
Hydrocracking of Polyethylene to Gasoline-Range Hydrocarbons over a Ruthenium-Zeolite Bifunctional Catalyst System with Optimal Synergy of Metal and Acid Sites


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
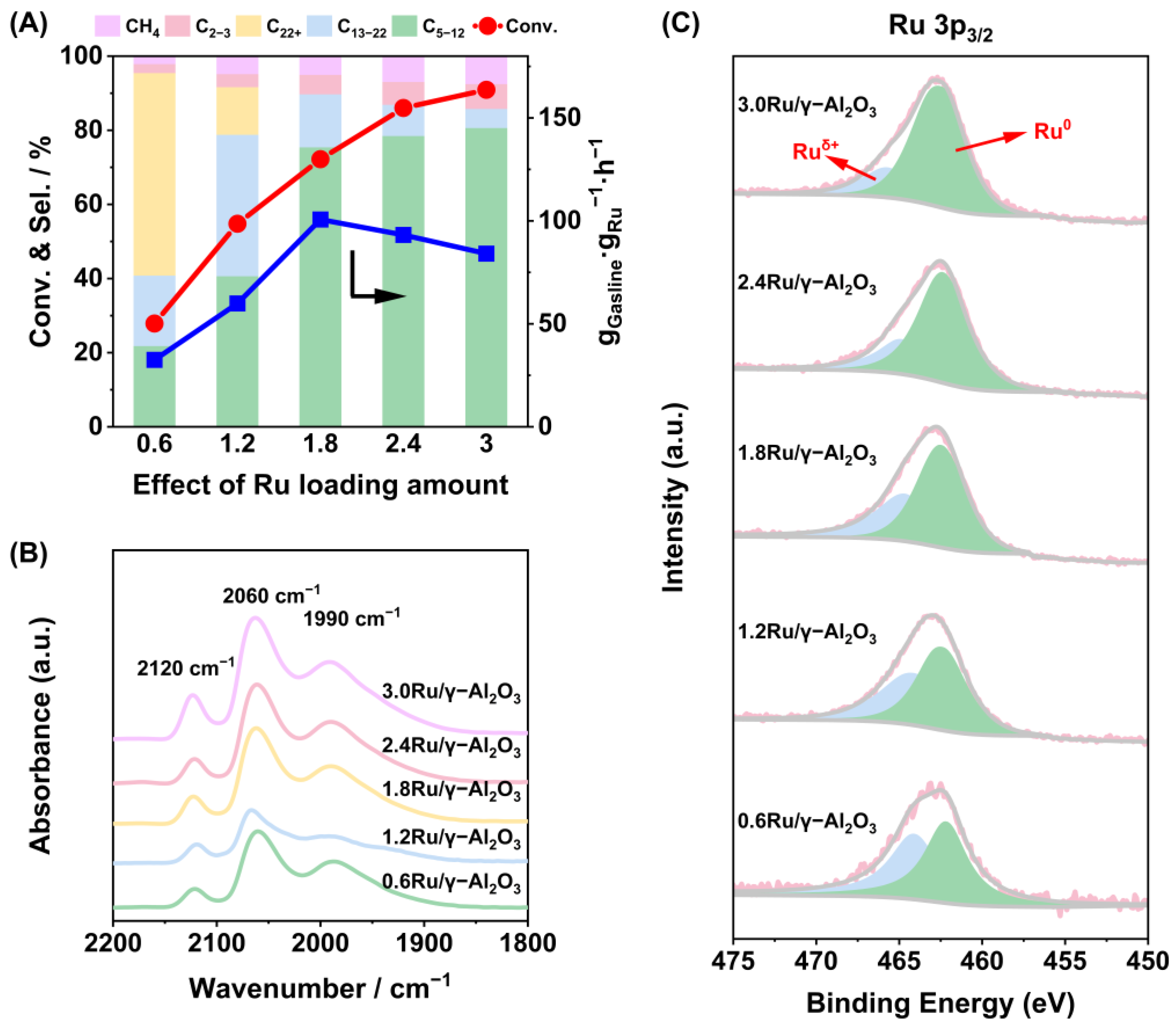
2.1. Catalytic Performance
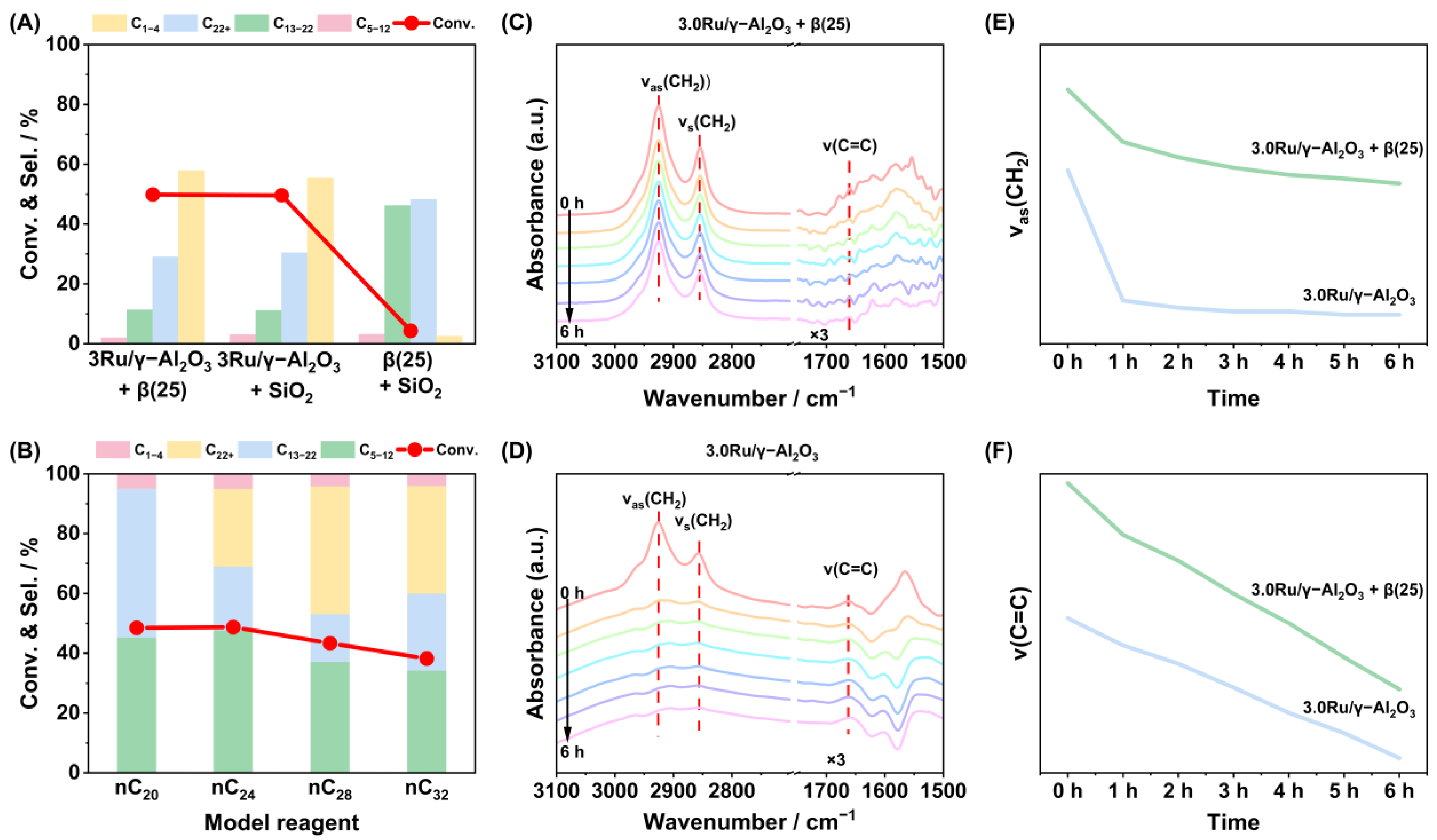
2.2. Effects of Metal–Acid Synergy
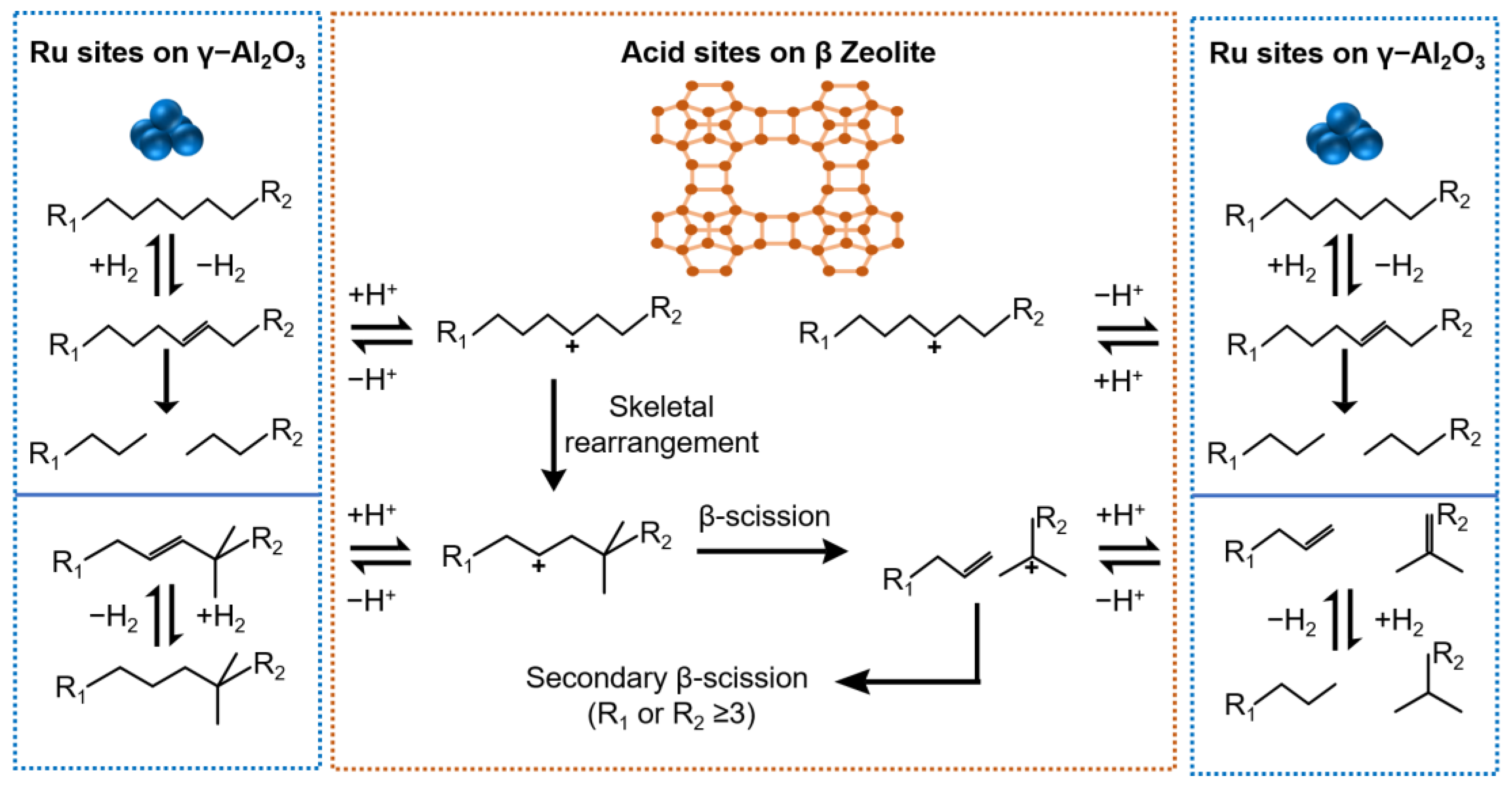
2.3. Reaction Mechanisms
3. Materials and Methods
3.1. Materials
3.2. Synthesis of Catalysts
3.3. Reaction Test
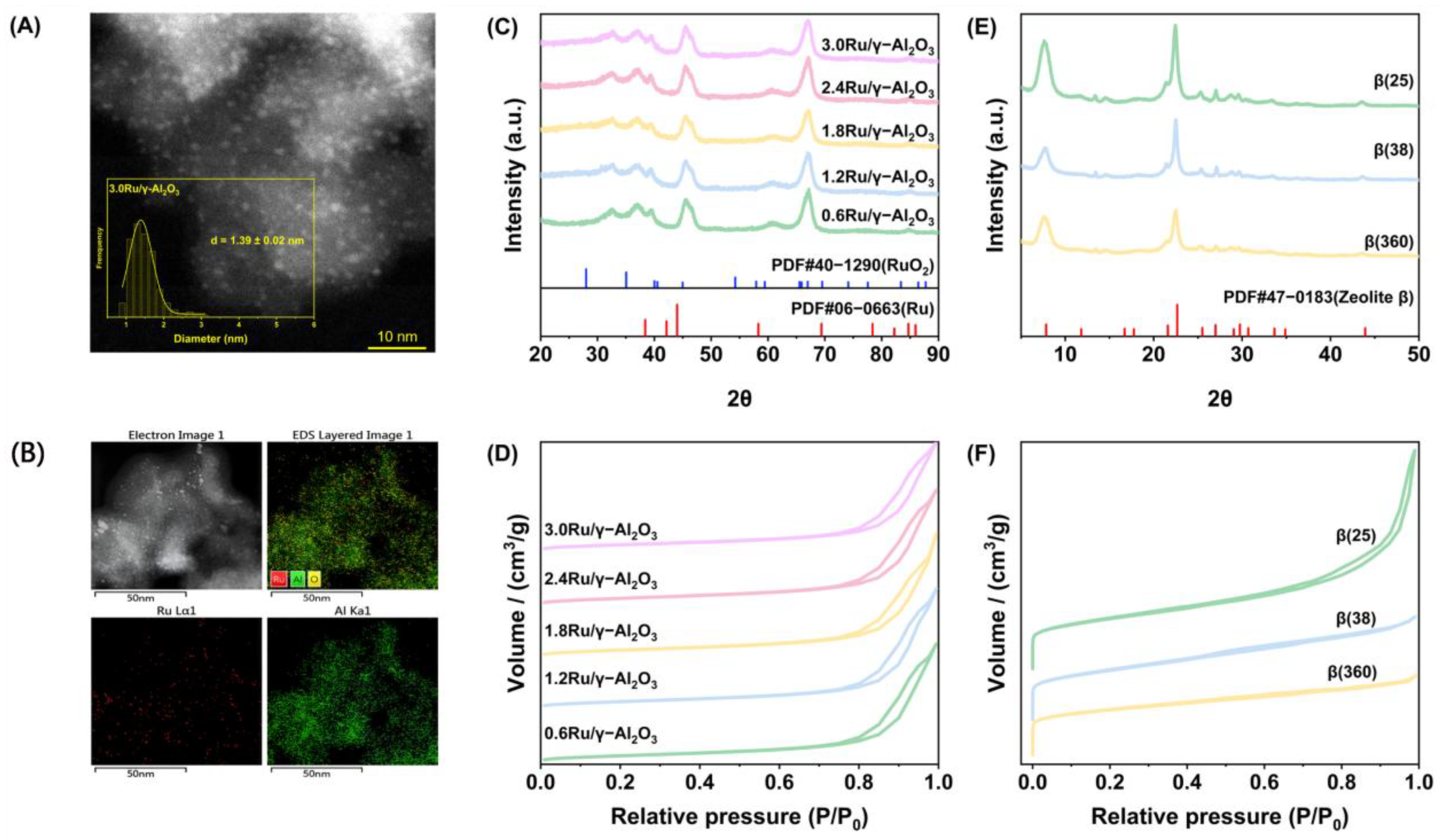
3.4. Catalyst Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Microplastics are everywhere—We need to understand how they affect human health. Nat. Med. 2024, 30, 913.
- Geyer, R.; Jambeck, J.R.; Law, K.L. Production, use, and fate of all plastics ever made. Sci. Adv. 2017, 3, e1700782. [Google Scholar] [CrossRef] [PubMed]
- Kots, P.A. Close-loop chemical recycling unlocked via waste polyolefins-ethylene co-metathesis. Chem Catal. 2024, 4, 101194. [Google Scholar]
- Cen, Z.; Han, X.; Lin, L.; Yang, S.; Han, W.; Wen, W.; Yuan, W.; Dong, M.; Ma, Z.; Li, F.; et al. Upcycling of polyethylene to gasoline through a self-supplied hydrogen strategy in a layered self-pillared zeolite. Nat. Chem. 2024, 16, 871–880. [Google Scholar]
- Selvam, E.; Yu, K.; Ngu, J.; Najmi, S.; Vlachos, D.G. Recycling polyolefin plastic waste at short contact times via rapid joule heating. Nat. Commun. 2024, 15, 5662. [Google Scholar] [CrossRef]
- Odenweller, A.; Ueckerdt, F. The green hydrogen ambition and implementation gap. Nat. Energy 2025, 10, 110–123. [Google Scholar]
- Aydogdu, A.C.; Erkmen, B.; Suerkan, A.; Ezdesir, A.; Guliyev, B.; Celik, G. Chemical upcycling of polyolefins into liquid refinery feedstock from the circularity and chemical engineering aspects. J. Environ. Chem. Eng. 2024, 12, 113430. [Google Scholar] [CrossRef]
- Munyaneza, N.E.; Ji, R.; DiMarco, A.; Miscall, J.; Stanley, L.; Rorrer, N.; Qiao, R.; Liu, G. Chain-length-controllable upcycling of polyolefins to sulfate detergents. Nat. Sustain. 2024, 7, 1681–1690. [Google Scholar]
- Tan, Y.; Cheng, Y.; Xu, J.; Wang, H. Catalytic chemical recycling and upcycling of polyolefin plastics. Giant 2024, 19, 100307. [Google Scholar] [CrossRef]
- Sun, J.A.; Kots, P.A.; Hinton, Z.R.; Marinkovic, N.S.; Ma, L.; Ehrlich, S.N.; Zheng, W.; Epps, T.H.; Korley, L.T.J.; Vlachos, D.G. Size and Structure Effects of Carbon-Supported Ruthenium Nanoparticles on Waste Polypropylene Hydrogenolysis Activity, Selectivity, and Product Microstructure. ACS Catal. 2024, 14, 3228–3240. [Google Scholar]
- Hu, Q.; Qian, S.; Wang, Y.; Zhao, J.; Jiang, M.; Sun, M.; Huang, H.; Gan, T.; Ma, J.; Zhang, J.; et al. Polyethylene hydrogenolysis by dilute RuPt alloy to achieve H2-pressure-independent low methane selectivity. Nat. Commun. 2024, 15, 10573. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Phu, H.; Kwon, T.; Kim, T.; Thi Do, L.; Hyuk Kang, K.; Ro, I. Investigating the influence of Ru structures and supports on hydrogenolysis of polyethylene plastic waste. Chem. Eng. J. 2023, 475, 146076. [Google Scholar] [CrossRef]
- Wu, X.; Tennakoon, A.; Yappert, R.; Esveld, M.; Ferrandon, M.S.; Hackler, R.A.; LaPointe, A.M.; Heyden, A.; Delferro, M.; Peters, B.; et al. Size-Controlled Nanoparticles Embedded in a Mesoporous Architecture Leading to Efficient and Selective Hydrogenolysis of Polyolefins. J. Am. Chem. Soc. 2022, 144, 5323–5334. [Google Scholar] [PubMed]
- Nakagawa, Y.; Oya, S.; Kanno, D.; Nakaji, Y.; Tamura, M.; Tomishige, K. Regioselectivity and Reaction Mechanism of Ru-Catalyzed Hydrogenolysis of Squalane and Model Alkanes. ChemSusChem 2016, 10, 189–198. [Google Scholar]
- Cheng, L.; Tian, S.; Liang, D.; Gu, J.; Chen, R.; Chen, X.; Yuan, H.; Chen, Y. Enhanced hydroconversion of polyethylene via dual-functional catalysis: Exploiting ZSM-22 pore-mouth catalysis and Ru electronic effect. Chem. Eng. J. 2024, 486, 150332. [Google Scholar] [CrossRef]
- Sun, C.; Wang, J.; Wang, J.; Shakouri, M.; Shi, B.; Liu, X.; Guo, Y.; Hu, Y.; Wu, X.-P.; Wang, Y. Pt enhanced C–H bond activation for efficient and low-methane-selectivity hydrogenolysis of polyethylene over alloyed RuPt/ZrO2. Appl. Catal. B 2024, 353, 124046. [Google Scholar] [CrossRef]
- Nadhifah, N.; Jovita, S.; Subagyo, R.; Utami, D.I.; Agustina, K.R.; Holilah, H.; Asikin-Mijan, N.; Bahruji, H.; Nugraha, R.E.; Jalil, A.A.; et al. Mesoporous aluminosilicate from metakaolin with natural surfactants for waste cooking oil conversion to hydrocarbon fuels via low-temperature pyrolysis. Case Stud. Chem. Environ. Eng. 2025, 11, 101111. [Google Scholar] [CrossRef]
- Ezenwa, S.; Gounder, R. Advances and challenges in designing active site environments in zeolites for Brønsted acid catalysis. Chem. Commun. 2024, 60, 12118–12143. [Google Scholar]
- Al Aslani, I.; Lezcano, G.; Colom, J.M.; Alahmadi, A.; Shoinkhorova, T.; Dikhtiarenko, A.; Cui, M.; Alfilfil, L.; Morales Osorio, I.; Almajnouni, K.; et al. Reaction network and kinetic modeling for the direct catalytic cracking of Arabian light crude oil to chemicals. Chem. Eng. J. 2024, 498, 154981. [Google Scholar] [CrossRef]
- Wu, X.; Wang, X.; Zhang, L.; Wang, X.; Song, S.; Zhang, H. Polyethylene Upgrading to Liquid Fuels Boosted by Atomic Ce Promoters. Angew. Chem. Int. Ed. 2024, 63, e202317594. [Google Scholar] [CrossRef]
- Liu, S.; Kots, P.A.; Vance, B.C.; Danielson, A.; Vlachos, D.G. Plastic waste to fuels by hydrocracking at mild conditions. Sci. Adv. 2021, 7, eabf8283. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, W.; Chu, M.; Gao, D.; Wang, Y.; Lv, Y.; Wang, R.; Song, L.; Zhao, H.; Chen, J.; et al. Ultra-Narrow Alkane Product Distribution in Polyethylene Waste Hydrocracking by Zeolite Micro-Mesopore Diffusion Optimization. Angew. Chem. Int. Ed. 2024, 63, e202409288. [Google Scholar] [CrossRef]
- Li, L.; Luo, H.; Shao, Z.; Zhou, H.; Lu, J.; Chen, J.; Huang, C.; Zhang, S.; Liu, X.; Xia, L.; et al. Converting Plastic Wastes to Naphtha for Closing the Plastic Loop. J. Am. Chem. Soc. 2023, 145, 1847–1854. [Google Scholar] [PubMed]
- Zhang, Z.; Wang, J.; Ge, X.; Wang, S.; Li, A.; Li, R.; Shen, J.; Liang, X.; Gan, T.; Han, X.; et al. Mixed Plastics Wastes Upcycling with High-Stability Single-Atom Ru Catalyst. J. Am. Chem. Soc. 2023, 145, 22836–22844. [Google Scholar]
- Zhao, M.; Chu, X.; Wang, F.; Fang, Y.; Sun, L.; Xie, Q.; Zhang, L.-l.; Song, S.; Zhang, H.; Wang, X. Enhancing the Conversion Efficiency of Polyethylene to Methane through Codoping of Mn Atoms into Ru Centers and CeO2 Supports. J. Am. Chem. Soc. 2024, 146, 33104–33111. [Google Scholar]
- Zhang, X.; Gan, Q.; Zhou, P.; Chen, Z.; Zhang, Z.; Lu, G.-P. Boosting strong metal-support interactions between Ru and sodium titanate nanowire for hydrogenolysis of polyolefins under mild conditions. Appl. Catal. B 2024, 344, 123626. [Google Scholar] [CrossRef]
- Conk, R.J.; Hanna, S.; Shi, J.X.; Yang, J.; Ciccia, N.R.; Qi, L.; Bloomer, B.J.; Heuvel, S.; Wills, T.; Su, J.; et al. Catalytic deconstruction of waste polyethylene with ethylene to form propylene. Science 2022, 377, 1561–1566. [Google Scholar]
- Jia, X.; Qin, C.; Friedberger, T.; Guan, Z.; Huang, Z. Efficient and selective degradation of polyethylenes into liquid fuels and waxes under mild conditions. Sci. Adv. 2016, 2, e1501591. [Google Scholar] [CrossRef]
- Chen, L.; Meyer, L.C.; Kovarik, L.; Meira, D.; Pereira-Hernandez, X.I.; Shi, H.; Khivantsev, K.; Gutiérrez, O.Y.; Szanyi, J. Disordered, Sub-Nanometer Ru Structures on CeO2 are Highly Efficient and Selective Catalysts in Polymer Upcycling by Hydrogenolysis. ACS Catal. 2022, 12, 4618–4627. [Google Scholar]
- Chen, L.; Zhu, Y.; Meyer, L.C.; Hale, L.V.; Le, T.T.; Karkamkar, A.; Lercher, J.A.; Gutiérrez, O.Y.; Szanyi, J. Effect of reaction conditions on the hydrogenolysis of polypropylene and polyethylene into gas and liquid alkanes. React. Chem. Eng. 2022, 7, 844–854. [Google Scholar]
- Rorrer, J.E.; Troyano-Valls, C.; Beckham, G.T.; Román-Leshkov, Y. Hydrogenolysis of Polypropylene and Mixed Polyolefin Plastic Waste over Ru/C to Produce Liquid Alkanes. ACS Sustain. Chem. Eng. 2021, 9, 11661–11666. [Google Scholar] [CrossRef]
- Tamura, M.; Miyaoka, S.; Nakaji, Y.; Tanji, M.; Kumagai, S.; Nakagawa, Y.; Yoshioka, T.; Tomishige, K. Structure-activity relationship in hydrogenolysis of polyolefins over Ru/support catalysts. Appl. Catal. B 2022, 318, 121870. [Google Scholar] [CrossRef]
- Rorrer, J.E.; Ebrahim, A.M.; Questell-Santiago, Y.; Zhu, J.; Troyano-Valls, C.; Asundi, A.S.; Brenner, A.E.; Bare, S.R.; Tassone, C.J.; Beckham, G.T.; et al. Role of Bifunctional Ru/Acid Catalysts in the Selective Hydrocracking of Polyethylene and Polypropylene Waste to Liquid Hydrocarbons. ACS Catal. 2022, 12, 13969–13979. [Google Scholar] [CrossRef]
- Kots, P.A.; Liu, S.; Vance, B.C.; Wang, C.; Sheehan, J.D.; Vlachos, D.G. Polypropylene Plastic Waste Conversion to Lubricants over Ru/TiO2 Catalysts. ACS Catal. 2021, 11, 8104–8115. [Google Scholar] [CrossRef]
- Rorrer, J.E.; Beckham, G.T.; Román-Leshkov, Y. Conversion of Polyolefin Waste to Liquid Alkanes with Ru-Based Catalysts under Mild Conditions. JACS Au 2020, 1, 8–12. [Google Scholar] [CrossRef]
- Nakaji, Y.; Tamura, M.; Miyaoka, S.; Kumagai, S.; Tanji, M.; Nakagawa, Y.; Yoshioka, T.; Tomishige, K. Low-temperature catalytic upgrading of waste polyolefinic plastics into liquid fuels and waxes. Appl. Catal. B 2021, 285, 119805. [Google Scholar] [CrossRef]
- Lee, W.-T.; van Muyden, A.; Bobbink, F.D.; Mensi, M.D.; Carullo, J.R.; Dyson, P.J. Mechanistic classification and benchmarking of polyolefin depolymerization over silica-alumina-based catalysts. Nat. Commun. 2022, 13, 4850. [Google Scholar] [CrossRef]
- Wang, C.; Xie, T.; Kots, P.A.; Vance, B.C.; Yu, K.; Kumar, P.; Fu, J.; Liu, S.; Tsilomelekis, G.; Stach, E.A.; et al. Polyethylene Hydrogenolysis at Mild Conditions over Ruthenium on Tungstated Zirconia. JACS Au 2021, 1, 1422–1434. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, L.; Wang, H.; Ma, X.; Yu, S.; Yan, Y.; Bu, H. Comparative Study on the Power Consumption and Flow Field Characteristics of a Three-Blade Combined Agitator. Processes 2021, 9, 1962. [Google Scholar] [CrossRef]
- Zhang, X.; Pan, L.; Wang, L.; Zou, J.-J. Review on synthesis and properties of high-energy-density liquid fuels: Hydrocarbons, nanofluids and energetic ionic liquids. Chem. Eng. Sci. 2018, 180, 95–125. [Google Scholar]
- Ma, Q.; Cheng, J.; Wu, X.; Xie, J.; Zhang, R.; Mao, Z.; Yang, H.; Fan, W.; Zeng, J.; Bitter, J.H.; et al. C–C bond coupling with sp3 C–H bond via active intermediates from CO2 hydrogenation. Nat. Commun. 2025, 16, 140. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, K.; Regalbuto, J.R. High Sensitivity Silicon Slit Detectors for 1 nm Powder XRD Size Detection Limit. Catal. Lett. 2015, 145, 777–783. [Google Scholar] [CrossRef]
- Meng, Q.; Liu, H.; Xu, K.; Wang, W.; Jia, C. CeO2 modified Ru/γ-Al2O3 catalysts for ammonia decomposition reaction. J. Rare Earths 2023, 41, 801–809. [Google Scholar] [CrossRef]
- Chin, S.Y.; Williams, C.T.; Amiridis, M.D. FTIR Studies of CO Adsorption on Al2O3- and SiO2-Supported Ru Catalysts. J. Phys. Chem. B 2005, 110, 871–882. [Google Scholar] [CrossRef]
- Xiao, H.; Zhang, J.; Du, C.; Zheng, Y.; Fang, J.; Li, S.; Zhang, C. Tailoring the Electronic Ru-Al2O3 Interaction to Regulate Reaction Barriers for Selective Hydrogenolysis of Aromatic Ether. ACS Sustain. Chem. Eng. 2023, 11, 1305–1310. [Google Scholar] [CrossRef]
- Zhou, J.; Gao, Z.; Xiang, G.; Zhai, T.; Liu, Z.; Zhao, W.; Liang, X.; Wang, L. Interfacial compatibility critically controls Ru/TiO2 metal-support interaction modes in CO2 hydrogenation. Nat. Commun. 2022, 13, 327. [Google Scholar] [CrossRef]
- Chen, M.; Liu, L.; Chen, X.; Qin, X.; Li, K.; Zhang, J.; Bao, X.; Ma, L.; Zhang, C. Effects of Ru particle size over TiO2 on the catalytic performance of CO2 hydrogenation. Appl. Surf. Sci. 2024, 654, 159460. [Google Scholar] [CrossRef]
- Lin, L.; Pei, C.; Ding, R.; Li, Y.; Park, H.S.; Yu, X. Ru nanoclusters coupling with hierarchical phosphorus and oxygen dual-doped carbon nanotube architectures for effective hydrogen evolution reaction. Int. J. Hydrogen Energy 2023, 48, 20350–20358. [Google Scholar] [CrossRef]
- Flaherty, D.W.; Hibbitts, D.D.; Iglesia, E. Metal-Catalyzed C–C Bond Cleavage in Alkanes: Effects of Methyl Substitution on Transition-State Structures and Stability. J. Am. Chem. Soc. 2014, 136, 9664–9676. [Google Scholar] [CrossRef]
- Tran, V.T.; Gurak, J.A.; Yang, K.S.; Engle, K.M. Activation of diverse carbon–heteroatom and carbon–carbon bonds via palladium(ii)-catalysed β-X elimination. Nat. Chem. 2018, 10, 1126–1133. [Google Scholar] [CrossRef]
- Jiang, W.; Cao, J.-P.; Zhu, C.; Zhao, M.; Ni, Z.-H.; Zhao, X.-Y.; Xie, J.-X.; Zhao, L.; Zhao, Y.-P.; Bai, H.-C. Catalytic hydrogenation of aromatic ring over ruthenium nanoparticles supported on α-Al2O3 at room temperature. Appl. Catal. B 2022, 307, 121137. [Google Scholar] [CrossRef]
- Ahmadi, M.; Mistry, H.; Roldan Cuenya, B. Tailoring the Catalytic Properties of Metal Nanoparticles via Support Interactions. J. Phys. Chem. Lett. 2016, 7, 3519–3533. [Google Scholar] [PubMed]
- Ran, H.; Zhang, S.; Ni, W.; Jing, Y. Precise activation of C–C bonds for recycling and upcycling of plastics. Chem. Sci. 2024, 15, 795–831. [Google Scholar] [PubMed]
- Li, R.; Ban, T.; Zhao, D.; Lin, J.; Liu, Z.; Wang, L.; Huang, X.; Tao, Z.; Wang, G. Defect engineering and Ni promoter synergistically accelerating electron transfer to Ru0 sites in UiO-66(Ce) for dicyclopentadiene hydrogenation under mild condition. Nano Res. 2024, 17, 9550–9563. [Google Scholar] [CrossRef]
- Tu, W.; Chu, M.; Wang, X.; Wang, X.; Li, Y.; Yang, W.; Cao, M.; Wang, L.; Li, Y.; Sham, T.-K.; et al. SMSI-induced charge transfer for selective hydrogenolysis of polyolefins. Appl. Catal. B 2023, 339, 123122. [Google Scholar] [CrossRef]
- Lu, S.; Jing, Y.; Jia, S.; Shakouri, M.; Hu, Y.; Liu, X.; Guo, Y.; Wang, Y. Enhanced Production of Liquid Alkanes from Waste Polyethylene via the Electronic Effect-Favored Csecondary−Csecondary Bond Cleavage. ChemCatChem 2023, 15, e202201375. [Google Scholar] [CrossRef]
- Zhong, X.; Liu, J.; Gao, L.; Chen, J.; Wang, X.; Zhang, Y.; Wu, Y.A.; Shakeri, M.; Zhang, X.; Zhang, B. Constructing the Al deficiency in Si-O(H)-Al units based on Pt/ZSM-5 for enhanced hydrocracking of polyethylene into high-quality liquid fuel. Nano Res. 2024, 17, 10088–10098. [Google Scholar]
- Blomsma, E.; Martens, J.A.; Jacobs, P.A. Reaction Mechanisms of Isomerization and Cracking of Heptane on Pd/H-Beta Zeolite. J. Catal. 1995, 155, 141–147. [Google Scholar]
- Du, B.; Chen, X.; Ling, Y.; Niu, T.; Guan, W.; Meng, J.; Hu, H.; Tsang, C.W.; Liang, C. Hydrogenolysis-Isomerization of Waste Polyolefin Plastics to Multibranched Liquid Alkanes. ChemSusChem 2023, 16, e202202035. [Google Scholar] [CrossRef]
- Zhou, Q.M.; Wang, D.L.; Wang, Q.Y.; He, K.L.; Lim, K.H.; Yang, X.; Wang, W.J.; Li, B.G.; Liu, P.W. Mechanistic Understanding of Efficient Polyethylene Hydrocracking over Two-Dimensional Platinum-Anchored Tungsten Trioxide. Angew. Chem. Int. Ed. 2023, 62, e202305644. [Google Scholar] [CrossRef]
- Albahar, M.; Li, C.; Zholobenko, V.L.; Garforth, A.A. The effect of ZSM-5 zeolite crystal size on p-xylene selectivity in toluene disproportionation. Microporous Mesoporous Mater. 2020, 302, 110221. [Google Scholar] [CrossRef]
- Wu, H.; Duan, A.; Zhao, Z.; Xu, C.; Jiang, G.; Liu, J.; Wei, Y.; Li, J.; Chi, K.; Guo, J. Restricted diffusion of model sulfides over a NiMo/BK catalyst under hydrodesulfurization reaction conditions. RSC Adv. 2017, 7, 44340–44347. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, Q.; Shang, X.; Yuan, Y.; Su, X.; Huang, Y. Hydrocracking of Polyethylene to Gasoline-Range Hydrocarbons over a Ruthenium-Zeolite Bifunctional Catalyst System with Optimal Synergy of Metal and Acid Sites. Catalysts 2025, 15, 335. https://doi.org/10.3390/catal15040335
Du Q, Shang X, Yuan Y, Su X, Huang Y. Hydrocracking of Polyethylene to Gasoline-Range Hydrocarbons over a Ruthenium-Zeolite Bifunctional Catalyst System with Optimal Synergy of Metal and Acid Sites. Catalysts. 2025; 15(4):335. https://doi.org/10.3390/catal15040335
Chicago/Turabian StyleDu, Qing, Xin Shang, Yangyang Yuan, Xiong Su, and Yanqiang Huang. 2025. "Hydrocracking of Polyethylene to Gasoline-Range Hydrocarbons over a Ruthenium-Zeolite Bifunctional Catalyst System with Optimal Synergy of Metal and Acid Sites" Catalysts 15, no. 4: 335. https://doi.org/10.3390/catal15040335
APA StyleDu, Q., Shang, X., Yuan, Y., Su, X., & Huang, Y. (2025). Hydrocracking of Polyethylene to Gasoline-Range Hydrocarbons over a Ruthenium-Zeolite Bifunctional Catalyst System with Optimal Synergy of Metal and Acid Sites. Catalysts, 15(4), 335. https://doi.org/10.3390/catal15040335