Preparation of Oxygen Reduction Catalyst Electrodes by an Efficient Electrodeposition Method on HNO3-Activated Carbon Paper


Abstract
:1. Introduction
2. Results and Discussions
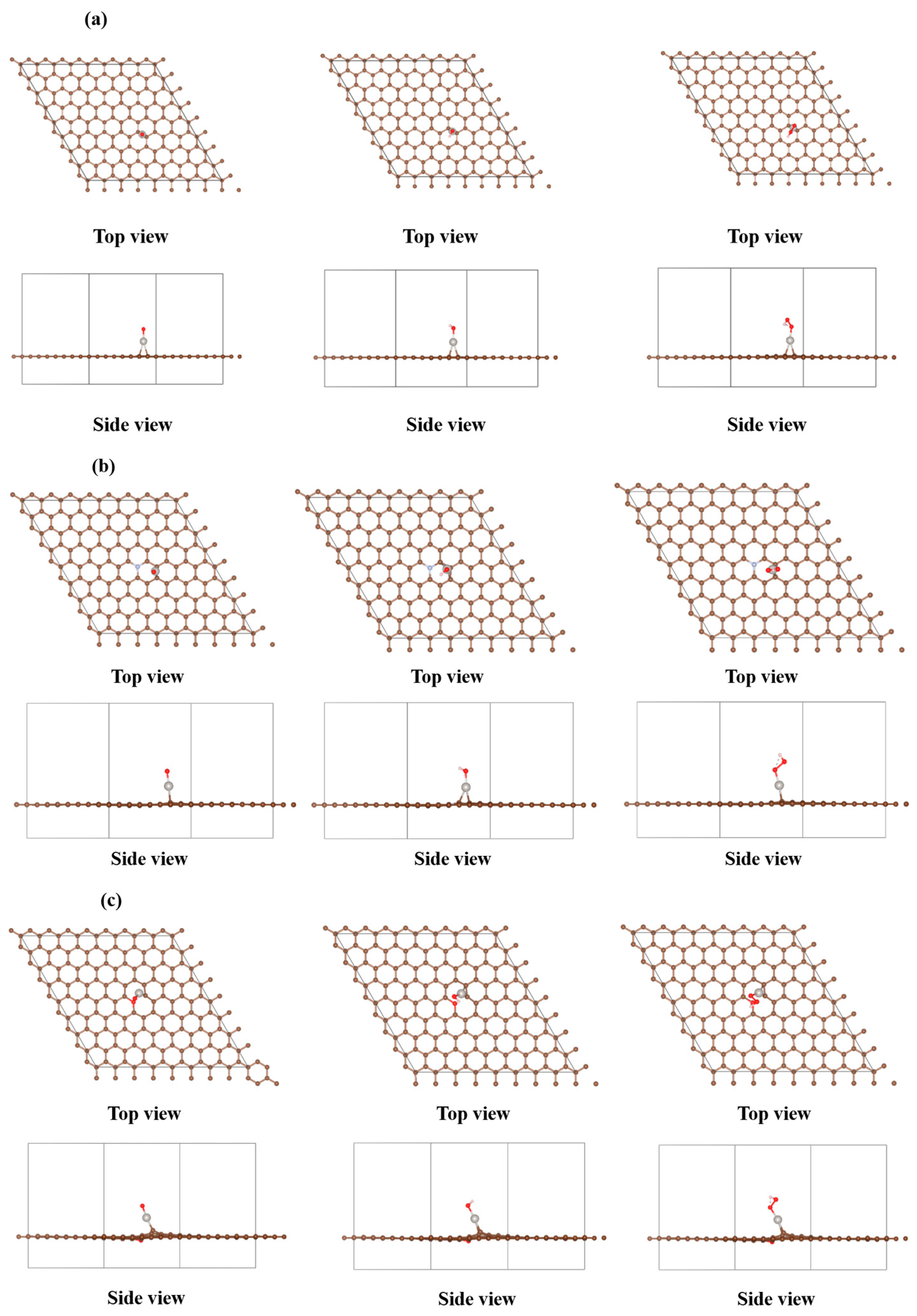
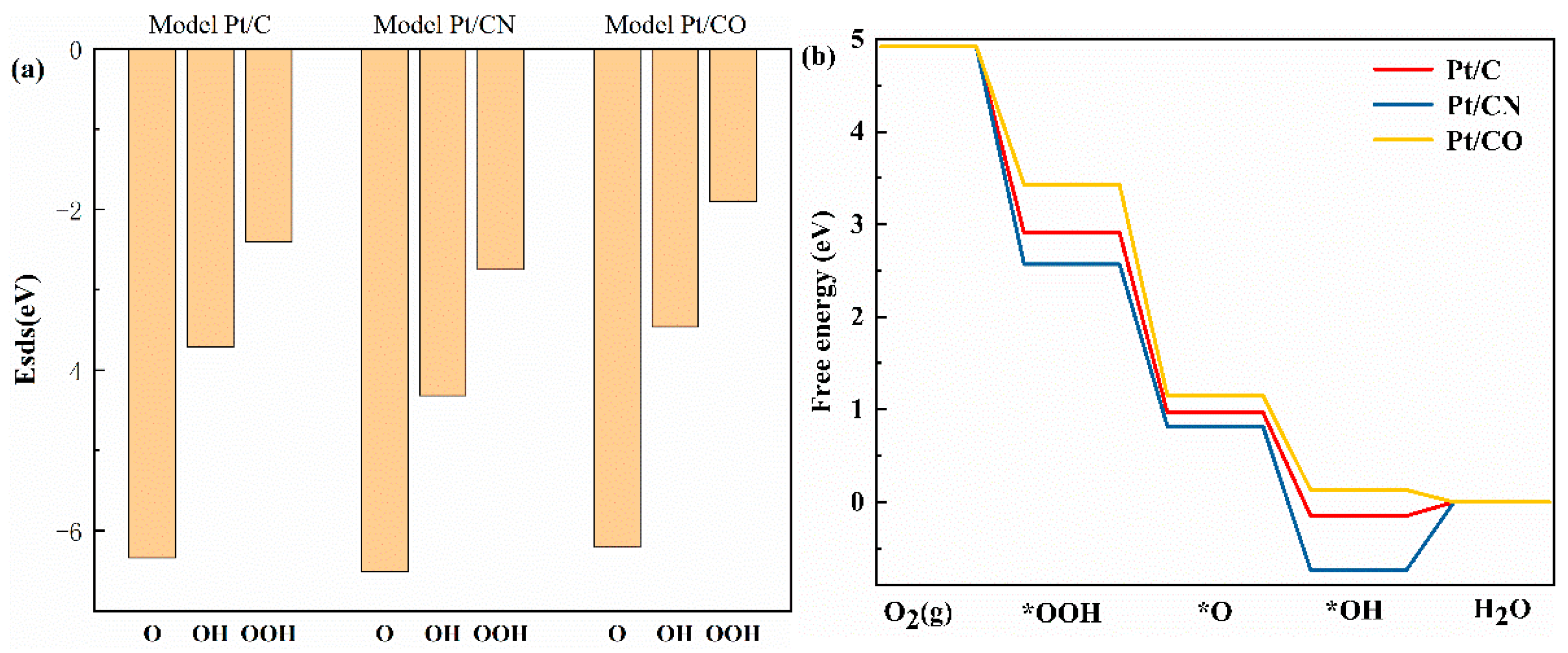
2.1. DFT Investigation on the Influence of Pt Loaded on N/O-Doped Carbon Supports in the Oxygen Reduction Reaction
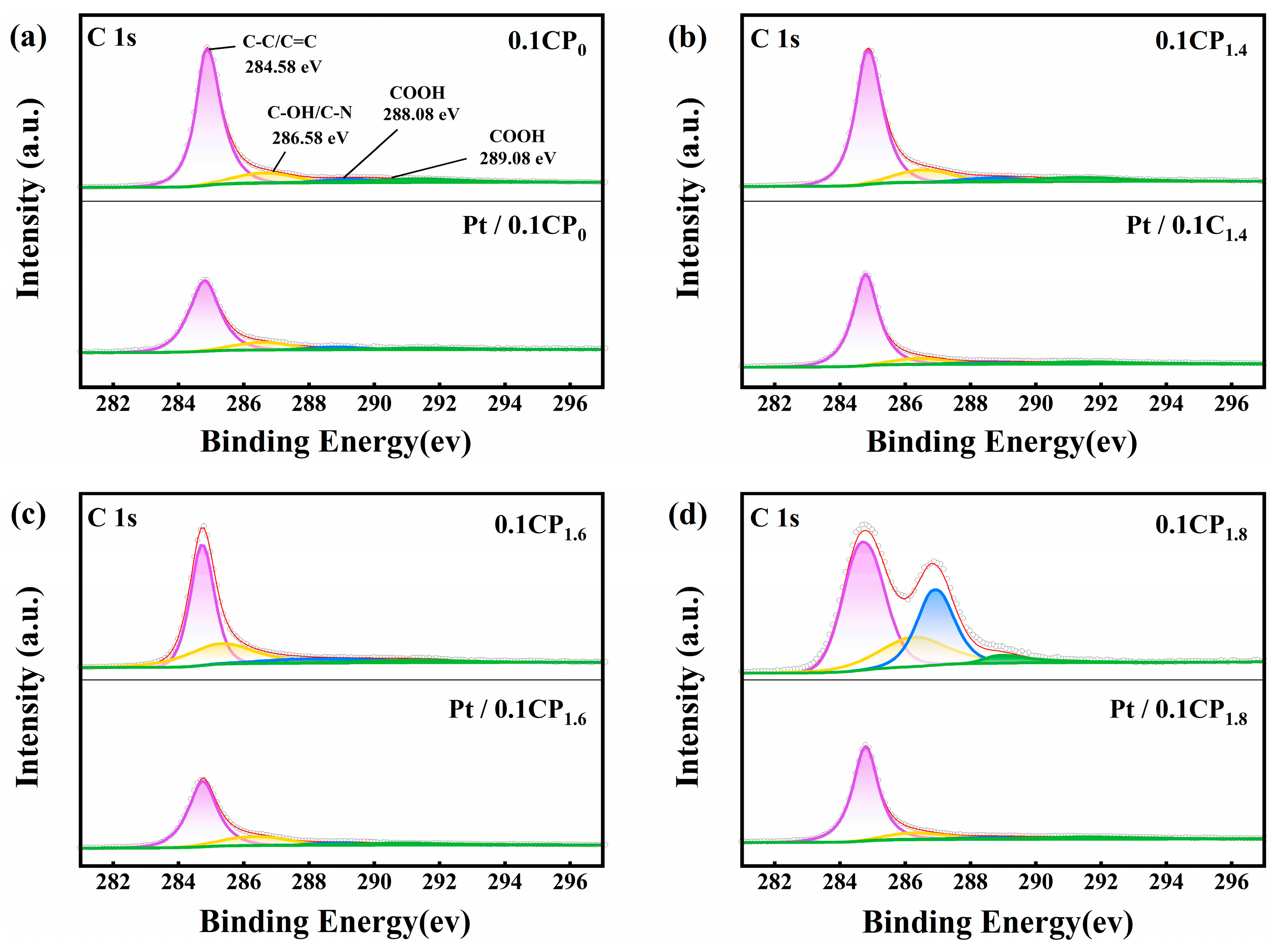
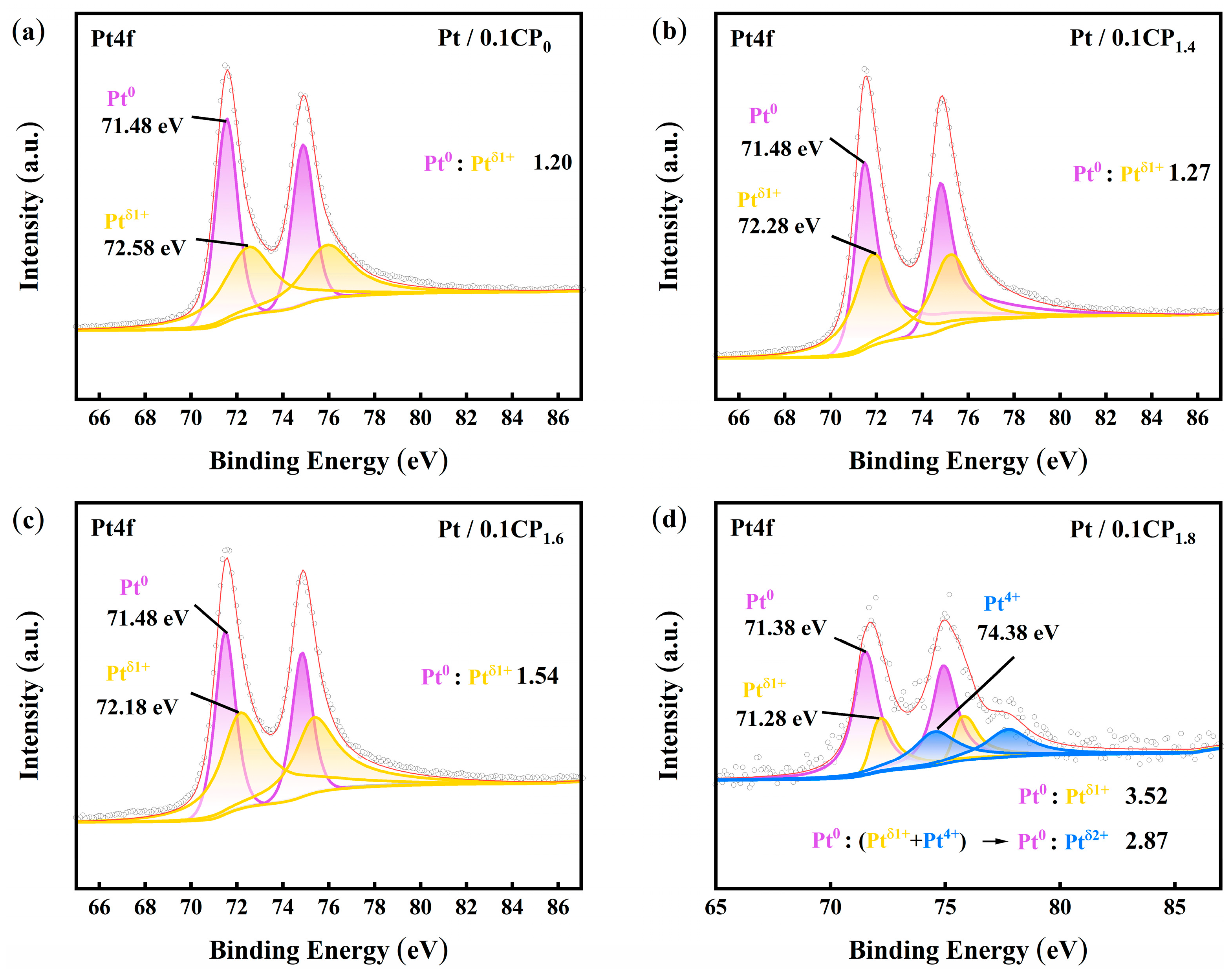
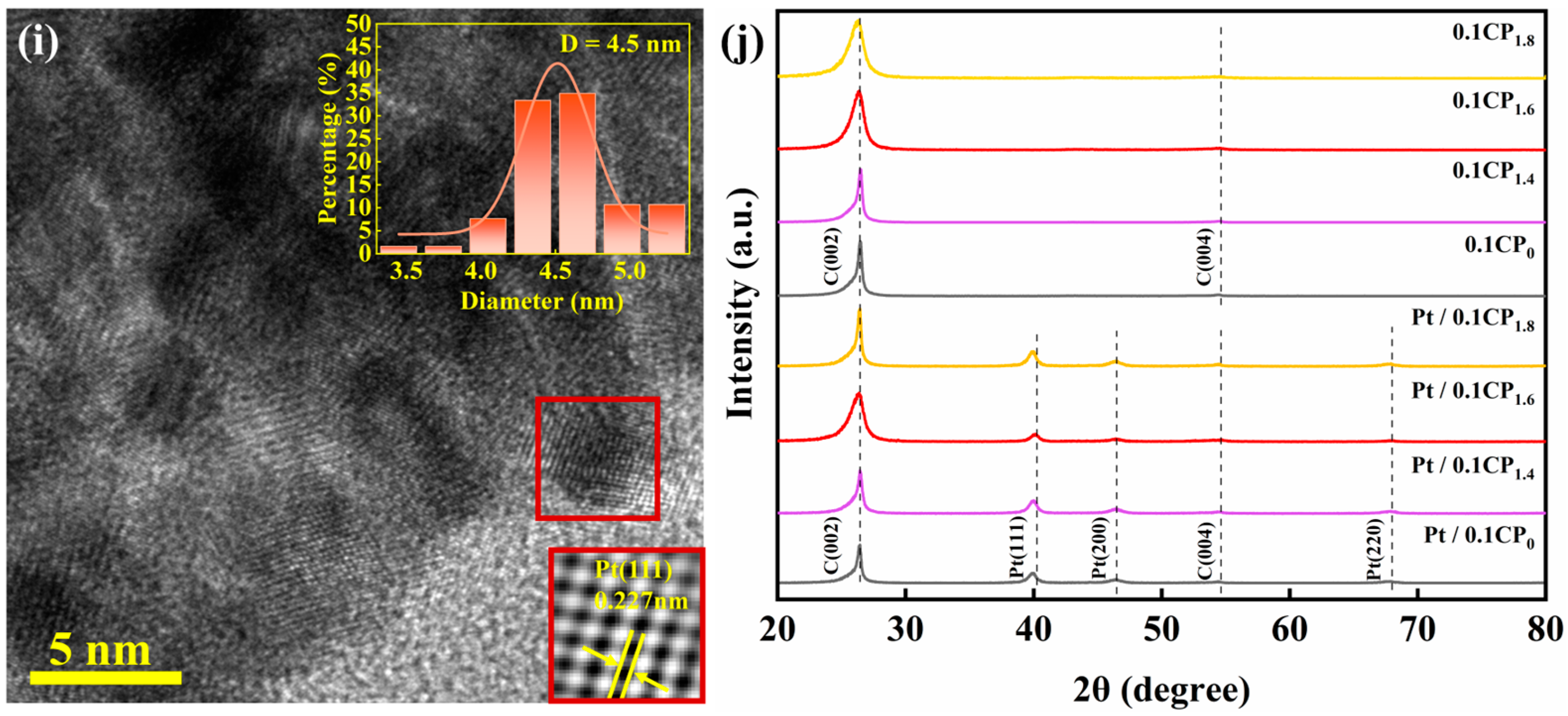
2.2. Physical Characterization and Half-Cell Performance Testing of Carbon-Supported Platinum Catalysts Regulated by Different Activation Voltages
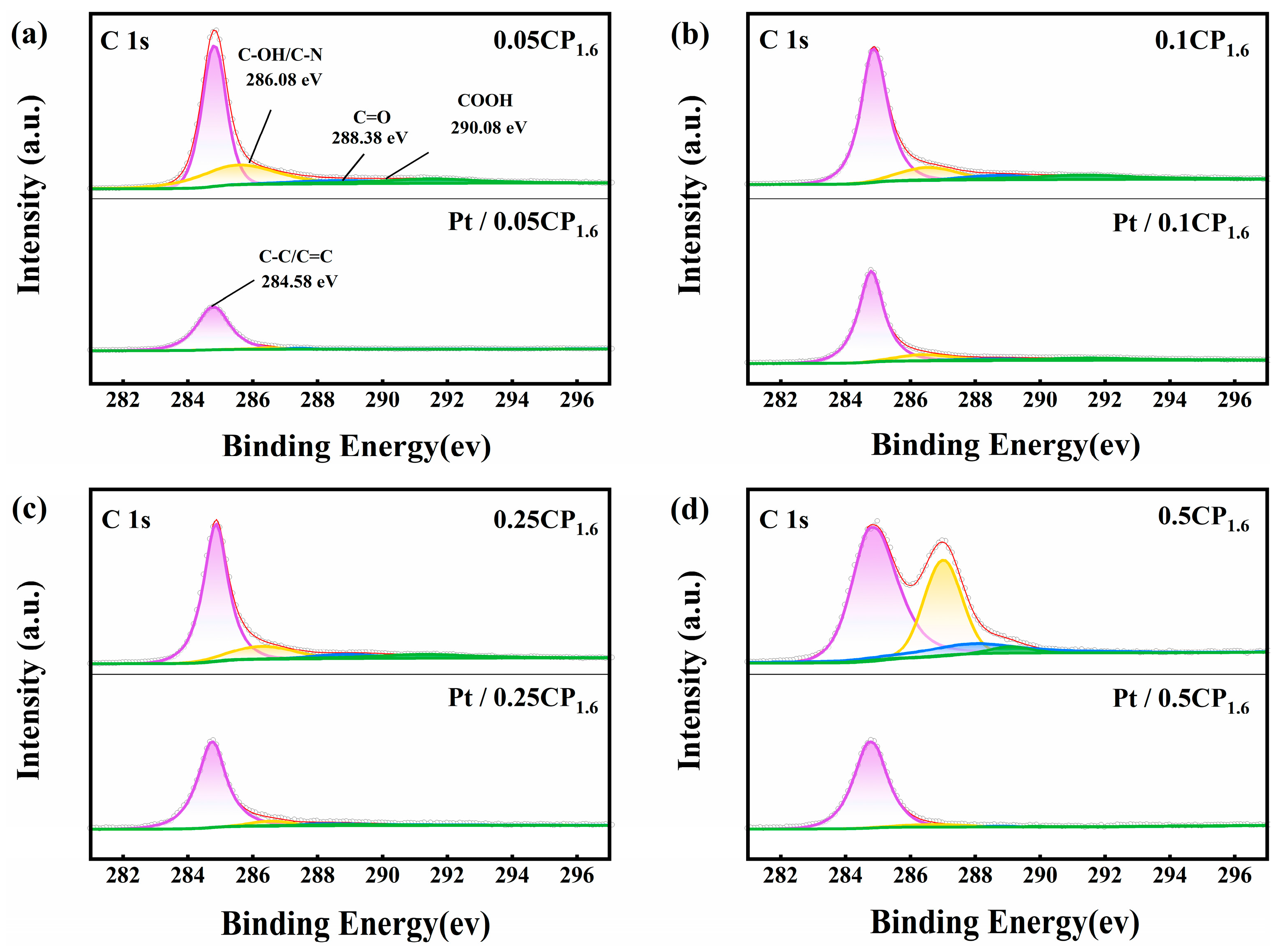
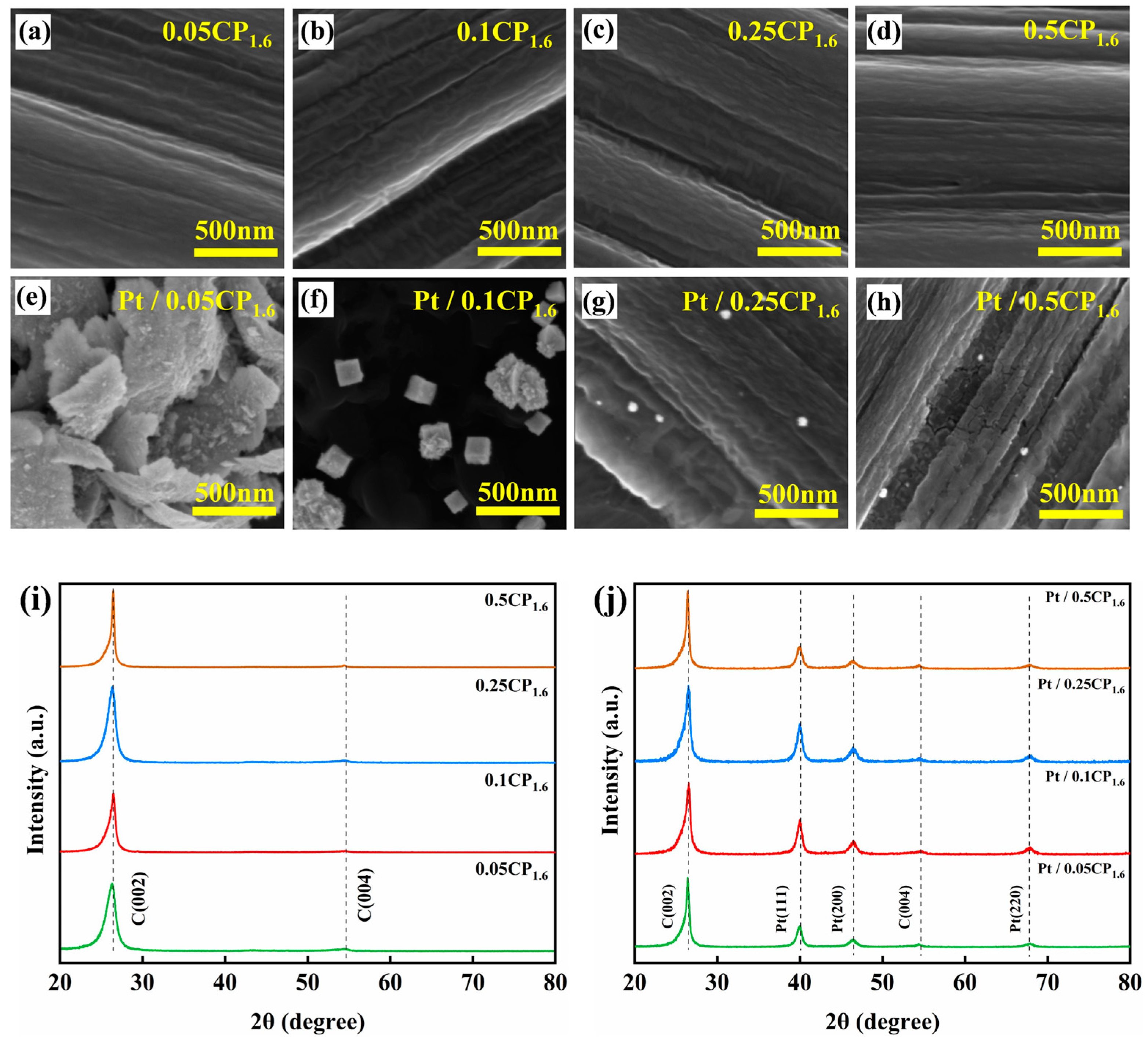
2.3. Physical Characterization and Half-Cell Performance Testing of the Carbon-Supported Platinum Catalysts Regulated by Different Activation Concentrations
3. Materials and Methods
3.1. Materials and Chemicals
3.2. Preparation of the Catalyst Electrodes
3.3. Electrochemical Test
3.4. Characterizations and Measurements
3.5. DFT Investigation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Chen, Y.; Huang, Z.; Yu, J.; Wang, H.; Qin, Y.; Xing, L.; Du, L. Research Progress of Pt-Based Catalysts toward Cathodic Oxygen Reduction Reactions for Proton Exchange Membrane Fuel Cells. Catalysts 2024, 14, 569. [Google Scholar] [CrossRef]
- Zhang, X.; Li, H.; Yang, J.; Lei, Y.; Wang, C.; Wang, J.; Tang, Y.; Mao, Z. Recent Advances in Pt-Based Electrocatalysts for PEMFCs. RSC Adv. 2021, 11, 13316–13328. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Guo, Y.; Xu, Y.; Li, P.; Zhang, Q. Preparation of Oxygen Reduction Catalyst Electrodes by Electrochemical Acidification and Synergistic Electrodeposition. Catalysts 2024, 14, 300. [Google Scholar] [CrossRef]
- Li, T.; Chen, D.; Gu, L.; Chen, S.; Li, C.; Liao, J.; Zhou, Y.; Xu, Y.; Sun, C.; Yang, Z.; et al. Single-Source Precursor Synthesis of Nitrogen-Doped Porous Carbon for High-Performance Electrocatalytic ORR Application. Ceram. Int. 2019, 45, 8354–8361. [Google Scholar] [CrossRef]
- Pajootan, E.; Alolabi, S.A.; Omanovic, S.; Coulombe, S. Radiofrequency Plasma-Assisted Pulsed Laser Deposited Pt/TiOxNy Coatings on Multi-Walled Carbon Nanotubes as Gas Diffusion Electrodes for the Oxygen Reduction Reaction. Adv. Mater. Technol. 2022, 7, 2200196. [Google Scholar] [CrossRef]
- Zhao, H.; Zhang, Y.; Li, L.; Geng, X.; Yang, H.; Zhou, W.; Sun, C.; An, B. Synthesis of an Ordered Porous Carbon with the Dual Nitrogen-Doped Interfaces and Its ORR Catalysis Performance. Chin. Chem. Lett. 2021, 32, 140–145. [Google Scholar] [CrossRef]
- Liang, J.; Yuan, Q. Effects of Graphitic- and Pyridinic-N Co-Doping on Structure Regulation and ORR Activity of N-Doped Graphene. Appl. Surf. Sci. 2024, 648, 159025. [Google Scholar] [CrossRef]
- Lan, Q.; Ye, D.; Zhu, X.; Yang, Y.; Chen, R.; Wang, S.; Zhang, T.; Liao, Q. High-Performance Direct Liquid Fuel Cells Benefited from Highly N-Doped and Defect-Rich Carbon Paper Cathode with Carbon Nanosheets. Carbon 2023, 207, 95–104. [Google Scholar] [CrossRef]
- Egetenmeyer, A.; Radev, I.; Durneata, D.; Baumgärtner, M.; Peinecke, V.; Natter, H.; Hempelmann, R. Pulse Electrodeposited Cathode Catalyst Layers for PEM Fuel Cells. Int. J. Hydrogen Energy 2017, 42, 13649–13660. [Google Scholar] [CrossRef]
- Eiler, K.; Mølmen, L.; Fast, L.; Leisner, P.; Sort, J.; Pellicer, E. Oxygen Reduction Reaction and Proton Exchange Membrane Fuel Cell Performance of Pulse Electrodeposited Pt–Ni and Pt–Ni–Mo(O) Nanoparticles. Mater. Today Energy 2022, 27, 101023. [Google Scholar] [CrossRef]
- Shen, S.; Li, F.; Luo, L.; Guo, Y.; Yan, X.; Ke, C.; Zhang, J. DMF-Coordination Assisted Electrodeposition of Highly Active PtCo Alloy Catalysts for the Oxygen Reduction Reaction. J. Electrochem. Soc. 2018, 165, D43–D49. [Google Scholar] [CrossRef]
- Hornberger, E.; Merzdorf, T.; Schmies, H.; Hübner, J.; Klingenhof, M.; Gernert, U.; Kroschel, M.; Anke, B.; Lerch, M.; Schmidt, J.; et al. Impact of Carbon N-Doping and Pyridinic-N Content on the Fuel Cell Performance and Durability of Carbon-Supported Pt Nanoparticle Catalysts. ACS Appl. Mater. Interfaces 2022, 14, 18420–18430. [Google Scholar] [CrossRef]
- Lin, J.F.; Wertz, J.; Ahmad, R.; Thommes, M.; Kannan, A.M. Effect of Carbon Paper Substrate of the Gas Diffusion Layer on the Performance of Proton Exchange Membrane Fuel Cell. Electrochim. Acta 2010, 55, 2746–2751. [Google Scholar] [CrossRef]
- Xie, Z.; Tang, X.; Yang, P.; Sun, M.; Yang, K.; Huang, Q. Carboxyl-Terminated Butadiene-Acrylonitrile-Modified Carbon Paper for Use of Gas Diffusion Layer in PEM Fuel Cells. Int. J. Hydrogen Energy 2015, 40, 14345–14352. [Google Scholar] [CrossRef]
- Wang, Y.; Li, C.; Li, J.; Liu, Z.; Liu, C.; Song, M.; Guo, J.; Gao, X. Electrophoretic Deposition of Graphene Oxide Modified Carbon Paper: Enhance the Interface Strength and Decrease the Interface Resistance. Colloids Surf. Physicochem. Eng. Asp. 2024, 703, 135312. [Google Scholar] [CrossRef]
- He, Z.; Su, A.; Gao, C.; Zhou, Z.; Pan, C.; Liu, S. Carbon Paper Modified by Hydrothermal Ammoniated Treatment for Vanadium Redox Battery. Ionics 2013, 19, 1021–1026. [Google Scholar] [CrossRef]
- Liu, C.; Li, S. Performance Enhancement of Proton Exchange Membrane Fuel Cell through Carbon Nanofibers Grown In Situ on Carbon Paper. Molecules 2023, 28, 2810. [Google Scholar] [CrossRef]
- Park, Y.-C.; Tokiwa, H.; Kakinuma, K.; Watanabe, M.; Uchida, M. Effects of Carbon Supports on Pt Distribution, Ionomer Coverage and Cathode Performance for Polymer Electrolyte Fuel Cells. J. Power Sources 2016, 315, 179–191. [Google Scholar] [CrossRef]
- Ahn, C.-Y.; Ahn, J.; Kang, S.Y.; Kim, O.-H.; Lee, D.W.; Lee, J.H.; Shim, J.G.; Lee, C.H.; Cho, Y.-H.; Sung, Y.-E. Enhancement of Service Life of Polymer Electrolyte Fuel Cells through Application of Nanodispersed Ionomer. Sci. Adv. 2020, 6, eaaw0870. [Google Scholar] [CrossRef]
- De Bruijn, F.A.; Dam, V.A.T.; Janssen, G.J.M. Review: Durability and Degradation Issues of PEM Fuel Cell Components. Fuel Cells 2008, 8, 3–22. [Google Scholar] [CrossRef]
- Komini Babu, S.; Spernjak, D.; Dillet, J.; Lamibrac, A.; Maranzana, G.; Didierjean, S.; Lottin, O.; Borup, R.L.; Mukundan, R. Spatially Resolved Degradation during Startup and Shutdown in Polymer Electrolyte Membrane Fuel Cell Operation. Appl. Energy 2019, 254, 113659. [Google Scholar] [CrossRef]
- More, K.; Borup, R.; Reeves, K. Identifying Contributing Degradation Phenomena in PEM Fuel Cell Membrane Electride Assemblies Via Electron Microscopy. ECS Trans. 2006, 3, 717–733. [Google Scholar] [CrossRef]
- Zhang, Y.; Antonietti, M. Photocurrent Generation by Polymeric Carbon Nitride Solids: An Initial Step towards a Novel Photovoltaic System. Chem. Asian J. 2010, 5, 1307–1311. [Google Scholar] [CrossRef] [PubMed]
- Jia, N.; Martin, R.B.; Qi, Z.; Lefebvre, M.C.; Pickup, P.G. Modification of Carbon Supported Catalysts to Improve Performance in Gas Diffusion Electrodes. Electrochim. Acta 2001, 46, 2863–2869. [Google Scholar] [CrossRef]
- Wang, Z.; Han, Y.; Zeng, Y.; Qie, Y.; Wang, Y.; Zheng, D.; Lu, X.; Tong, Y. Activated Carbon Fiber Paper with Exceptional Capacitive Performance as a Robust Electrode for Supercapacitors. J. Mater. Chem. A 2016, 4, 5828–5833. [Google Scholar] [CrossRef]
- Sullivan, M.G.; Schnyder, B.; Bärtsch, M.; Alliata, D.; Barbero, C.; Imhof, R.; Kötz, R. Electrochemically Modified Glassy Carbon for Capacitor Electrodes Characterization of Thick Anodic Layers by Cyclic Voltammetry, Differential Electrochemical Mass Spectrometry, Spectroscopic Ellipsometry, X-Ray Photoelectron Spectroscopy, FTIR, and AFM. J. Electrochem. Soc. 2000, 147, 2636. [Google Scholar] [CrossRef]
- Ci, S.; Wen, Z.; Chen, J.; He, Z. Decorating Anode with Bamboo-like Nitrogen-Doped Carbon Nanotubes for Microbial Fuel Cells. Electrochem. Commun. 2012, 14, 71–74. [Google Scholar] [CrossRef]
- Achour, A.; Vizireanu, S.; Dinescu, G.; Le Brizoual, L.; Djouadi, M.-A.; Boujtita, M. Electrochemical Anodic Oxidation of Nitrogen Doped Carbon Nanowall Films: X-Ray Photoelectron and Micro-Raman Spectroscopy Study. Appl. Surf. Sci. 2013, 273, 49–57. [Google Scholar] [CrossRef]
- Wang, J.; Yin, G.; Shao, Y.; Zhang, S.; Wang, Z.; Gao, Y. Effect of Carbon Black Support Corrosion on the Durability of Pt/C Catalyst. J. Power Sources 2007, 171, 331–339. [Google Scholar] [CrossRef]
- Shang, N.G.; Au, F.C.K.; Meng, X.M.; Lee, C.S.; Bello, I.; Lee, S.T. Uniform Carbon Nanoflake Films and Their Field Emissions. Chem. Phys. Lett. 2002, 358, 187–191. [Google Scholar] [CrossRef]
- Zhang, G.; Yang, D.; Sacher, E. X-Ray Photoelectron Spectroscopic Analysis of Pt Nanoparticles on Highly Oriented Pyrolytic Graphite, Using Symmetric Component Line Shapes. J. Phys. Chem. C 2007, 111, 565–570. [Google Scholar] [CrossRef]
- Wakisaka, M.; Suzuki, H.; Mitsui, S.; Uchida, H.; Watanabe, M. Increased Oxygen Coverage at Pt−Fe Alloy Cathode for the Enhanced Oxygen Reduction Reaction Studied by EC−XPS. J. Phys. Chem. C 2008, 112, 2750–2755. [Google Scholar] [CrossRef]
- Balbuena, P.B.; Altomare, D.; Vadlamani, N.; Bingi, S.; Agapito, L.A.; Seminario, J.M. Adsorption of O, OH, and H2O on Pt-Based Bimetallic Clusters Alloyed with Co, Cr, and Ni. J. Phys. Chem. A 2004, 108, 6378–6384. [Google Scholar] [CrossRef]
- Cao, F.; Zang, Z.; Sun, S.; Sun, X.; Li, X.; Liu, T.; Wu, J. The influence of deposited potential on the ORR activity of Pt catalysts on glassy carbon electrode. RSC Adv. 2017, 7, 25429–25436. [Google Scholar] [CrossRef]
- Guan, D.; Zhong, J.; Xu, H.; Huang, Y.-C.; Hu, Z.; Chen, B.; Zhang, Y.; Ni, M.; Xu, X.; Zhou, W.; et al. A Universal Chemical-Induced Tensile Strain Tuning Strategy to Boost Oxygen-Evolving Electrocatalysis on Perovskite Oxides. Appl. Phys. Rev. 2022, 9, 011422. [Google Scholar] [CrossRef]
- Darling, R.M.; Meyers, J.P. Kinetic Model of Platinum Dissolution in PEMFCs. J. Electrochem. Soc. 2003, 150, A1523. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Ou, L.; Chen, S. Comparative Study of Oxygen Reduction Reaction Mechanisms on the Pd(111) and Pt(111) Surfaces in Acid Medium by DFT. J. Phys. Chem. C 2013, 117, 1342–1349. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrates | C:O | C:N | Catalysts | C:O | C:N | Pt Wt% |
|---|---|---|---|---|---|---|
| 0.1CP0 | 14.0 | - | Pt/0.1CP0 | 10.8 | - | 83.7 |
| 0.1CP1.4 | 4.9 | 84.8 | Pt/0.1CP1.4 | 6.2 | 77.8 | 69.7 |
| 0.1CP1.6 | 3.5 | 38.4 | Pt/0.1CP1.6 | 10.6 | 64.2 | 64.4 |
| 0.1CP1.8 | 2.6 | 29.8 | Pt/0.1CP1.8 | 11.5 | 59.4 | 47.8 |
| Substrates | C:O | C:N | Catalysts | C:O | C:N | Pt Wt% |
|---|---|---|---|---|---|---|
| 0.05CP1.6 | 4.9 | 94.7 | Pt/0.05CP1.6 | 14.9 | 99.6 | 79.6 |
| 0.1CP1.6 | 3.2 | 38.4 | Pt/0.1CP1.6 | 10.6 | 64.2 | 64.4 |
| 0.25CP1.6 | 3.4 | 40.4 | Pt/0.25CP1.6 | 11.2 | 63.0 | 53.8 |
| 0.5CP1.6 | 2.6 | 29.8 | Pt/0.5CP1.6 | 14.2 | 55.8 | 47.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Y.; Zhou, L.; Zhang, W.; Zhang, Q. Preparation of Oxygen Reduction Catalyst Electrodes by an Efficient Electrodeposition Method on HNO3-Activated Carbon Paper. Catalysts 2025, 15, 403. https://doi.org/10.3390/catal15040403
Guo Y, Zhou L, Zhang W, Zhang Q. Preparation of Oxygen Reduction Catalyst Electrodes by an Efficient Electrodeposition Method on HNO3-Activated Carbon Paper. Catalysts. 2025; 15(4):403. https://doi.org/10.3390/catal15040403
Chicago/Turabian StyleGuo, Yongjian, Liheng Zhou, Wenwen Zhang, and Qi Zhang. 2025. "Preparation of Oxygen Reduction Catalyst Electrodes by an Efficient Electrodeposition Method on HNO3-Activated Carbon Paper" Catalysts 15, no. 4: 403. https://doi.org/10.3390/catal15040403
APA StyleGuo, Y., Zhou, L., Zhang, W., & Zhang, Q. (2025). Preparation of Oxygen Reduction Catalyst Electrodes by an Efficient Electrodeposition Method on HNO3-Activated Carbon Paper. Catalysts, 15(4), 403. https://doi.org/10.3390/catal15040403