Palladium-Catalyzed Cross-Coupling Reactions of Perfluoro Organic Compounds

Abstract
:1. Introduction
2. Results and Discussion
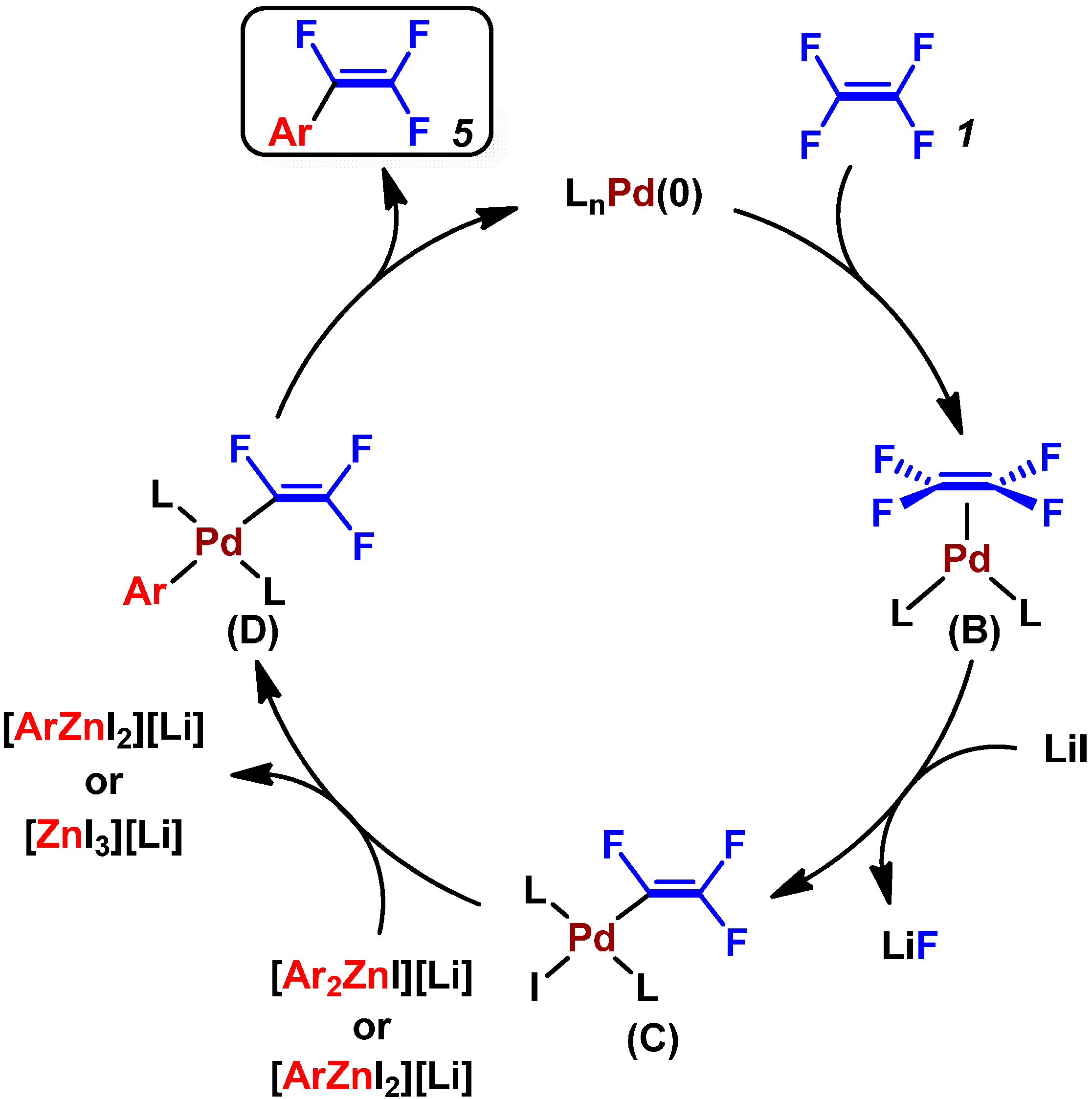
2.1. Pd(0)-Catalyzed Cross-Coupling Reactions of Tetrafluoroethylene with Diarylzinc Reagents




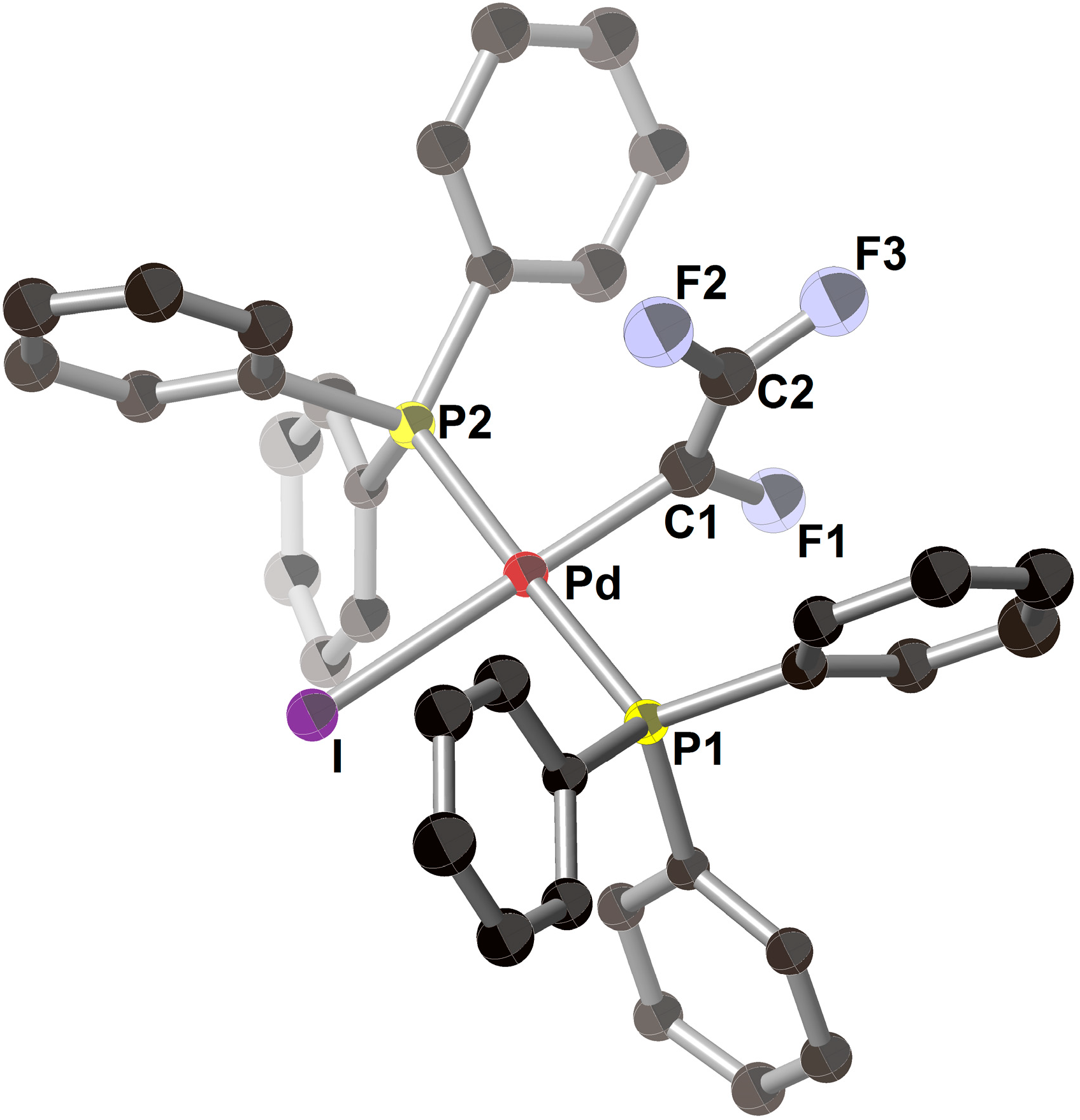{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
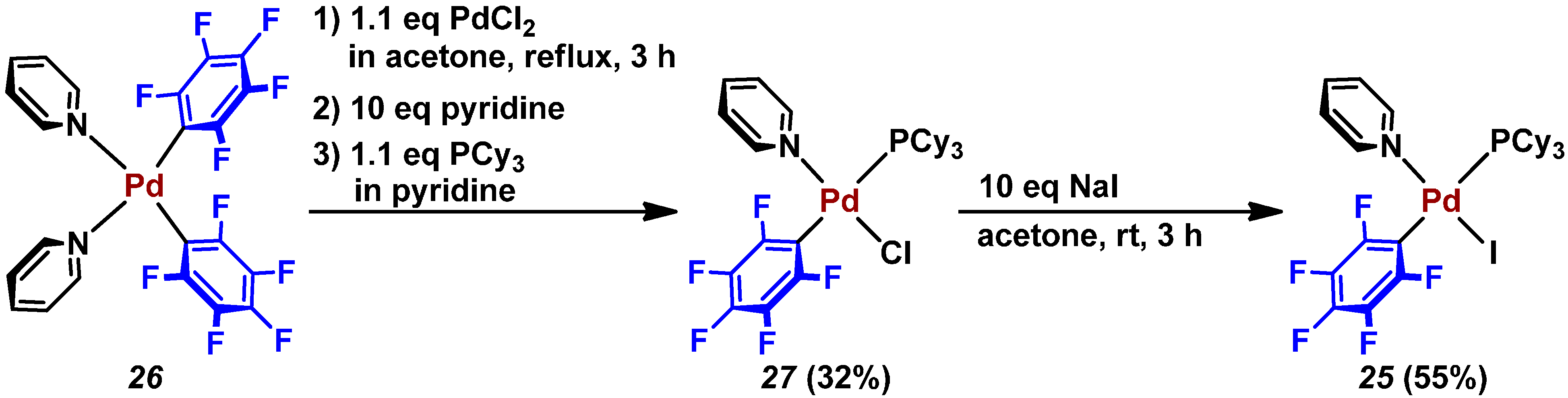{kind=link}
{kind=link}
{kind=link}

| Entry | Pd2(dba)3/mol% | PPh3/mol% | Preparation of ZnPh2 (4a) | LiI/mmol | Time/h | Yield/% |
|---|---|---|---|---|---|---|
| 1 | 2.5 | 10.0 | ZnCl2 + 2 PhMgBr | – | 24 | 48 |
| 2 | 2.5 | 10.0 | isolated ZnPh2 | – | 24 | 19 |
| 3 | 2.5 | 10.0 | ZnCl2 + 2 PhMgBr | 0.240 | 9 | 61 |
| 4 | 0.01 | 0.04 | ZnCl2 + 2 PhMgBr | 0.240 | 24 | 72 |
| 5 | 0.01 | – | ZnCl2 + 2 PhMgBr | 0.240 | 4 | 73 |
| 6 | – | – | ZnCl2 + 2 PhMgBr | 0.240 | 24 | 3 |
| 7 | – | – | ZnCl2 + 2 PhMgBr | – | 24 | 9 |


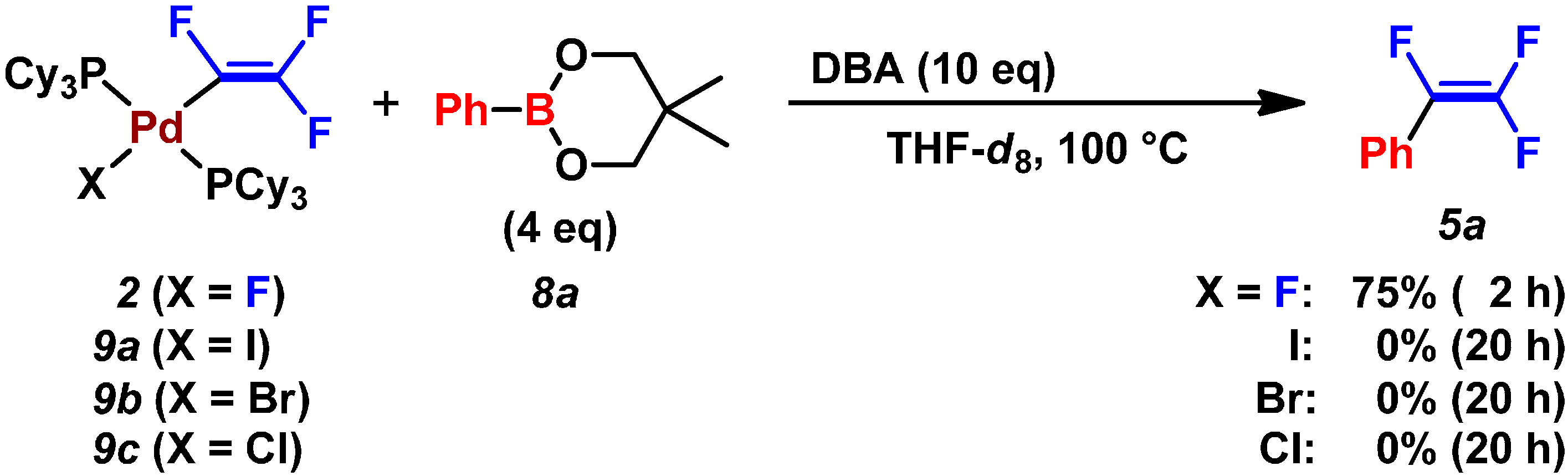
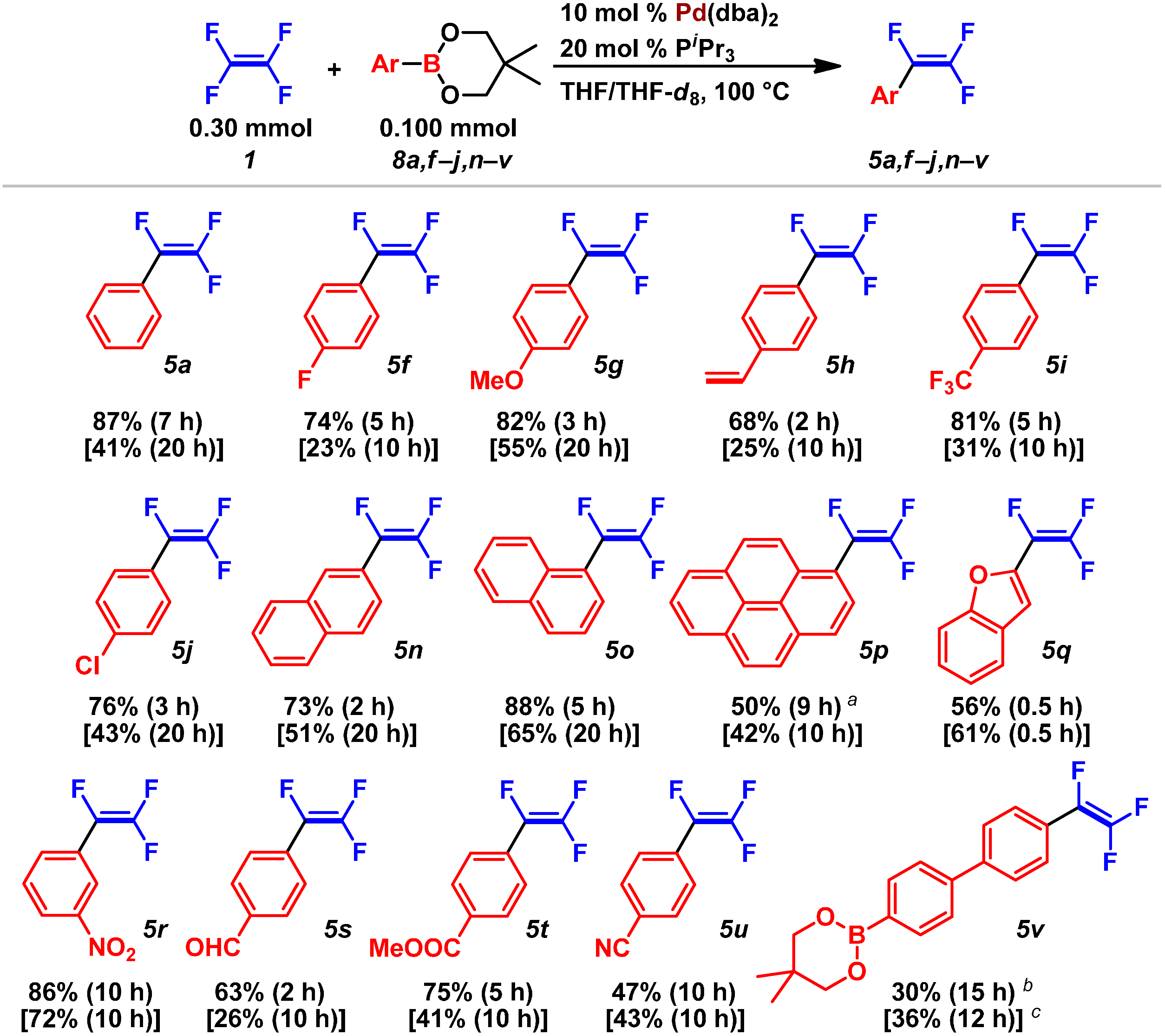
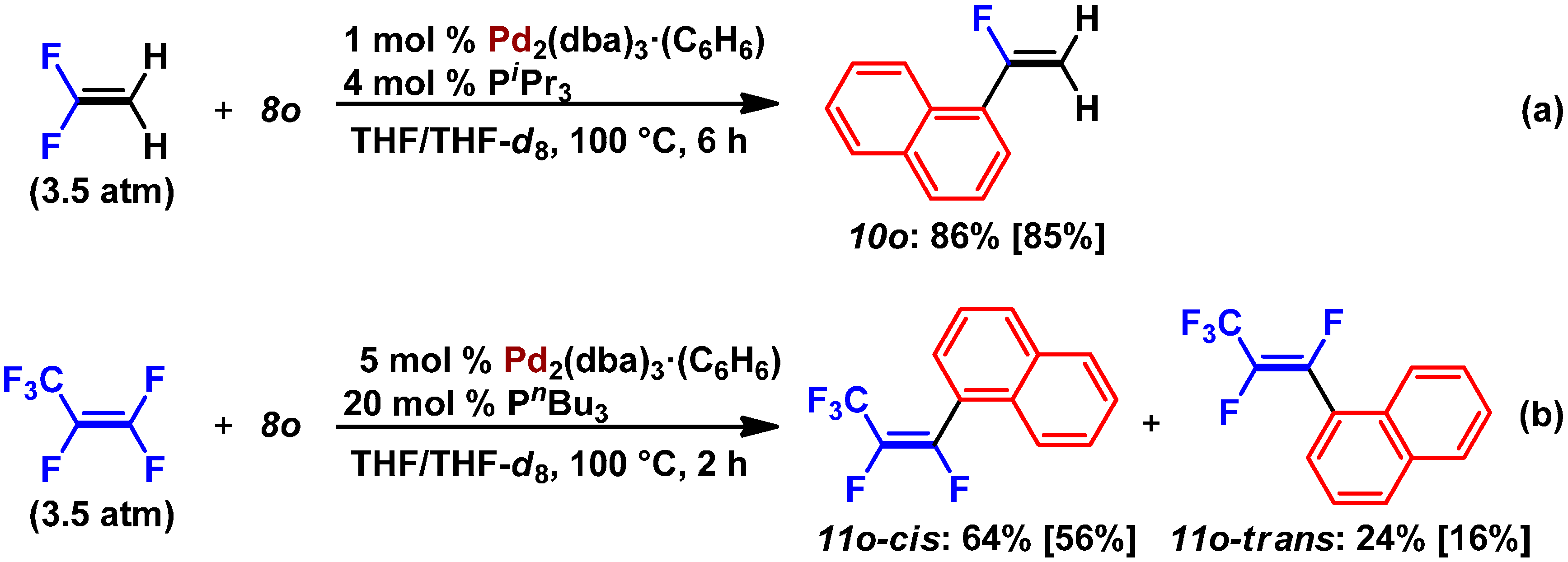
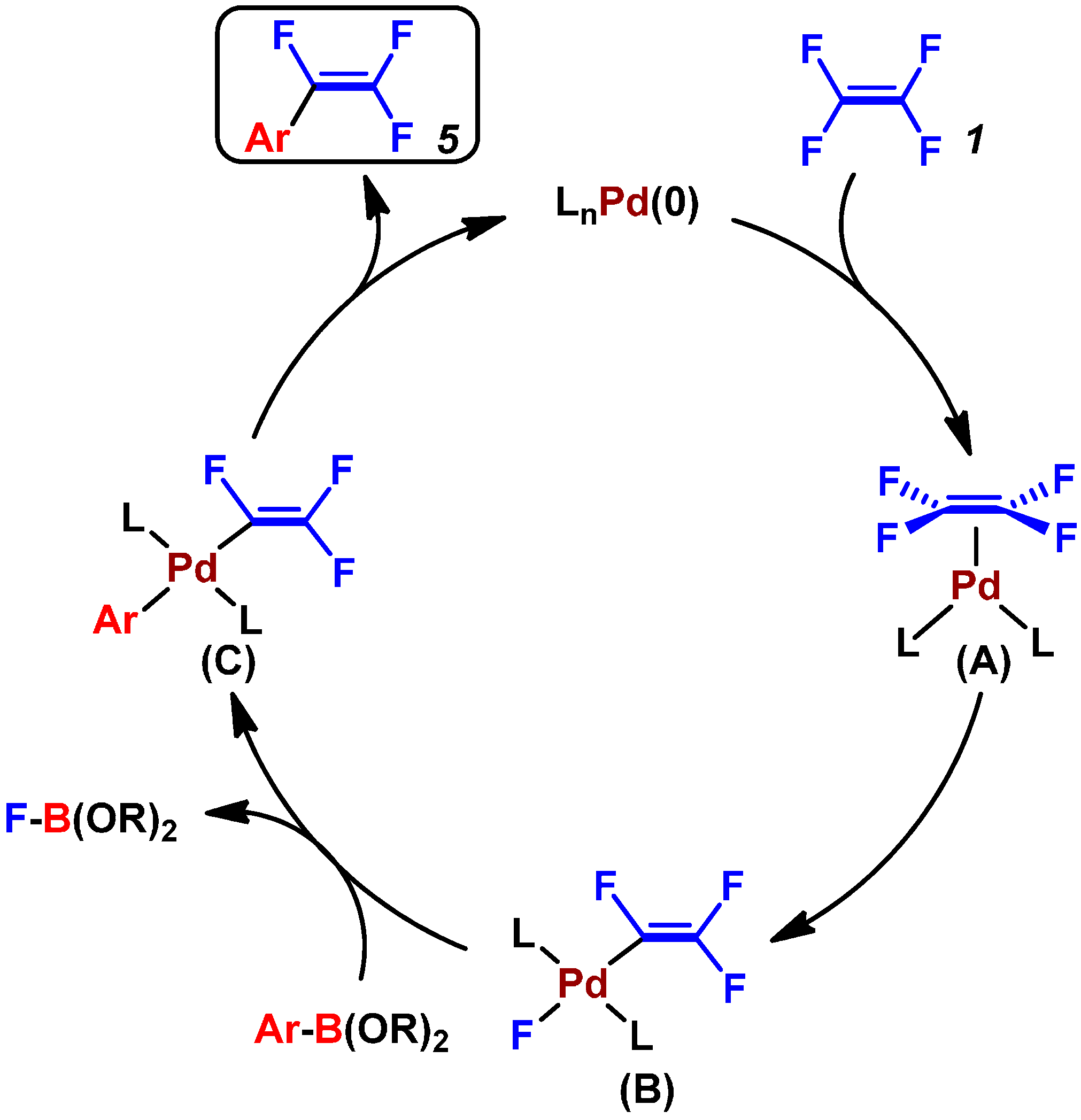
2.2. Pd(0)-Catalyzed Cross-Coupling Reactions of Tetrafluoroethylene with Arylboronates


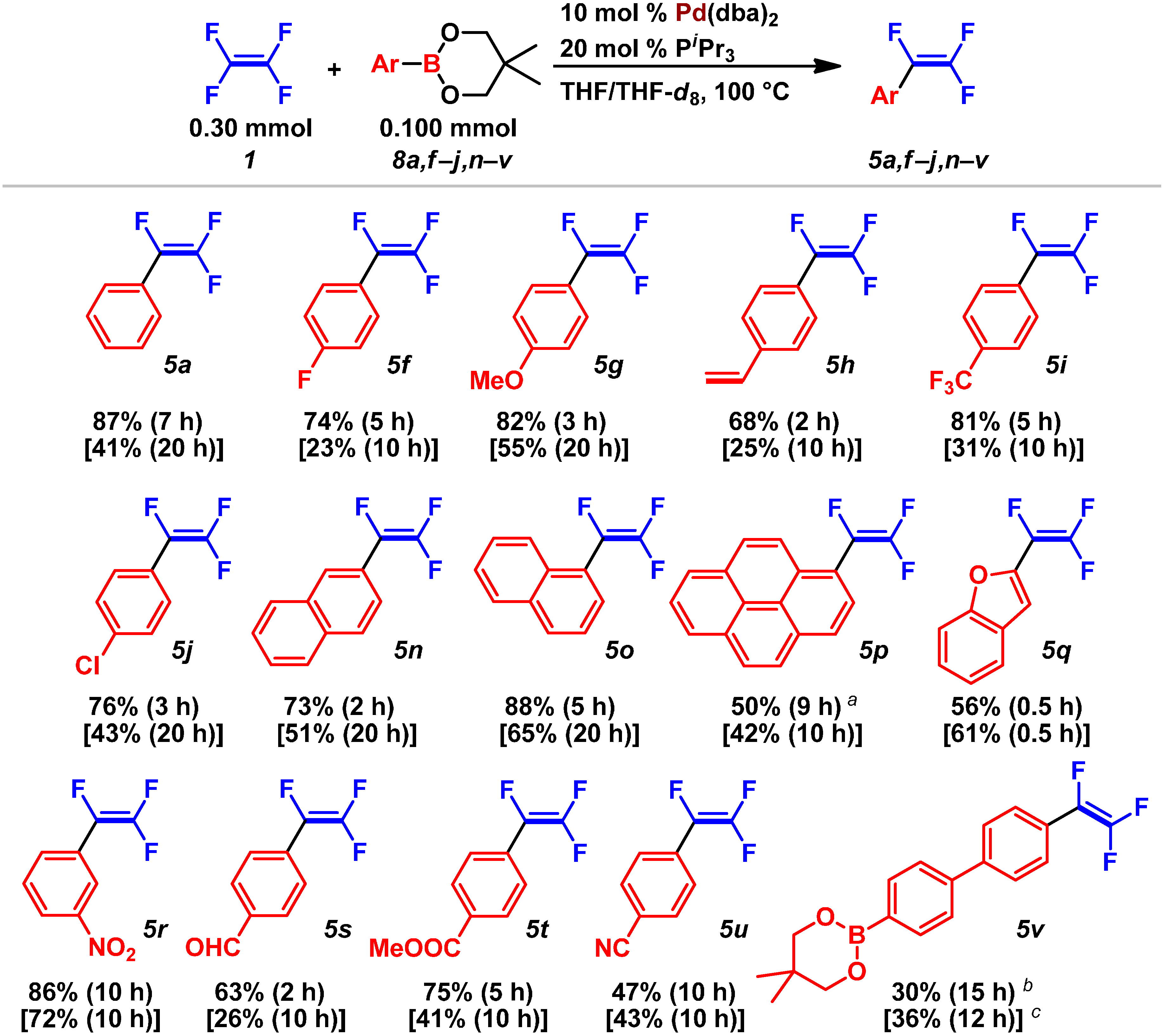


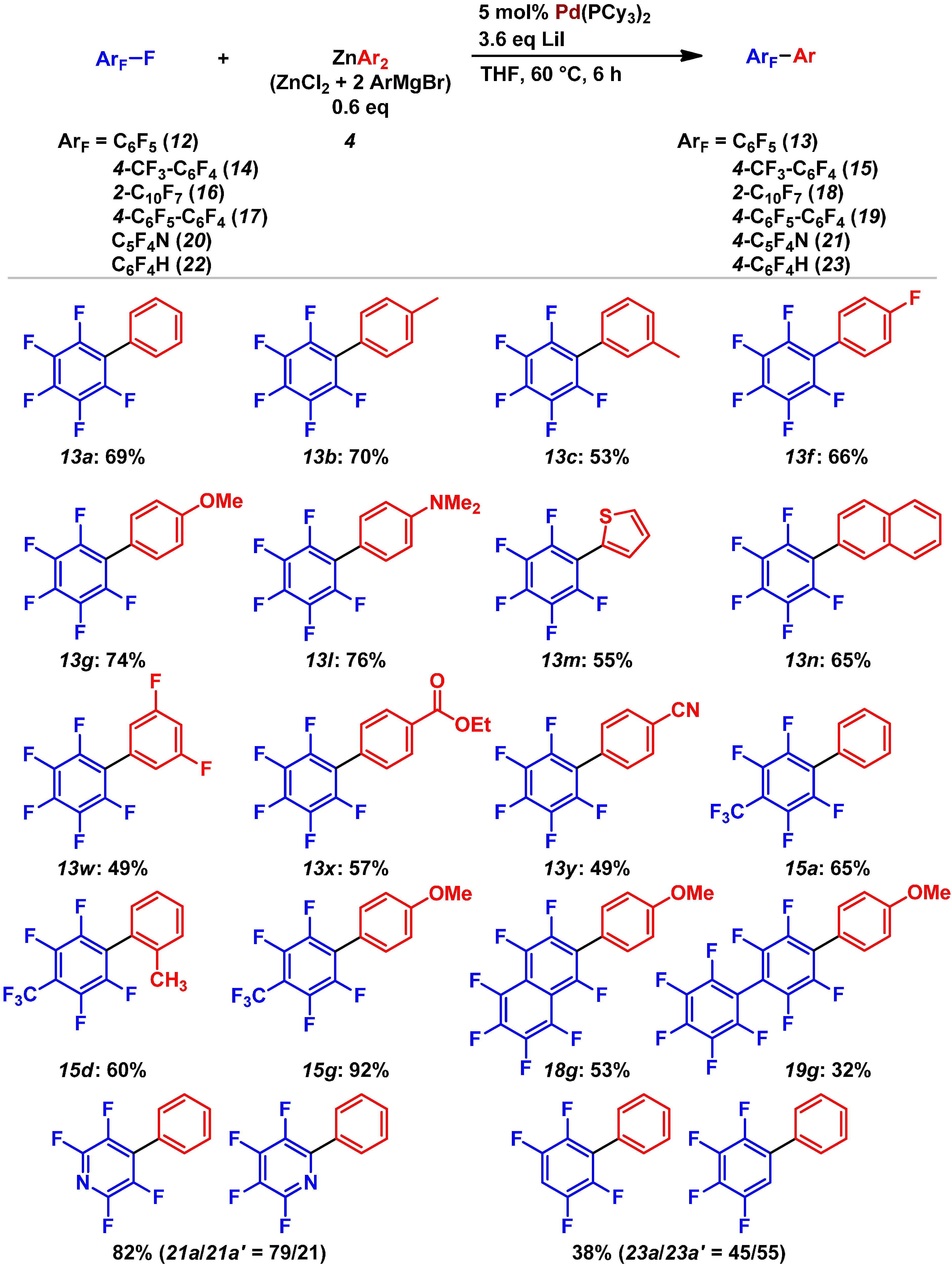
2.3. Pd(0)-Catalyzed Cross-Coupling Reaction of Perfluoroarenes with Diarylzinc Reagents

| Entry | Catalyst/mol% | Preparation of ZnPh2 (4a) | LiI/mmol | Time/h | Yield/% |
|---|---|---|---|---|---|
| 1 | Pd2(dba)3 (5)/PPh3 (20) | ZnCl2 + 2 PhMgBr | 0.240 | 10 | trace |
| 2 | Pd(PCy3)2 (5) | ZnCl2 + 2 PhMgBr | 0.240 | 4 | 70 |
| 3 | Pd(PCy3)2 (10) | isolated ZnPh2 | 0.200 | 4 | 63 |
| 4 | – | ZnCl2 + 2 PhMgBr | 0.240 | 21 | – |
| 5 | Pd(PCy3)2 (5) | ZnCl2 + 2 PhMgBr | 0.360 | 6 | 75 |
| 6 | Pd(PCy3)2 (5) | ZnCl2 + 2 PhMgBr | – | 10 | 5 |
| 7 a | Pd(OAc)2 (5)/PCy3 (10) | ZnCl2 + 2 PhMgBr | 0.240 | 4 | 65 |
| 8 | Pd2(dba)3 (5)/PCy3 (20) | ZnCl2 + 2 PhMgBr | 0.360 | 4 | 77 |
| 9 a | Pd(OAc)2 (5)/DCPE (5) | ZnCl2 + 2 PhMgBr | 0.240 | 9 | 13 |
| 10 a | Pd(OAc)2 (5)/DCPB (5) | ZnCl2 + 2 PhMgBr | 0.240 | 15 | trace |







3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Banks, R.E.; Sharp, D.W.A.; Tatlow, J.C. Fluorines—The First Hundred Years (1886–1986); Elsevier: New York, NY, USA, 1986. [Google Scholar]
- Banks, R.E.; Smart, B.E.; Tatlow, J.C. Organofluorine Chemistry: Principles and Commercial Applications; Plenum Press: New York, NY, USA, 2000. [Google Scholar]
- Filler, R.; Kobayashi, Y.; Yagupolskii, L.N. Organofluorine Compounds in Medicinal and Biomedical Applications; Elsevier: Amsterdam, The Netherlands, 1993. [Google Scholar]
- Ojima, I.; MaCaethy, J.R.; Welch, J.T. Biomedical Frontiers of Fluorine Chemistry; American Chemical Society: Washington, DC, USA, 1996. [Google Scholar]
- Hiyama, T.; Kanie, K.; Kusumoto, T.; Morizawa, Y.; Shimizu, M. Organofluorine Compounds: Chemistry and Application; Springer-Verlag: Berlin, Germany, 2000. [Google Scholar]
- Kirsh, P. Modern Fluoroorganic Chemistry; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Chambers, R.D. Fluorine in Organic Chemistry; Blackwell: Oxford, UK, 2004. [Google Scholar]
- Uneyama, K. Organofluorine Chemistry; Blackwell: Oxford, UK, 2006. [Google Scholar]
- Bégué, J.-P.; Bonnet-Delpon, D. Bioorganic and Medicinal Chemistry of Fluorine; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008. [Google Scholar]
- Souzy, R.; Ameduri, B.; Boutevin, B. Synthesis and (co)polymerization of monofluoro, difluoro, trifluorostyrene and ((trifluorovinyl)oxy)benzene. Prog. Polym. Sci. 2004, 29, 75–106. [Google Scholar]
- Souzy, R.; Ameduri, B.; Boutevin, B.; Gebel, G.; Capron, P. Functional fluoropolymers for fuel cell membranes. Solid State Ion. 2005, 176, 2839–2848. [Google Scholar] [CrossRef]
- Hougham, G.; Cassidy, P.E.; Johns, K.; Davidson, T. Fluoropolymers 1: Synthesis in Topics in Applied Chemistry; Kluwer Academic/Plenum Publisher: New York, NY, USA, 2002. [Google Scholar]
- Cohen, S.G.; Wolosinski, H.T.; Scheuer, P.J. α,β,β-trifluorostyrene and α-Chloro-β,β-difluorostyrene. J. Am. Chem. Soc. 1949, 71, 3439–3440. [Google Scholar] [CrossRef]
- Prober, M. The Synthesis and Polymerization of Some Fluorinated Styrenes. J. Am. Chem. Soc. 1953, 75, 968–973. [Google Scholar] [CrossRef]
- Dvornikova, K.V.; Platonov, V.E.; Yakobson, G.G. Thermolytic transformations of polyfluoroorganic compounds XXIX. Formation of fluorine-containing styrenes by reactions of benzotrichloride and related compounds with dihalocarbene sources. J. Fluor. Chem. 1985, 28, 99–113. [Google Scholar]
- Anilkumar, R.; Burton, D.J. A highly efficient room temperature non-organometallic route for the synthesis of α,β,β-trifluorostyrenes by dehydrohalogenation. Tetrahedron Lett. 2003, 44, 6661–6664. [Google Scholar] [CrossRef]
- Dixon, S. Elimination Reaction of Fluoroölefines with Organolithium Compounds. J. Org. Chem. 1956, 21, 400–403. [Google Scholar] [CrossRef]
- Tarrant, P.; Heyes, J. Fluoro Olefins. XII. The Reaction of Allylmagnesium Bromide with Fluoro Olefins. J. Org. Chem. 1965, 30, 1485–1487. [Google Scholar]
- Jiang, X.; Wu, C.; Wu, Z. Chemistry of trifluorostyrenes and their dimmers. Acta Chem. Sin. 1983, 1, 42–49. [Google Scholar] [CrossRef]
- Sigalov, A.B.; Rybakova, L.F.; Beletskaya, I.P. Arylation of fluoroolefins by phenyl derivatives of ytterbium(II) and europium(II). Izv. Akad. Nauk. SSSR Ser. Khim. 1983, 1208. [Google Scholar]
- Sigalov, A.B.; Beletskaya, I.P. Reactions of phenyl derivatives of lanthanides with fluoro olefins. Izv. Akad. Nauk. SSSR, Ser. Khim. 1988, 445–450. [Google Scholar]
- Crouse, G.D.; Webster, J.D. Addition of fluorinated olefins to ester enolates. Synthesis of fluorinated carboxylic esters and tetrafluorocyclobutanes. J. Org. Chem. 1992, 57, 6643–6646. [Google Scholar]
- Ohashi, M.; Kamura, R.; Doi, R.; Ogoshi, S. Preparation of Trifluorovinyl Compounds by Lithium Salt-promoted Monoalkylation of Tetrafluoroethene. Chem. Lett. 2013, 42, 933–935. [Google Scholar] [CrossRef]
- Beletskaya, I.P. The cross-coupling reactions of organic halides with organic derivatives of tin, mercury and copper catalyzed by palladium. J. Organomet. Chem. 1983, 250, 551–564. [Google Scholar] [CrossRef]
- Gillet, J.-P.; Sauvetre, R.; Normant, J.-F. Preparation et reactivite de fluorovinylzincs. Tetrahedron Lett. 1985, 26, 3999–4002. [Google Scholar]
- Heinze, P.L.; Burton, D.J. Palladium catalyzed coupling of F-cinyl zinc reagents with aryl iodides. An improved synthesis of α,β,β-trifluorostyrenes and the stereospecific preparation of 1-phenyl-F-propenes. J. Fluor. Chem. 1986, 31, 115–119. [Google Scholar]
- Tellier, F.; Sauvetre, R.; Normant, J.F.; Dromzee, Y.; Jeannin, Y. Réactivité des diènes et styrènes fluorés obtenus par réaction de couplage au palladium. J. Organomet. Chem. 1987, 331, 281–298. (In French) [Google Scholar]
- Heinze, P.L.; Burton, D.J. Palladium-catalyzed cross-coupling of perfluoroalkenylzinc reagents with aryl iodides. A new, simple synthesis of α,β,β-trifluorostyrenes and the stereoselective preparation of 1-arylperfluoropropenes. J. Org. Chem. 1988, 53, 2714–2720. [Google Scholar]
- Anilkumar, R.; Burton, D.J. A remarkable room temperature preparation of the trifluorovinylzinc reagent from HFC-134a. A cost-effective, high yield synthesis of α,β,β-trifluorostyrenes. Tetrahedron Lett. 2002, 43, 2731–2733. [Google Scholar]
- Raghavanpillai, A.; Burton, D.J. Room Temperature Preparation of Trifluoroethenylzinc Reagent by Metalation of the Readily Available Halocarbon HFC-134a and an Efficient, Economically Viable Synthesis of 1,2,2-Trifluorostyrenes. J. Org. Chem. 2004, 69, 7083–7091. [Google Scholar] [CrossRef] [PubMed]
- Lechel, T.; Dash, J.; Hommes, P.; Lentz, D.; Reissig, H.-U. Three-Component Synthesis of Perfluoroalkyl- or Perfluoroaryl-Substituted 4-Hydroxypyridine Derivatives and Their Palladium-Catalyzed Coupling Reactions. J. Org. Chem. 2010, 75, 726–732. [Google Scholar] [CrossRef] [PubMed]
- Roemer, M.; Lentz, D. Autocatalytic formation of fluorinated ferrocenophanes from 1,1'-bis(trifluorovinyl)ferrocene. Chem. Commun. 2011, 47, 7239–7241. [Google Scholar] [CrossRef]
- Roemer, M.; Schmiel, P.; Lentz, D. Cymantrene- and Ferrocene-Based Complexes with Perfluorinated Bridging Moieties. Organometallics 2011, 30, 2063–2066. [Google Scholar] [CrossRef]
- Duric, S.; Schmidt, B.M.; Ninnemann, N.M.; Lentz, D.; Tzschucke, C.C. Synthesis of Trifluorostyrene Derivatives by Palladium-Catalyzed Cross-Coupling of Lithium Trimethoxy(trifluorovinyl)borate with Aryl Bromides. Chem. Eur. J. 2012, 18, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Frohn, H.J.; Adonin, N.Y.; Bardin, V.V.; Starichenko, V.F. Highly efficient cross-coupling reactions with the perfluoroorganotrifluoroborate salts K[RFBF3] (RF = C6F5, CF2 = CF). Tetrahedron Lett. 2002, 43, 8111–8114. [Google Scholar] [CrossRef]
- Yamamoto, T.; Yamakawa, T. Pd-Catalyzed Arylation of Chlorotrifluoroethylene Using Arylboronic Acids. Org. Lett. 2012, 14, 3454–3457. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Chen, S.; Lu, L.; Shen, Q. Preparation of Trifluorostyrenes via Palladium-Catalyzed Coupling of Arylboronic Acids with Chloro- and Bromotrifluoroethylene. J. Org. Chem. 2012, 77, 10314–10320. [Google Scholar] [CrossRef] [PubMed]
- Park, J.D.; Benning, A.F.; Downing, F.B.; Laucius, J.F.; McHarness, R.C. Synthesis of Tetrafluorethylene—Pyrolisis of monochlorodifluoromethane. Ind. Eng. Chem. 1947, 39, 354–358. [Google Scholar] [CrossRef]
- Arcella, V.; Troglia, C.; Ghielmi, A. Hyflon Ion Membranes for Fuel Cells. Ind. Eng. Chem. Res. 2005, 44, 7646–7651. [Google Scholar] [CrossRef]
- Ameduri, B.; Boutevin, B. Copolymerization of fluorinated monomers: Recent developments and future trends. J. Fluor. Chem. 2000, 104, 53–62. [Google Scholar] [CrossRef]
- Ohashi, M.; Kambara, T.; Hatanaka, T.; Saijo, H.; Doi, R.; Ogoshi, S. Palladium-Catalyzed Coupling Reactions of Tetrafluoroethylene with Arylzinc Compounds. J. Am. Chem. Soc. 2011, 133, 3256–3259. [Google Scholar] [CrossRef] [PubMed]
- Richmond, T.G. Organometallic Transformations Demonstrate That Fluorocarbons Are Reactive Molecules. Angew. Chem. Int. Ed. 2000, 39, 3241–3244. [Google Scholar] [CrossRef]
- Braun, T.; Perutz, R.N. Routes to fluorinated organic derivatives by nickel mediated C–F activation of heteroaromatics. Chem. Commun. 2002. [Google Scholar] [CrossRef]
- Jones, W.D. Activation of C–F bonds using Cp*2ZrH2: A diversity of mechanisms. Dalton Trans. 2003. [Google Scholar] [CrossRef]
- Torrens, H. Carbon–fluorine bond activation by platinum group metal complexes. Coord. Chem. Rev. 2005, 249, 1957–1985. [Google Scholar] [CrossRef]
- Meier, G.; Braun, T. Catalytic C–F Activation and Hydrodefluorination of Fluoroalkyl Groups. Angew. Chem. Int. Ed. 2009, 48, 1546–1548. [Google Scholar] [CrossRef]
- Amii, H.; Uneyama, K. C–F Bond Activation in Organic Synthesis. Chem. Rev. 2009, 109, 2119–2183. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.D.; Love, J.A. Cross coupling reactions of polyfluoroarenes via C–F activation. Dalton Trans. 2010, 39, 10362–10374. [Google Scholar] [CrossRef] [PubMed]
- Braun, T.; Wehmeier, F. C–F Bond Activation of Highly Fluorinated Molecules at Rhodium: From Model Reactions to Catalysis. Eur. J. Inorg. Chem. 2011. [Google Scholar] [CrossRef]
- Clot, E.; Eisenstein, O.; Jasim, N.; Macgregor, S.A.; McGrady, J.E.; Perutz, R.N. C–F and C–H Bond Activation of Fluorobenzenes and Fluoropyridines at Transition Metal Centers: How Fluorine Tips the Scales. Acc. Chem. Res. 2011, 44, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Klahn, M.; Rosenthal, U. An Update on Recent Stoichiometric and Catalytic C–F Bond Cleavage Reactions by Lanthanide and Group 4 Transition-Metal Complexes. Organometallics 2012, 31, 1235–1244. [Google Scholar] [CrossRef]
- Nova, A.; Mas-Ballesté, R.; Lledós, A. Breaking C–F Bonds via Nucleophilic Attack of Coordinated Ligands: Transformations from C–F to C–X Bonds (X = H, N, O, S). Organometallics 2012, 31, 1245–1256. [Google Scholar] [CrossRef]
- Kuehnel, M.F.; Lentz, D.; Braun, T. Synthesis of Fluorinated Building Blocks by Transition-Metal-Mediated Hydrodefluorination Reactions. Angew. Chem. Int. Ed. 2013, 52, 3328–3348. [Google Scholar] [CrossRef]
- Kemmitt, R.D.W.; Moore, R.D. Tetrafluoroethylene complexes of platinum(0). J. Chem. Soc. A 1971. [Google Scholar] [CrossRef]
- Booth, B.L.; Casey, G.C.; Haszeldine, R.N. Reactions involving transition metals: XII. Some attemps to prepare alkulidynetrirhodium cluster compounds. J. Organomet. Chem. 1981, 219, 401–408. [Google Scholar]
- Anderson, D.J.; McDonald, R.; Cowie, M. Carbon–Fluorine Bond Activation in Fluoroolefins: Clear Documentation of Cooperative C–F Bond Activation by Adjacent Metal Centers. Angew. Chem. Int. Ed. 2007, 46, 3741–3744. [Google Scholar] [CrossRef]
- Ohashi, M.; Shibata, M.; Saijo, H.; Kambara, T.; Ogoshi, S. Carbon-Fluorine Bond Activation of Tetrafluoroethylene on Palladium(0) and Nickel(0): Heat or Lewis Acidic Additive Promoted Oxidative Addition. Organometallics 2013, 32, 3631–3639. [Google Scholar] [CrossRef]
- Ohashi, M.; Saijo, H.; Shibata, M.; Ogoshi, S. Palladium-Catalyzed Base-Free Suzuki-Miyaura Coupling Reaction of Fluorinated Alkenes and Arenes via Palladium Fluoride Key Intermediate. Eur. J. Org. Chem. 2013. [Google Scholar] [CrossRef]
- Schaub, T.; Backes, M.; Radius, U. Catalytic C–C Bond Formation Accomplished by Selective C–F Activation of Perfluorinated Arenes. J. Am. Chem. Soc. 2006, 128, 15964–15965. [Google Scholar] [CrossRef] [PubMed]
- Schaub, T.; Radius, U. Efficient C–F and C–C Activation by a Novel N-Heterocyclic Carbene–Nickel(0) Complex. Chem. Eur. J. 2005, 11, 5024–5030. [Google Scholar] [CrossRef] [PubMed]
- Schaub, T.; Fischer, P.; Steffen, A.; Braun, T.; Radius, U.; Mix, A. C–F Activation of Fluorinated Arenes using NHC-Stabilized Nickel(0) Complexes: Selectivity and Mechanistic Investigations. J. Am. Chem. Soc. 2008, 130, 9304–9317. [Google Scholar] [CrossRef] [PubMed]
- Fischer, P.; Götz, K.; Eichhorn, A.; Radius, U. Decisive Steps of the Hydrodefluorination of Fluoroaromatics using [Ni(NHC)2]. Organometallics 2012, 31, 1374–1383. [Google Scholar] [CrossRef]
- Guo, H.; Kong, F.; Kanno, K.-I.; He, J.; Nakajima, K.; Takahashi, T. Early Transition Metal-Catalyzed Cross-Coupling Reaction of Aryl Fluorides with a Phenethyl Grignard Reagent Accompanied by Rearrangement of the Phenethyl Group. Organometallics 2006, 25, 2045–2048. [Google Scholar] [CrossRef]
- Nakamura, Y.; Yoshikai, N.; Ilies, L.; Nakamura, E. Nickel-Catalyzed Monosubstitution of Polyfluoroarenes with Organozinc Reagents Using Alkoxydiphosphine Ligand. Org. Lett. 2012, 14, 3316–3319. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, M.; Doi, R.; Ogoshi, S. Palladium-Catalyzed Coupling Reaction of Perfluoroarenes with Diarylzinc Compounds. Chem. Eur. J. 2014, 20, 2040–2048. [Google Scholar] [CrossRef] [PubMed]
- Recently, Lu and Shen reported the molecular structure of the related trifluorovinyl palladim(II) chloride, trans-(PPh3)2Pd(Cl)(CF=CF2), whereas the trifluorovinyl complex was prepared via oxidative addition of the C–F bond of chlorotrifluoroethylene. See Ref. [37].
- Bartlett, P.D.; Cohen, G.M. Dimers of α,β,β-trifluorostyrene. J. Am. Chem. Soc. 1973, 95, 7923–7925. [Google Scholar] [CrossRef] [PubMed]
- Achonduh, G.T.; Hadei, N.; Valente, C.; Avola, S.; O’Brien, C.J.; Organ, M.G. On the role of additives in alkyl–alkyl Negishi cross-couplings. Chem. Commun. 2010, 46, 4109–4111. [Google Scholar] [CrossRef]
- Burton et al. claimed that some of (α,β,β-trifluoro)styrene derivatives are prone to cyclodimerize under a higher concentration (especially in pure form) condition even at room temperature or below. See also Ref. [28].
- Miyaura, N.; Suzuki, A. Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef]
- Suzuki, A.; Brown, H.C. Organic Syunthesis via Boranes; Aldrich: Milwaukee, WI, USA, 2003. [Google Scholar]
- Hall, D.G. Boronic Acids; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- Miyaura, N. Metal-Catalyzed Reactions of Organoboronic Acids and Esters. Bull. Chem. Soc. Jpn. 2008, 81, 1535–1553. [Google Scholar] [CrossRef]
- Suzuki, A. Cross-Coupling Reactions Of Organoboranes: An Easy Way To Construct C–C Bonds (Nobel Lecture). Angew. Chem. Int. Ed. 2011, 50, 6722–6737. [Google Scholar] [CrossRef]
- Widdowson, D.A.; Wilhelm, R. Palladium catalysed cross-coupling of (fluoroarene)tricarbonyl−chromium(0) complexes. Chem. Commun. 1999. [Google Scholar] [CrossRef]
- Widdowson, D.A.; Wilhelm, R. Palladium catalysed Suzuki reactions of fluoroarenes. Chem. Commun. 2003. [Google Scholar] [CrossRef]
- Kim, Y.M.; Yu, S. Palladium(0)-Catalyzed Amination, Stille Coupling, and Suzuki Coupling of Electron-Deficient Aryl Fluorides. J. Am. Chem. Soc. 2003, 125, 1696–1697. [Google Scholar] [CrossRef] [PubMed]
- Mikami, K.; Miyamoto, T.; Hatano, M. A highly efficient asymmetric Suzuki–Miyaura coupling reaction catalyzed by cationic chiral palladium(II) complexes. Chem. Commun. 2004. [Google Scholar] [CrossRef]
- Steffen, A.; Sladek, M.; Braun, T.; Neumann, B.; Stammler, H.-G. Catalytic C–C Coupling Reactions at Nickel by C–F Activation of a Pyrimidine in the Presence of a C–Cl Bond: The Crucial Role of Highly Reactive Fluoro Complexes. Organometallics 2005, 24, 4057–4064. [Google Scholar] [CrossRef]
- Bahmanyar, S.; Borer, B.C.; Kim, Y.M.; Kurtz, D.M.; Yu, S. Proximity Effects in the Palladium-Catalyzed Substitution of Aryl Fluorides. Org. Lett. 2005, 7, 1011–1014. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Robins, M.J. Fluoro, Alkylsulfanyl, and Alkylsulfonyl Leaving Groups in Suzuki Cross-Coupling Reactions of Purine 2'-Deoxynucleosides and Nucleosides. Org. Lett. 2005, 7, 1149–1151. [Google Scholar] [CrossRef] [PubMed]
- Cargill, M.R.; Sandford, G.; Tadeusiak, A.J.; Yufit, D.S.; Howard, J.A.K.; Kilickiran, P.; Nelles, G. Palladium-Catalyzed C–F Activation of Polyfluoronitrobenzene Derivatives in Suzuki–Miyaura Coupling Reactions. J. Org. Chem. 2010, 75, 5860–5866. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.D.; Love, J.A. Nickel-Catalyzed Selective Defluorination to Generate Partially Fluorinated Biaryls. Org. Lett. 2011, 13, 2750–2753. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Shen, Q.; Lu, L. Selective Palladium-Catalyzed C–F Activation/Carbon–Carbon Bond Formation of Polyfluoroaryl Oxazolines. J. Org. Chem. 2012, 77, 1798–1804. [Google Scholar] [CrossRef] [PubMed]
- Breyer, D.; Braun, T.; Kläring, P. Synthesis and Reactivity of the Fluoro Complex trans-[Pd(F)(4-C5NF4)(iPr2PCH2CH2OCH3)2]: C–F Bond Formation and Catalytic C–F Bond Activation Reactions. Organometallics 2012, 31, 1417–1424. [Google Scholar] [CrossRef]
- Miyaura, N. Cross-coupling reaction of organoboron compounds via base-assisted transmetalation to palladium(II) complexes. J. Organomet. Chem. 2002, 653, 54–57. [Google Scholar] [CrossRef]
- Carrow, B.P.; Hartwig, J.F. Distinguishing Between Pathways for Transmetalation in Suzuki–Miyaura Reactions. J. Am. Chem. Soc. 2011, 133, 2116–2119. [Google Scholar] [CrossRef] [PubMed]
- Gooßen, L.J.; Ghosh, K. Palladium-Catalyzed Synthesis of Aryl Ketones from Boronic Acids and Carboxylic Acids or Anhydrides. Angew. Chem. Int. Ed. 2001, 40, 3458–3460. [Google Scholar]
- Tsukamoto, H.; Sato, M.; Kondo, Y. Palladium(0)-catalyzed direct cross-coupling reaction of allyl alcohols with aryl- and vinyl-boronic acids. Chem. Commun. 2004. [Google Scholar] [CrossRef]
- Tobisu, M.; Xu, T.; Shimasaki, T.; Chatani, N. Nickel-Catalyzed Suzuki–Miyaura Reaction of Aryl Fluorides. J. Am. Chem. Soc. 2011, 133, 19505–19511. [Google Scholar] [CrossRef] [PubMed]
- Morken, P.A.; Campbell, R.F.; Burton, D.J. Preparation of the E- and Z-heptafluorobutenyl-2-zinc reagent by zinc-induced dehalogenation/metallation of 2,2-dibromo-octafluorobutane. J. Fluor. Chem. 1994, 66, 81–85. [Google Scholar] [CrossRef]
- Jasim, N.A.; Perutz, R.N.; Whitwood, A.C.; Braun, T.; Izundu, J.; Neumann, B.; Rothfeld, S.; Stammler, H.-G. Contrasting Reactivity of Fluoropyridines at Palladium and Platinum: C−F Oxidative Addition at Palladium, P−C and C−F Activation at Platinum. Organometallics 2004, 23, 6140–6149. [Google Scholar] [CrossRef]
- Macgregor, S.A.; Roe, D.C.; Marshall, W.J.; Bloch, K.M.; Bakhmutov, V.I.; Grushin, V.V. The F/Ph Rearrangement Reaction of [(Ph3P)3RhF], the Fluoride Congener of Wilkinson’s Catalyst. J. Am. Chem. Soc. 2005, 127, 15304–15321. [Google Scholar] [CrossRef] [PubMed]
- Braun, T.; Steffen, A.; Schorlemer, V.; Neumann, B.; Stammler, H.-G. Routes to unique palladium A-frame complexes with a bridging fluoro-ligand. Dalton Trans. 2005. [Google Scholar] [CrossRef]
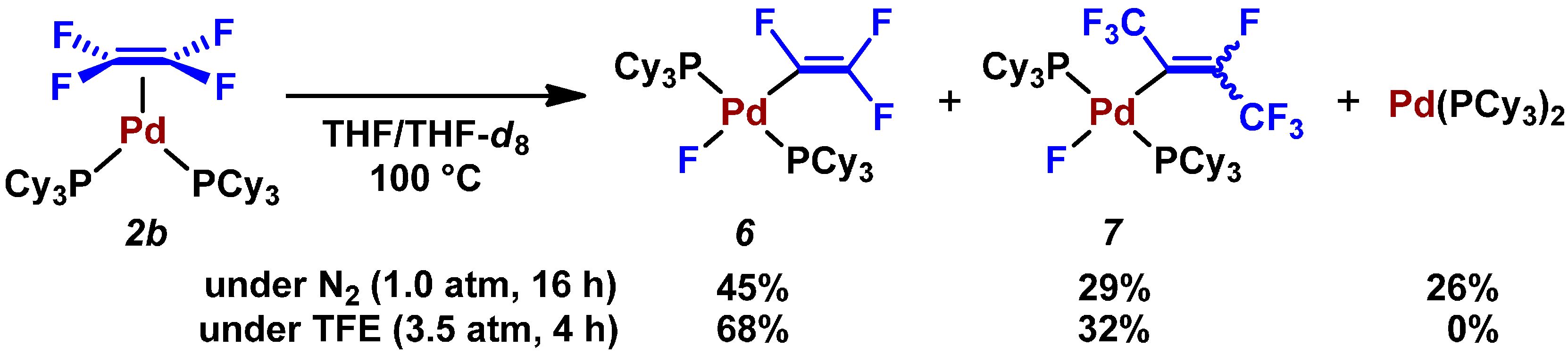
- GC-MS analysis of the crude product revealed this catalytic reaction gave a small percentage of a coupling product with a formula of C10H5F7 (m/z = 256). This undesired coupling product might generate via 7.
- Dmowski, W. An improved synthesis of 1-phenylpentafluoropropenes. J. Fluor. Chem. 1981, 18, 25–30. [Google Scholar] [CrossRef]
- Amatore, C.; Jutand, A.; le Duc, G. The Triple Role of Fluoride Ions in Palladium-Catalyzed Suzuki–Miyaura Reactions: Unprecedented Transmetalation from [ArPdFL2] Complexes. Angew. Chem. Int. Ed. 2012, 51, 1379–1382. [Google Scholar] [CrossRef]
- Mankad, N.P.; Toste, F.D. C–C Coupling Reactivity of an Alkylgold(III) Fluoride Complex with Arylboronic Acids. J. Am. Chem. Soc. 2010, 132, 12859–12861. [Google Scholar] [CrossRef] [PubMed]
- Krasovskiy, A.; Malakhov, V.; Gavryushin, A.; Knochel, P. Efficient Synthesis of Functionalized Organozinc Compounds by the Direct Insertion of Zinc into Organic Iodides and Bromides. Angew. Chem. Int. Ed. 2006, 45, 6040–6044. [Google Scholar] [CrossRef]
- Kashiwabara, T.; Tanaka, M. Decarbonylative Coupling of Fluorobenzoyl Chlorides with Hexamethyldisilane in the Presence of a Palladium Complex Catalyst: Extremely Facile Decarbonylation of Pentafluorobenzoyl–Pd Complex Relevant to C6F5SiMe3 Formation. Organometallics 2006, 25, 4648–4652. [Google Scholar] [CrossRef]
- Tao, S.; Weng, Z.; Hor, T.S.A. 1,1'-P/O-Ferrocenyl Ligands in Palladium-Catalyzed Suzuki Coupling of Aryl Chlorides. Organometallics 2006, 25, 1199–1205. [Google Scholar] [CrossRef]
- Barrios-Landeros, F.; Carrow, B.P.; Hartwig, J.F. Effect of Ligand Steric Properties and Halide Identity on the Mechanism for Oxidative Addition of Haloarenes to Trialkylphosphine Pd(0) Complexes. J. Am. Chem. Soc. 2009, 131, 8141–8154. [Google Scholar]
- Uson, R.; Fornies, J.; Gimeno, J.; Espinet, P.; Navarro, R. Bis(pentafluorophenyl)complexes of palladium(II) and of platinum(II). J. Organomet. Chem. 1974, 81, 115–122. [Google Scholar] [CrossRef]
- Uson, R.; Fornies, J.; Navarro, R. A new route for the synthesis of binuclear organometallic and inorganic palladium(II) complexes. Inorg. Chim. Acta 1979, 33, 69–75. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ohashi, M.; Ogoshi, S. Palladium-Catalyzed Cross-Coupling Reactions of Perfluoro Organic Compounds. Catalysts 2014, 4, 321-345. https://doi.org/10.3390/catal4030321
Ohashi M, Ogoshi S. Palladium-Catalyzed Cross-Coupling Reactions of Perfluoro Organic Compounds. Catalysts. 2014; 4(3):321-345. https://doi.org/10.3390/catal4030321
Chicago/Turabian StyleOhashi, Masato, and Sensuke Ogoshi. 2014. "Palladium-Catalyzed Cross-Coupling Reactions of Perfluoro Organic Compounds" Catalysts 4, no. 3: 321-345. https://doi.org/10.3390/catal4030321
APA StyleOhashi, M., & Ogoshi, S. (2014). Palladium-Catalyzed Cross-Coupling Reactions of Perfluoro Organic Compounds. Catalysts, 4(3), 321-345. https://doi.org/10.3390/catal4030321