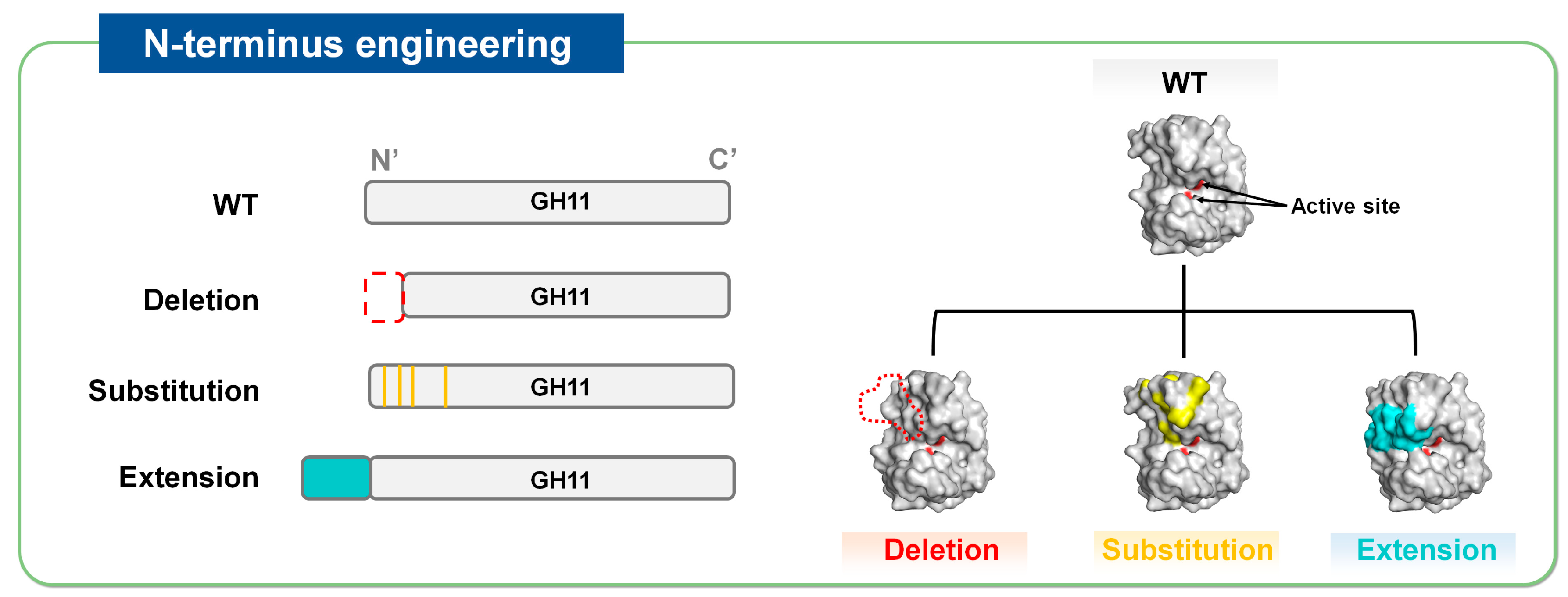
The N-terminal region is closely associated with the catalytic activity and thermal stability of GH11 xylanases [20]. Thermostable GH11 xylanases tend to have extended N-terminal sequences that form strong interactions via disulfide bonds [21]. In contrast, their mesophilic counterparts often possess a nonstructured and flexible N-terminal region (i.e., a loop) [22]. Molecular dynamics analysis revealed that the structural destabilization (i.e., denaturation) of GH11 xylanases starts from the N-terminal region under high-temperature conditions, highlighting a potential target (or hotspot) for mutation [22]. Therefore, N-terminal modification, in which elements in this region are removed, replaced, or extended by adding new segments, is considered a promising strategy for improving the thermal stability of GH11 xylanases (Figure 3).
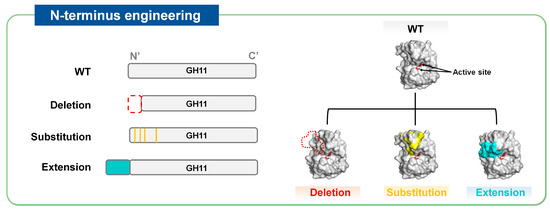
Figure 3.
Strategies of N-terminus engineering to improve thermostability of GH11 xylanases, represented by N-terminus deletion, substitution, and extension.
3.1.1. N-Terminal Deletion
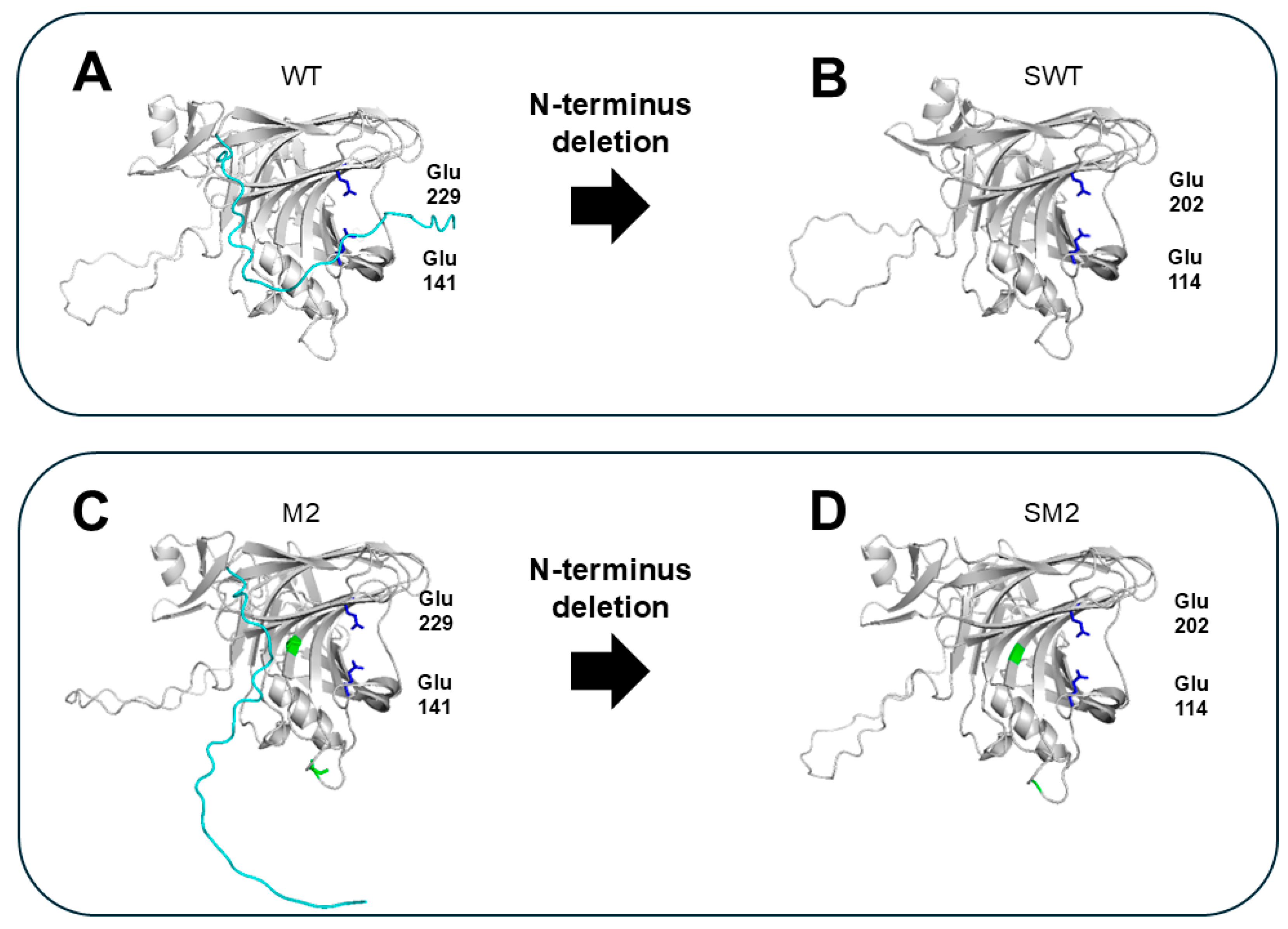
Non-structured amino acids at the N-terminus (without forming secondary structures (i.e., α-helix or β-sheet)), may confer flexibility to GH11 xylanases. The protein loops, located closely to the cleft of the active center, could be extended to the catalytic site forming a surface of the cleft, restricting access of the substrate to the active site (Figure 4A,C). Accordingly, various studies have attempted to delete the N-terminal sequences of GH11 xylanases to improve their thermostability and activity. The exact N-terminal amino acid residues responsible for flexibility (the targets for deletion) can be effectively determined using in silico analysis [23]. For example, deleting the highly flexible N-terminal tail (1–27 amino acid residues; based on B-factor analysis in silico) of the GH11 xylanase from Orpinomyces sp. PC-2 greatly enhanced the thermostability of two variants (SWT and SM2 generated from wild-type (WT) and M2 mutants (V135A and A226T), respectively), showing increased half-lives (t1/2) at 50 °C compared to the WT (0.8 h vs. 2.3 h for SWT and 29.5 h for SM2). Consequently, the activities of the two variants increased more than 10-fold. This experimentally proves that the N-terminus is the major factor that is responsible for the low activity and low thermal stability of the enzyme possibly attributed to its location close to the catalytic center on one hand and its high flexibility on the other hand and that its deletion is an effective RD strategy for improving the thermal stability and activity of GH11 xylanases.
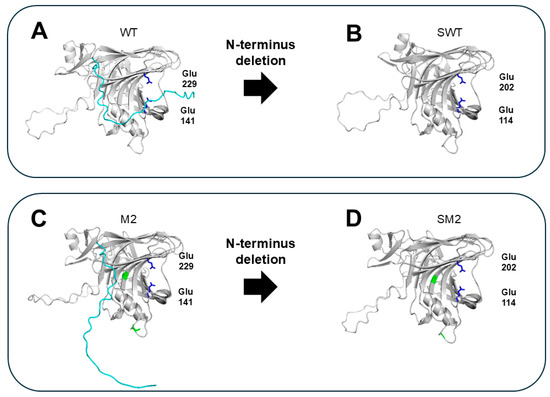
Figure 4.
Structural comparison of GH11 xylanases with intact and deleted unstructured N-terminal region. The model structures were predicted using enzyme sequences from Orpinomyces sp. PC-2 through AlphaFold [23]. N-terminal sequence is shown in cyan. The catalytic residues Glu141 and Glu229 are shown in blue. The point mutations V135A and A226T are shown in green. (A) WT: native enzyme with intact N-terminal tail. (B) SWT: WT with the N-terminal tail removed. (C) M2: a variant with point mutations in WT (V135A and A226T). (D) SM2: an M2 variant with N-terminal truncation.
The presence of the N-terminus, albeit non-structured, is not always deleterious to thermostability. In a GH11 xylanase from Aspergillus niger (Xyn), the deletion of five N-terminal residues (XynΔN) decreased thermostability, exhibiting decreased t1/2 at 50 °C compared to that of WT (33.9 h vs. 15.7 min for XynΔN) [24]. In addition, the optimal temperature (Topt) shifted from 48 °C to 46 °C. In contrast, its thermal properties were enhanced by the removal of the C-terminal tail (XynΔC), which increased Topt to 50 °C and produced a t1/2 at 50 °C of 73.9 min. The thermal properties lost by the N-terminal deletion were not recovered by the C-terminal deletion. This selective effect can be explained in terms of N-terminal rigidity and C-terminal flexibility. The authors posit that the N-terminal residues in Xyn, located upstream of the protein, may be crucial for maintaining structural stability and ensuring proper folding, whereas the C-terminus, located downstream, appears to contribute less to protein folding and may instead introduce unwanted flexibility.
Notably, the impact of terminus modifications on the stability of GH11 xylanase does not follow a general rule but is instead highly dependent on structural context.
3.1.2. N-Terminal Substitution
Given that the flexible N-terminal region frequently has a detrimental effect on the thermal integrity of GH11 xylanases [9,17,25,26], the entire sequence or key residues in this region have been modified to improve thermostability [27]. Some thermophilic GH11 xylanases exhibit conserved N-terminal amino acid sequences, suggesting that these residues are evolutionary hallmarks [28]. The critical region (or site) associated with thermostability can be identified based on multiple sequence alignment and structural comparisons of mesophilic and thermophilic xylanases. Recently, Li et al. identified flexible N-(1–37) and C-terminal (179–188) sequences of xynA from Aspergillus niger AG11 through molecular docking (MD) simulations, which were then replaced with the corresponding regions in enzymes from eight thermophiles, Thermopolyspora flexuosa, Nesterenkonia xinjiangensis, Actinomadura sp., Bacillus amyloliquefaciens, Talaromyces funiculosus, Thermothelomyces heterothallicus, and Thermotoga maritima as well as a mutant enzyme (EvXyn11TS). Consequently, the variant replaced with N- and C-terminal sequences from EvXyn11TS and N. xinjiangensis xylanases (EV-xynA-NX), respectively, performed best in terms of stability, with remarkably increased residual activity and t1/2 at 50 °C (1043.6 min) compared to xynA (18.2 min). Notably, the catalytic properties (Km, kcat/Km, and specific activity) of EV-xynA-NX were not significantly sacrificed. Since the rigidity required for stability can limit the flexibility required for efficient catalysis, enhanced protein stability is often achieved at the expense of catalytic activity. Structural analysis in the study proved that the mutation sites of EV-xynA-NX were far from the substrate-binding and catalytic sites, hardly affecting the catalytic properties. The key mechanism underlying its stabilization may be associated with increased interactions, such as disulfide bonds, hydrogen bonds, and electrostatic interactions, particularly at the N-terminus. Notably, a hydrophobic cluster consisting of three aromatic residues was newly formed between its N- and C-termini. Current evidence suggests that the N- and C-terminal interactions caused by adjacent residues may be important for enzyme stability as previously demonstrated [29,30].
3.1.3. N-Terminal Extension
Thermophilic GH11 xylanases tend to have longer N-termini than their mesophilic counterparts, which can increase stability through additional interactions. Thus, replacement with the corresponding N-terminal sequences from thermophilic xylanases can produce an extended N-terminus [28,31]. The mesophilic xylanases AEXyn11A and EV-xynA-NX, in which the N-termini were replaced with those of EvXyn11TS (one of the most heat-resistant xylanases), exhibited additional disulfide bonds (in AEXyn11A) or new interactions, including hydrophobic, electrostatic, and disulfide bonds (in EV-xynA-NX) [28,31]. The thermal stabilities of these variants with extended N-termini were significantly enhanced. Variants substituted with longer N-termini from thermophilic xylanases often possess new β-strands in this region instead of loops, as in AEXyn11A, further promoting interactions in the local region (i.e., N-terminus), in turn promoting protein structural integrity under high temperatures. This is consistent with the presence of β-strands at the N-terminus of native thermophilic xylanases, whereas native mesophilic xylanases frequently feature a long, flexible, and structurally disordered loop [22].
N-terminus extension was previously achieved by adding a few residues to existing terminal sequences in a thermolabile GH11 xylanase from Thermobacillus xylanilyticus with an unusually short N-terminus (Tx-Xyn) [26]. Contrary to expectations, the thermal stability was reduced when 17 N-terminal residues of the thermostable GH11 xylanase from Neocallimastix patriciarum (Np-Xyn) were added. Instead, catalytic activity was increased, possibly due to the formation of an extra subunit for substrate accommodation. N-terminal extension often generates an additional β-sheet in the palm domain in GH11 to provide additional substrate binding sites [26]. This suggests that, while many xylanases with high thermostability, either native or engineered, exhibit an extended N-terminus that reinforces structural interactions, this is not the rule and other mechanisms may be involved.
3.2. Chimera Construction
Natural GHs are often found with modular architectures comprising a catalytic domain (CD) and non-catalytic carbohydrate-binding module (CBM), each folding independently. The CD and CBM are connected by a flexible glycine- and proline-rich linker, thereby separating the two domains to perform their distinct functions [32]. In the CAZy database, CBMs are categorized into approximately 100 families based on their sequence similarity (www.cazy.org) [33]. Another classification system groups CBMs according to their structures into Type A (families 1, 2a, 3, 5, and 10), Type B (families 2b, 4, 6, 15, 17, 20, 22, 27, 28, 29, 34, and 36), and Type C (families 9, 13, and 14), which have distinct substrate specificities [34,35]. Type A CBMs with flat surfaces exhibit a strong affinity for crystalline and insoluble substrates, whereas Type B CBMs with deep clefts in their structures internally recognize linear oligosaccharides. Type C CBMs tend to bind small sugars at oligosaccharide termini [36]. With a high binding affinity towards xylan, the CBMs in GH11 promote (1) substrate targeting by identifying the specific binding site and (2) local enzyme concentrations, both of which determine the rate of hydrolysis [37]. In this reaction, CBMs are synergistic accessory proteins that enhance the hydrolytic efficiency of CD in GHs through non-hydrolytic mechanisms [35]. In addition to modulating catalytic activity, CBMs also contribute to the thermostability of GHs, as evidenced by the decreased optimal temperature and thermostability of native GH enzymes after CBM truncation [38]. The underlying mechanism is thought to involve the stabilization of the enzyme structure, possibly by facilitating substrate binding or reducing protein conformational flexibility under thermal stress.
CBMs from various families, including CBM2, -5, -9, and -22, have been artificially fused to the terminal region of the GH11 xylanase (Table 2). This strategy has proven effective in chimeric enzymes, such as CDBFV-C2 and Xyn2-A2 [39,40]. Notably, although CBMs derived from (hyper)thermophilic organisms, such as T. maritima are often selected [39,40], their impact on enzyme stability is influenced by multiple factors other than the source, including CBM size/number, fusion position, and linker sequence. Large CBMs composed of multiple submodules with distinct functions often not only fail to enhance thermostability but can even reduce it [41,42]. Interestingly, modification with a submodule from these full-length CBMs into GH11 significantly enhanced thermal stability [39]. Specifically, GH11 from Neocallimastix patriciarum fused at the N′-, C′-, or both termini with the C2 submodule from CBM9_1-2 of T. maritima GH10 (called C2-CDBFV, CDBFV-C2, and C2-CDBFV-C2, respectively) showed a higher t1/2 than the WT, specifically: CDBFV-C2 > C2-CDBFV-C2 > C2-CDBFV > WT. This revealed that the fusion position may be a crucial determinant of thermal stability and requires careful consideration in the construction of a stable variant. Depending on the fusion position, CBMs differentially influence the overall structural organization of the enzyme by modulating intermolecular interactions, in turn affecting its structural rigidity and thermal stability. Finally, the linker region may be another influential factor in enzyme thermostability. In addition to their well-known role in conferring flexibility for easier domain separation, linker regions with high variations in length and sequence can strongly influence linker conformation, structure, and thermal stability [43]. The crucial role of the linker region in thermostability, among other enzymatic features, has been demonstrated in GH11 xylanases from Paenibacillus campinasensis [32]. This study aimed to remove the CBM with deleterious effects while retaining the linker. The presence of the linker did more than merely maintain thermostability, it resulted in the highest thermostability reported to date. The underlying mechanism is highly related to its stabilizing effect on the CD structure. Considering that the repeat sequence (GGGNP) of the linker may influence the rigidity of the CD, future engineering strategies should focus on fine-tuning the linker length and sequence for application in other xylanases.
Table 2.
CBM-fusion or -truncation strategies for improving thermostability of GH11 xylanases.
3.3. Glycosylation
Engineering N-glycosylation sites is another useful approach for rationally designing thermostable enzymes [46,47]. Protein N-glycosylation is a posttranslational modification (PTM) prevalent in eukaryotes, conferring great complexity to protein structure and function. N-glycosylation confers various functional roles in enzymes, including enhanced protein folding and stability, thereby promoting proteolytic degradation resistance and protein solubility, preventing aggregation, modulating enzymatic activity, and affecting substrate specificity [48]. The relationship between N-glycosylation and protein thermostability is well recognized. Although the underlying mechanism remains elusive, it may be related to active site rigidification [47].
Several studies have attempted to engineer N-glycosylation sites in GH11 xylanases by site-directed mutagenesis to optimize the pattern or number of glycosylations [28,49]. Glycosylation sites have been introduced into the flexible region (HFR IV) of XynA from A. niger. Whereas the unglycosylated WT completely lost its activity at 70 °C, the glycosylated mutants (A55N/D57S, S61N, and A55N/D57S/S61N) showed considerable residual activity (41.6–94.5%). The specific activities were also higher than those of the WT, indicating that the new N-glycosylation contributed to both thermal stability and catalytic activity. The introduced glycosylation sites (A55N, D57S, and S61N) are positioned away from the catalytic residues of the enzyme, implying its indirect effect on activity. The authors suggested that the binding of xylan substrates rich in hydroxyl groups could be promoted through glycan attachment in mutants, leading to increased substrate recognition and activity [28]. In XynA from B. subtilis, extensive glycosylation was not necessarily beneficial for thermostability [49]. After identifying several N-glycosylation sites in XynA from P. pastoris (a eukaryotic host with an innate PTM system) using intact mass spectrometry, combinatorial mutants with specific N-glycosylation site deletions were constructed through site-directed mutagenesis. By comparing the t1/2 of the WT and several mutants, a crucial glycosylation site associated with thermostability was identified. Moreover, the findings suggested that a reduced level of glycosylation does not necessarily decrease thermostability but can instead enhance it. Maintaining the number of glycosylation sites at different positions can improve thermostability, implying that the position (rather than the number) of glycosylation sites is a key determinant of xylanase thermostability.
4. Recent Advances
4.1. AI Technologies
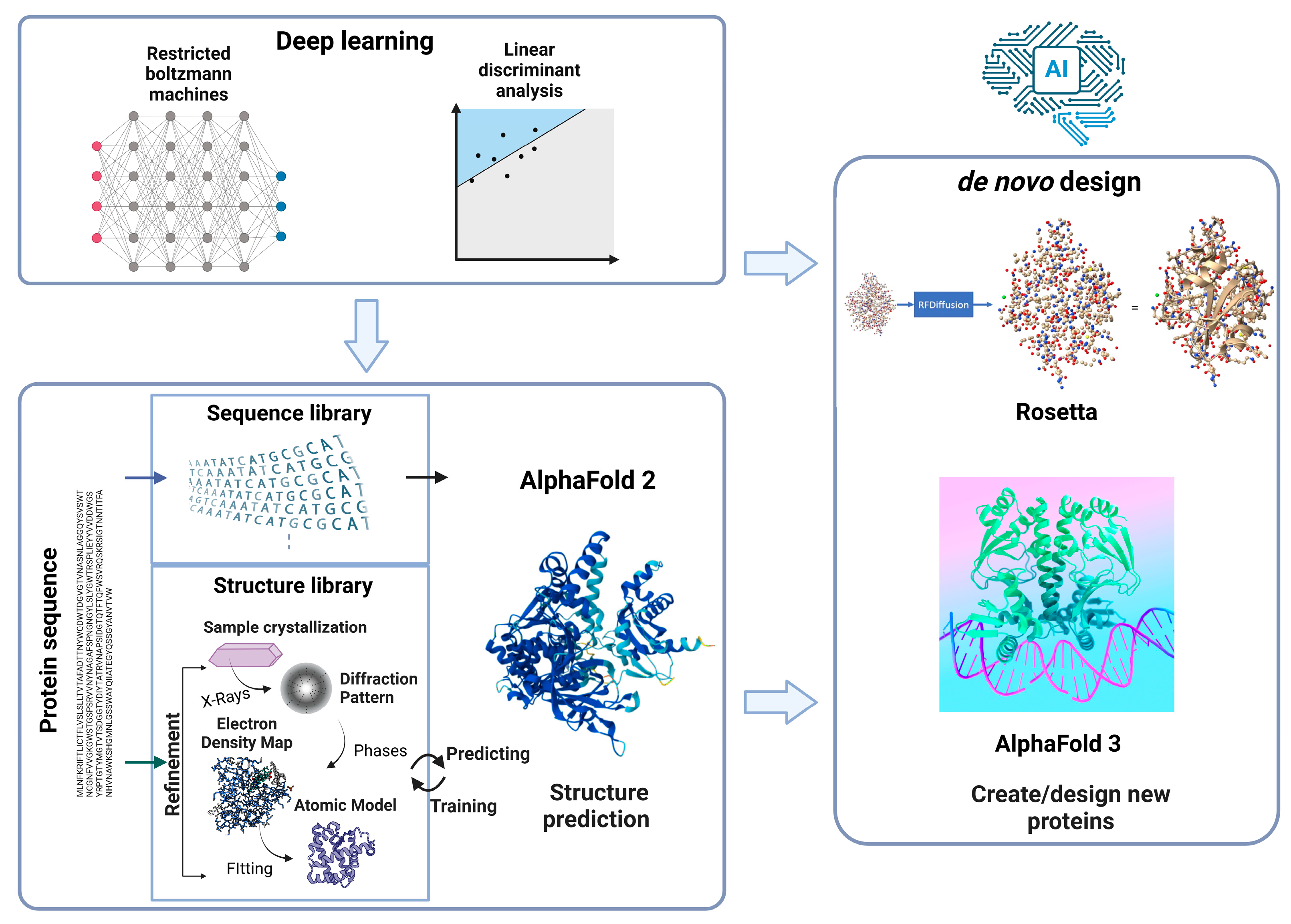
Recent progress in AI has greatly influenced all sectors of science. With their Nobel-winning contributions in Chemistry in 2024, Demis Hassabis, John Jumper, and David Baker incorporated AI technologies and protein structure/engineering, thereby revolutionizing research in various fields, such as structural biology and biotechnology (Figure 5).
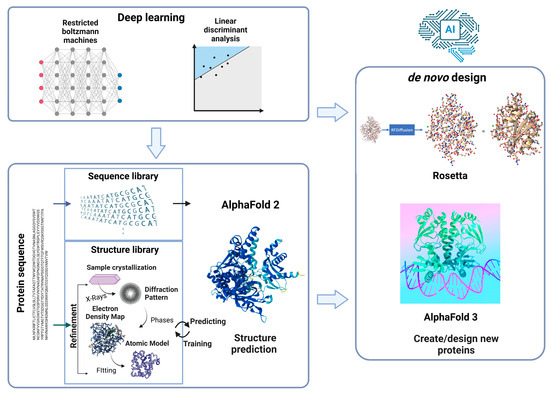
Figure 5.
Schematic illustration of AI-driven approach for protein engineering.
Classical protein engineering, especially RD-based engineering, relies on highly accurate 3D structures that have been experimentally verified using X-ray crystallography, nuclear magnetic resonance, and cryogenic electron microscopy [50]. However, the empirical investigation of complex protein structures requires labor- and time-intensive procedures in addition to highly specialized skills and costly equipment (i.e., beamline for X-ray crystallography). The recently developed AlphaFold, a key contributor to the 2024 Nobel Prize in Chemistry, is a powerful AI-based tool for efficiently predicting protein structures. In contrast to conventional homology modeling that relies on homologous templates, AlphaFold can build structural models from scratch (directly from amino acid sequences) with much higher speed and accuracy using deep learning algorithms trained to recognize patterns between amino acid sequences and structural features, which not only shortens development time but also expands its applicability to a wide range of proteins with experimentally unknown structures. AlphaFold has predicted structures for over 200 million proteins to date, far exceeding the approximately 200,000 structures determined through experimental methods, avoiding intensive experimental processes, and saving valuable resources and time. The increasing availability of protein structures can promote the rational design of customized biocatalysts with desirable properties.
AI approaches can advance protein engineering in different ways, such as through the rapid prediction of the properties of new protein sequences and the design of novel and potentially advantageous variants based on learned data patterns [51]. For this purpose, amino acid sequences that are readily accessible from public databases, such as BRENDA and InterPro are used as input. The predicted output data represent the protein properties of interest, such as thermostability, catalytic efficiency, and substrate specificity. Recently, an ML approach (freely available at arimees.com) was developed to efficiently predict and screen xylanases with the desired thermal properties from metagenomic sources [52], which can be further extended to engineer thermostable xylanases. Although AI-based protein engineering requires high initial computational costs compared with conventional RD and DE, it can significantly reduce experimental costs by minimizing trial-and-error processes and improving success rates. With its high predictive accuracy, the overall efficiency of protein engineering is enhanced.
4.2. Ancestral Sequence Reconstruction
ASR uses phylogenetic trees, multiple sequence alignment, and computational algorithms to infer the sequences of ancient proteins and trace their evolution over millions of years. Ancestral proteins are frequently more thermostable and functionally versatile than their contemporary counterparts. ASR offers valuable templates for engineering xylanases with enhanced stability and/or catalytic activity under harsh conditions without relying on prior knowledge of structure (as in the case of RD) or extensive protein expression or screening (as in the case of DE) [53,54,55,56]. Compared to DE, ASR shortens development time and reduces experimental costs by minimizing reduced screening but still requires the computational cost associated with sequence inference and reconstruction. ASR generally involves the construction of a phylogenetic tree of selected thermostable xylanases, from which the ancestral sequences are inferred using ASR tools, such as FireProtASR and FastML. Although the success rate for improving thermostability is high, mining and functional fine-tuning are required. MD simulations and sequence/structure analysis are useful for mining ancestral sequences with desirable properties. This is necessary because ancestral enzymes may be non-functional and versatile, requiring theoretical and experimental validation of their functions. Based on the ASR approach, Sun et al. obtained a robust GH11 xylanase (AncXyn18) that could withstand thermal and alkaline conditions [55]. Specifically, 25 GH11 xylanases with heat and alkaline tolerance have been reconstructed from 24 inferred ancestral sequences. Subsequent structural comparisons, MD, and expression tests identified three candidates (AncXyn03, AncXyn05, and AncXyn18), among which AncXyn18 displayed the highest performance with 50% residual activity at 70 °C after 20 h incubation or pH 10 after 10 h incubation. In another study, a xylanase from T. maritima (TmxB) and its homologs were used as the ASR template [56], from which three ancestral enzymes (TmxN1, TmxN2, and TmxN3) displaying a high number of H interactions with wheat xylan in MD simulations were finally chosen for experimental analysis. Among these, TmxN3 showed superior thermal (6-fold higher t1/2 at 100 °C) and kinetic properties (1.7-fold higher kcat/Km value) compared to the modern TmxB. Its robustness was further proven to enhance its performance in terms of bread quality, indicating its potential for industrial applications. Specifically, when TmxN3 is added at a concentration of 4.5 mg/kg of flour, the bread volume increases significantly from 3.5 cm to 4.5 cm. Additionally, TmxN3 inclusion reduces bread hardness, chewiness, and gumminess by 55.2%, 40.11%, and 53.52%, respectively.
5. Conclusions
This review investigates representative strategies and recent progress in the engineering of thermostable GH11 xylanases. Successful engineering strategies were associated with N-terminal modification, glycosylation, and chimera construction. We identified potential underlying mechanisms according to the detailed procedures of key studies and highlighted the complex roles and potential relationships of relevant factors. Finally, state-of-the-art AI and computational technologies have greatly advanced the engineering of GH11 xylanase with industrially beneficial traits and show great potential for reducing resource and time expenditures.
Author Contributions
Conceptualization, I.J.K.; writing—original draft preparation, B.S.K.; writing—review and editing, I.J.K.; visualization, B.S.K.; funding acquisition, I.J.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Research Foundation of Korea (NRF, Republic of Korea; grant number 2022R1I1A1A01072158) and the Development Fund Foundation of Gyeongsang National University, 2024.
Data Availability Statement
No new data were created or analyzed in this study.
Acknowledgments
Some figures were created from Biorender.com.
Conflicts of Interest
The authors declare they have no competing interests.
References
- Gault, S.; Higgins, P.M.; Cockell, C.S.; Gillies, K. A meta-analysis of the activiy, stability, and mutational characteristics of temperature-adapted enzymes. Biosci. Rep. 2021, 41, BSR20210336. [Google Scholar] [CrossRef]
- Bornscheuer, U.T.; Huisman, G.W.; Kazlauskas, R.J.; Lutz, S.; Moore, J.C.; Robins, K. Engineering the third wave of biocatalysis. Nature 2012, 485, 185–194. [Google Scholar] [CrossRef]
- Deng, L.; Li, C.; Lan, Y.; Sun, G.; Shang, C. Differential evolution with dynamic combination based mutation operator and two-level parameter adaptation strategy. Expert Syst. Appl. 2022, 192, 116298. [Google Scholar] [CrossRef]
- Wu, S.; Snajdrova, R.; Moore, J.C.; Baldenius, K.; Bornscheuer, U.T. Biocatalysis: Enzymatic synthesis for industrial applications. Angew. Chem. Int. Ed. 2020, 60, 88–119. [Google Scholar] [CrossRef]
- Ao, Y.F.; Dörr, M.; Menke, M.J.; Born, S.; Heuson, E.; Bornscheuer, U.T. Data-Driven protein engineering for improving catalytic activity and selectivity. ChemBioChem 2023, 25, e202300754. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Nam, K.H. Recognition of a Single β-D-xylopyranose molecule by xylanase GH11 from Thermoanaerobacterium saccharolyticum. Crystals 2024, 14, 402. [Google Scholar] [CrossRef]
- Kim, I.J.; Kim, S.R.; Kim, K.H.; Bornscheuer, U.T.; Nam, K.H. Characterization and structural analysis of the endo-1,4-β-xylanase GH11 from the hemicellulose-degrading Thermoanaerobacterium saccharolyticum useful for lignocellulose saccharification. Sci. Rep. 2023, 13, 17332. [Google Scholar] [CrossRef]
- Briganti, L.; Capetti, C.; Pellegrini, V.O.A.; Ghio, S.; Campos, E.; Nascimento, A.S.; Polikarpov, I. Structural and molecular dynamics investigations of ligand stabilization via secondary binding site interactions in Paenibacillus xylanivorans GH11 xylanase. Comput. Struct. Biotechnol. J. 2021, 19, 1557–1566. [Google Scholar] [CrossRef]
- Kazlauskas, R. Engineering more stable proteins. Chem. Soc. Rev. 2018, 47, 9026–9045. [Google Scholar] [CrossRef]
- Han, N.; Ma, Y.; Mu, Y.; Tang, X.; Li, J.; Huang, Z. Enhancing thermal tolerance of a fungal GH11 xylanase guided by B-factor analysis and multiple sequence alignment. Enzym. Microb. Technol. 2019, 131, 109422. [Google Scholar] [CrossRef]
- Bu, Y.; Cui, Y.; Peng, Y.; Hu, M.; Tian, Y.e.; Tao, Y.; Wu, B. Engineering improved thermostability of the GH11 xylanase from Neocallimastix patriciarum via computational library design. Appl. Microbiol. Biotechnol. 2018, 102, 3675–3685. [Google Scholar] [CrossRef]
- Han, N.; Miao, H.; Ding, J.; Li, J.; Mu, Y.; Zhou, J.; Huang, Z. Improving the thermostability of a fungal GH11 xylanase via site-directed mutagenesis guided by sequence and structural analysis. Biotechnol. Biofuels 2017, 10, 133. [Google Scholar] [CrossRef]
- Li, Q.; Wu, T.; Duan, Y.; Pei, J.; Zhao, L. Improving the thermostability and pH stability of Aspergillus niger xylanase by site-directed mutagenesis. Appl. Biochem. Microbiol. 2019, 55, 136–144. [Google Scholar] [CrossRef]
- Tian, W.; Zhang, Z.; Yang, C.; Li, P.; Xiao, J.; Wang, R.; Du, P.; Li, N.; Wang, J. Engineering mesophilic GH11 xylanase from Cellulomonas flavigena by rational design of N-terminus substitution. Front. Bioeng. Biotechnol. 2022, 10, 1044291. [Google Scholar] [CrossRef]
- Li, T.; Yang, S.; Wang, X.; Cai, H.; Wang, Y.; Li, C.; Li, E. Improving thermostability of GH11 xylanase XynASP by the design of loop region. Crystals 2022, 12, 1228. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, C.; Lu, H.; Wu, Q.; Wu, Y.; Li, W.; Li, X. Improvement of thermostability and catalytic efficiency of xylanase from Myceliophthora thermophilar by N-terminal and C-terminal truncation. Front. Microbiol. 2024, 15, 1385329. [Google Scholar] [CrossRef]
- Hokanson, C.A.; Cappuccilli, G.; Odineca, T.; Bozic, M.; Behnke, C.A.; Mendez, M.; Coleman, W.J.; Crea, R. Engineering highly thermostable xylanase variants using an enhanced combinatorial library method. Protein Eng. Des. Sel. 2011, 24, 597–605. [Google Scholar] [CrossRef]
- Ruller, R.; Alponti, J.; Deliberto, L.A.; Zanphorlin, L.M.; Machado, C.B.; Ward, R.J. Concommitant adaptation of a GH11 xylanase by directed evolution to create an alkali-tolerant/thermophilic enzyme. Protein Eng. Des. Sel. 2014, 27, 255–262. [Google Scholar] [CrossRef]
- Wu, Q.; Zhang, C.; Dong, W.; Lu, H.; Yang, Y.; Li, W.; Xu, Y.; Li, X. Simultaneously enhanced thermostability and catalytic activity of xylanase from Streptomyces rameus L2001 by rigidifying flexible regions in loop regions of the N-terminus. J. Agric. Food Chem. 2023, 71, 12785–12796. [Google Scholar] [CrossRef]
- Jänis, J.; Turunen, O.; Leisola, M.; Derrick, P.J.; Rouvinen, J.; Vainiotalo, P. Characterization of mutant xylanases using fourier transform ion cyclotron resonance mass spectrometry: Stabilizing contributions of disulfide bridges and N-terminal extensions. Biochemistry 2004, 43, 9556–9566. [Google Scholar] [CrossRef] [PubMed]
- Purmonen, M.; Valjakka, J.; Takkinen, K.; Laitinen, T.; Rouvinen, J. Molecular dynamics studies on the thermostability of family 11 xylanases. Protein Eng. Des. Sel. 2007, 20, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Ventorim, R.Z.; de Oliveira Mendes, T.A.; Trevizano, L.M.; dos Santos Camargos, A.M.; Guimarães, V.M. Impact of the removal of N-terminal non-structured amino acids on activity and stability of xylanases from Orpinomyces sp. PC-2. Int. J. Biol. Macromol. 2018, 106, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Permyakov, E.A.; Liu, L.; Sun, X.; Yan, P.; Wang, L.; Chen, H. Non-structured amino-acid impact on GH11 differs from GH10 Xylanase. PLoS ONE 2012, 7, e45762. [Google Scholar] [CrossRef]
- Bhat, S.K.; Purushothaman, K.; Kini, K.R.; Gopala Rao Appu Rao, A.R. Design of mutants of GH11 xylanase from Bacillus pumilus for enhanced stability by amino acid substitutions in the N-terminal region: An in silico analysis. J. Biomol. Struct. Dyn. 2021, 40, 7666–7679. [Google Scholar] [CrossRef]
- Song, L.; Dumon, C.; Siguier, B.; André, I.; Eneyskaya, E.; Kulminskaya, A.; Bozonnet, S.; O’Donohue, M.J. Impact of an N-terminal extension on the stability and activity of the GH11 xylanase from Thermobacillus xylanilyticus. J. Biotechnol. 2014, 174, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Zhang, C.; Zhu, W.; Lu, H.; Li, X.; Yang, Y.; Xu, Y.; Li, W. Improved thermostability, acid tolerance as well as catalytic efficiency of Streptomyces rameus L2001 GH11 xylanase by N-terminal replacement. Enzym. Microb. Technol. 2023, 162, 110143. [Google Scholar] [CrossRef]
- Li, Y.; Li, C.; Huang, H.; Rao, S.; Zhang, Q.; Zhou, J.; Li, J.; Du, G.; Liu, S. Significantly enhanced thermostability of Aspergillus niger xylanase by modifying Its highly flexible regions. J. Agric. Food Chem. 2022, 70, 4620–4630. [Google Scholar] [CrossRef]
- Hofmann, A.; Bhardwaj, A.; Leelavathi, S.; Mazumdar-Leighton, S.; Ghosh, A.; Ramakumar, S.; Reddy, V.S. The critical role of N- and C-terminal contact in protein stability and folding of a family 10 xylanase under extreme conditions. PLoS ONE 2010, 5, e11347. [Google Scholar] [CrossRef]
- Mahanta, P.; Bhardwaj, A.; Kumar, K.; Reddy, V.S.; Ramakumar, S. Structural insights intoN-terminal to C-terminal interactions and implications for thermostability of a (β/α)8-triosephosphate isomerase barrel enzyme. FEBS J. 2015, 282, 3543–3555. [Google Scholar] [CrossRef]
- Zhang, H.; Li, J.; Wang, J.; Yang, Y.; Wu, M. Determinants for the improved thermostability of a mesophilic family 11 xylanase predicted by computational methods. Biotechnol. Biofuels Bioprod. 2014, 7, 3. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.; Chang, S.; Gao, Z.; Ma, J.; Wu, B.; He, B.; Wei, P. Identification and characterization of a thermostable GH11 xylanase from Paenibacillus campinasensis NTU-11 and the distinct roles of its carbohydrate-binding domain and linker sequence. Colloids Surf. B Biointerfaces 2022, 209, 112167. [Google Scholar] [CrossRef]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [PubMed]
- Hao, M.-S.; Mazurkewich, S.; Li, H.; Kvammen, A.; Saha, S.; Koskela, S.; Inman, A.R.; Nakajima, M.; Tanaka, N.; Nakai, H.; et al. Structural and biochemical analysis of family 92 carbohydrate-binding modules uncovers multivalent binding to β-glucans. Nat. Commun. 2024, 15, 3429. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.J.; Lee, H.J.; Choi, I.-G.; Kim, K.H. Synergistic proteins for the enhanced enzymatic hydrolysis of cellulose by cellulase. Appl. Microbiol. Biotechnol. 2014, 98, 8469–8480. [Google Scholar] [CrossRef]
- Kim, I.J.; Ko, H.J.; Kim, T.W.; Choi, I.G.; Kim, K.H. Characteristics of the binding of a bacterial expansin (BsEXLX1) to microcrystalline cellulose. Biotechnol. Bioeng. 2012, 110, 401–407. [Google Scholar] [CrossRef]
- Krska, D.; Mazurkewich, S.; Brown, H.A.; Theibich, Y.; Poulsen, J.-C.N.; Morris, A.L.; Koropatkin, N.M.; Lo Leggio, L.; Larsbrink, J. Structural and functional analysis of a multimodular hyperthermostable xylanase-glucuronoyl esterase from Caldicellulosiruptor kristjansonii. Biochemistry 2021, 60, 2206–2220. [Google Scholar] [CrossRef]
- You, Y.; Kong, H.; Li, C.; Gu, Z.; Ban, X.; Li, Z. Carbohydrate binding modules: Compact yet potent accessories in the specific substrate binding and performance evolution of carbohydrate-active enzymes. Biotechnol. Adv. 2024, 73, 108365. [Google Scholar] [CrossRef]
- Miao, H.; Ma, Y.; Zhe, Y.; Tang, X.; Wu, Q.; Huang, Z.; Han, N. Improving the thermostability of a fungal GH11 xylanase via fusion of a submodule (C2) from Hyperthermophilic CBM9_1-2. Int. J. Mol. Sci. 2021, 23, 463. [Google Scholar] [CrossRef]
- Jun, H.; Bing, Y.; Keying, Z.; Xuemei, D.; Daiwen, C. Thermostable carbohydrate binding module increases the thermostability and substrate-binding capacity of Trichoderma reesei xylanase 2. New Biotechnol. 2009, 26, 53–59. [Google Scholar] [CrossRef]
- Liu, L.; Zeng, L.; Wang, S.; Cheng, J.; Li, X.; Song, A.; Wu, K.; Chen, H. Activity and thermostability increase of xylanase following transplantation with modules sub-divided from hyper-thermophilic CBM9_1-2. Process Biochem. 2012, 47, 853–857. [Google Scholar] [CrossRef]
- Mamo, G.; Hatti-Kaul, R.; Mattiasson, B. Fusion of carbohydrate binding modules from Thermotoga neapolitana with a family 10 xylanase from Bacillus halodurans S7. Extremophiles 2006, 11, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, Z.; Stepnov, A.A.; Tesei, G.; Wang, Y.; Buchinger, E.; Kristiansen, S.K.; Aachmann, F.L.; Arleth, L.; Eijsink, V.G.H.; Lindorff-Larsen, K.; et al. The effect of linker conformation on performance and stability of a two-domain lytic polysaccharide monooxygenase. J. Biol. Chem. 2023, 299, 105262. [Google Scholar] [CrossRef]
- Pandey, C.; Sharma, P.; Gupta, N. Engineering to enhance thermostability of xylanase: For the new era of biotechnology. J. Appl. Biol. Biotechnol. 2022, 11, 41–54. [Google Scholar] [CrossRef]
- Yang, A.; Cheng, J.; Liu, M.; Shangguan, Y.; Liu, L. Sandwich fusion of CBM9_2 to enhance xylanase thermostability and activity. Int. J. Biol. Macromol. 2018, 117, 586–591. [Google Scholar] [CrossRef]
- Han, C.; Wang, Q.; Sun, Y.; Yang, R.; Liu, M.; Wang, S.; Liu, Y.; Zhou, L.; Li, D. Improvement of the catalytic activity and thermostability of a hyperthermostable endoglucanase by optimizing N-glycosylation sites. Biotechnol. Biofuels 2020, 13, 30. [Google Scholar] [CrossRef]
- Ramakrishnan, K.; Johnson, R.L.; Winter, S.D.; Worthy, H.L.; Thomas, C.; Humer, D.C.; Spadiut, O.; Hindson, S.H.; Wells, S.; Barratt, A.H.; et al. Glycosylation increases active site rigidity leading to improved enzyme stability and turnover. FEBS J. 2023, 290, 3812–3827. [Google Scholar] [CrossRef] [PubMed]
- Varki, A. Biological roles of glycans. Glycobiology 2017, 27, 3–49. [Google Scholar] [CrossRef]
- Fonseca-Maldonado, R.; Vieira, D.S.; Alponti, J.S.; Bonneil, E.; Thibault, P.; Ward, R.J. Engineering the pattern of protein glycosylation modulates the thermostability of a GH11 xylanase. J. Biol. Chem. 2013, 288, 25522–25534. [Google Scholar] [CrossRef]
- Nam, K.H. Real-time monitoring of large-scale crystal growth using batch crystallization for serial crystallography. J. Cryst. Growth 2023, 614, 127219. [Google Scholar] [CrossRef]
- Yang, J.; Li, F.-Z.; Arnold, F.H. Opportunities and challenges for machine learning-assisted enzyme engineering. ACS Cent. Sci. 2024, 10, 226–241. [Google Scholar] [CrossRef]
- Foroozandeh Shahraki, M.; Farhadyar, K.; Kavousi, K.; Azarabad, M.H.; Boroomand, A.; Ariaeenejad, S.; Hosseini Salekdeh, G. A generalized machine-learning aided method for targeted identification of industrial enzymes from metagenome: A xylanase temperature dependence case study. Biotechnol. Bioeng. 2020, 118, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Pinto, G.P.; Corbella, M.; Demkiv, A.O.; Kamerlin, S.C.L. Exploiting enzyme evolution for computational protein design. Trends Biochem. Sci. 2022, 47, 375–389. [Google Scholar] [CrossRef] [PubMed]
- Ngo, K.; Bruno da Silva, F.; Leite, V.B.P.; Contessoto, V.G.; Onuchic, J.N. Improving the thermostability of xylanase a from Bacillus subtilis by combining bioinformatics and electrostatic interactions optimization. J. Phys. Chem. B 2021, 125, 4359–4367. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Qi, H.; Dou, G.; Mao, S.; Lu, F.; Tian, K.; Qin, H.-M. Ancestral sequence reconstruction of a robust β-1,4-xylanase and efficient expression in Bacillus subtilis. Int. J. Biol. Macromol. 2024, 282, 137188. [Google Scholar] [CrossRef]
- Hu, G.; Hong, X.; Zhu, M.; Lei, L.; Han, Z.; Meng, Y.; Yang, J. Improving the quality of wheat flour bread by a thermophilic xylanase with ultra activity and stability reconstructed by ancestral sequence and computational-aided analysis. Molecules 2024, 29, 1895. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).