
Competition of CO and H2 for Active Oxygen Species during the Preferential CO Oxidation (PROX) on Au/TiO2 Catalysts

Abstract
:1. Introduction
2. Results and Discussions
2.1. Active Oxygen Removal by CO and H2

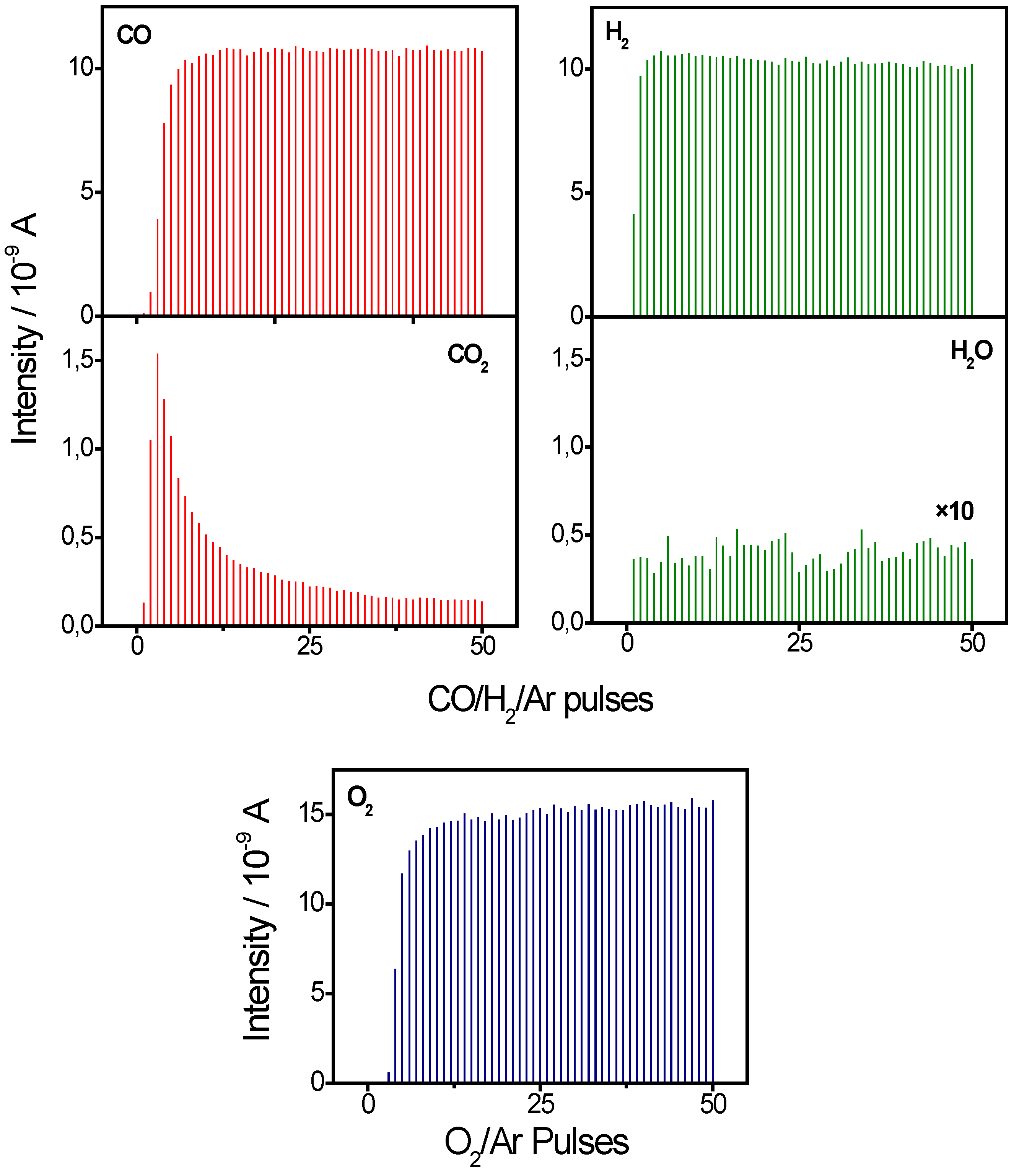{kind=link}
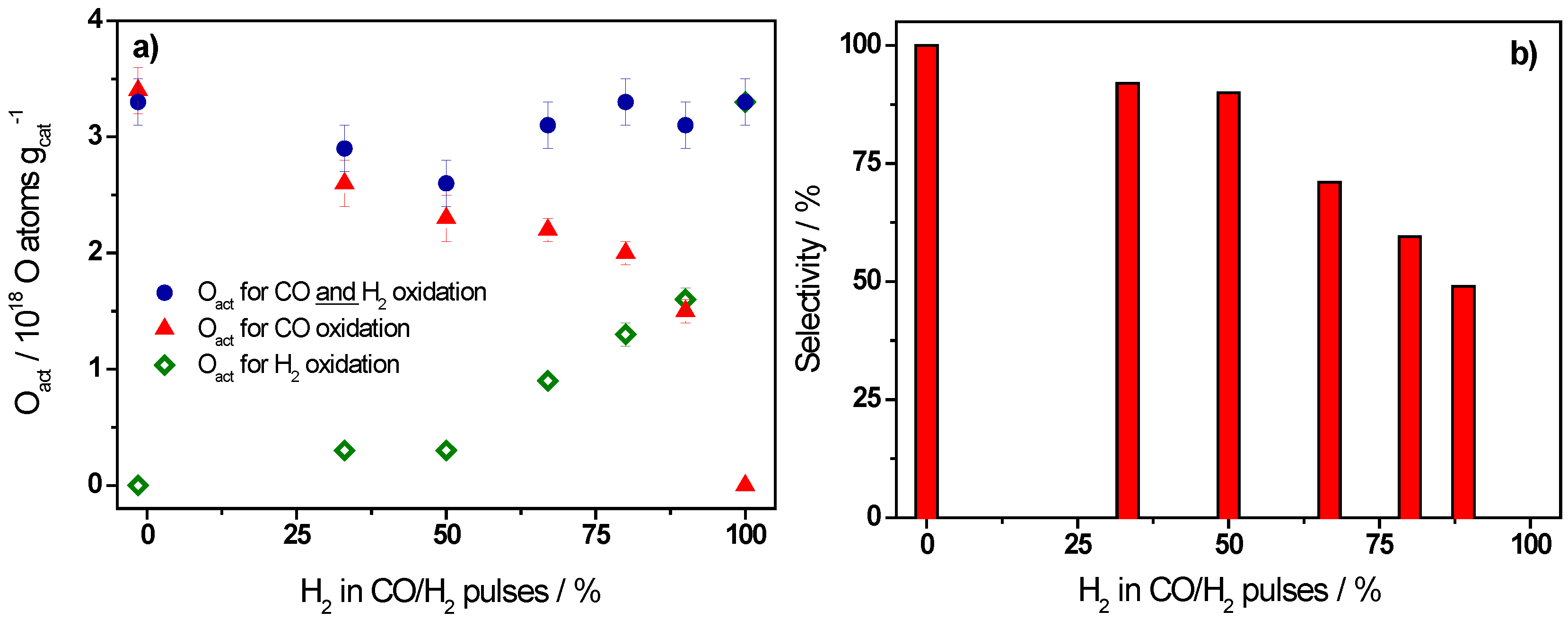{kind=link}
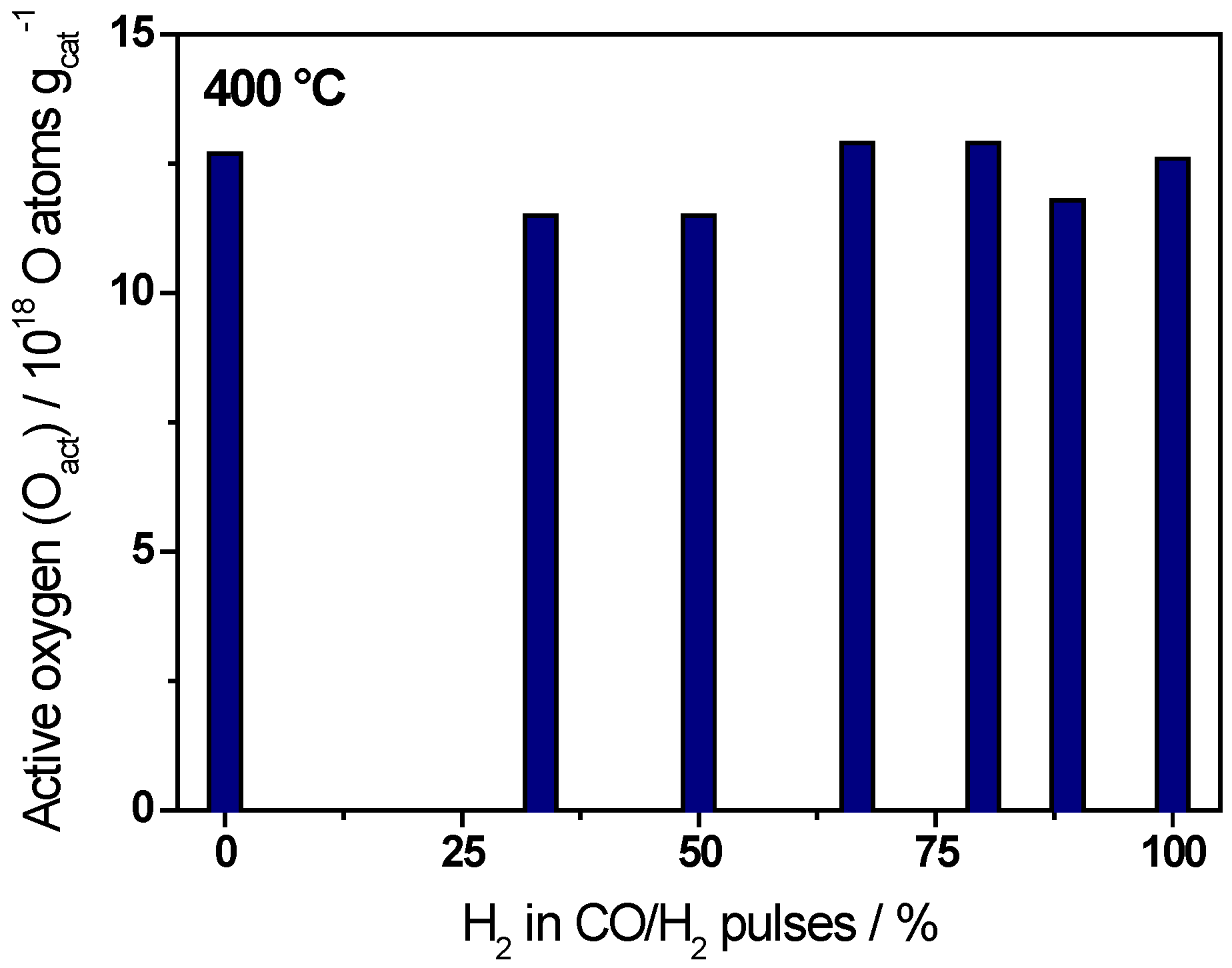{kind=link}
| Ratio CO/H2 | H2/(CO + H2)/% | Oact Removal */1018 O atoms·gcat−1 | CO2 Formation */1018 molecules·gcat−1 | Selectivity/% |
|---|---|---|---|---|
| CO only | 0 | 3.3 ± 0.2 | 3.4 ± 0.2 | 100 |
| 1/0.5 | 33 | 2.9 ± 0.2 | 2.6 ± 0.2 | 92 |
| 1/1 | 50 | 2.6 ± 0.2 | 2.3 ± 0.2 | 90 |
| 1/2 | 67 | 3.1 ± 0.2 | 2.2 ± 0.1 | 71 |
| 1/4 | 80 | 3.3 ± 0.2 | 2.0 ± 0.1 | 60 |
| 1/8 | 89 | 3.1 ± 0.2 | 1.5 ± 0.1 | 49 |
| H2 only | 100 | 3.3 ± 0.2 | 0 | 0 |
| Ratio CO/H2 | H2/(CO+H2)/% | Oact removal */1018 O atoms·gcat−1 | CO2 formation */1018 molecules·gcat−1 |
|---|---|---|---|
| CO only | 0 | 12.7 ± 0.6 | 10.0 ± 0.5 |
| 1/0.5 | 33 | 11.5 ± 0.6 | 9.3 ± 0.5 |
| 1/1 | 50 | 11.5 ± 0.6 | 8.8 ± 0.4 |
| 1/2 | 67 | 12.9 ± 0.6 | 8.7 ± 0.4 |
| 1/4 | 80 | 12.9 ± 0.6 | 7.7 ± 0.3 |
| 1/8 | 89 | 11.8 ± 0.6 | 6.1 ± 0.3 |
| H2 only | 100 | 12.6 ± 0.6 | 0 |
2.2. Active Oxygen Removal by CO/H2 Mixtures



3. Materials and Methods
3.1. Preparation and Characterization of Au/TiO2
3.2. TAP Reactor Measurements
3.3. Calculation of the Selectivity
4. Conclusions
- (1)
- CO and hydrogen are oxidized by the same stable active oxygen species under present reaction conditions, also in the simultaneous presence of CO and H2 in the reaction atmosphere, as evidenced by the similar amounts of active oxygen removal from a O2/Ar pulse oxidized Au/TiO2 catalyst upon exposure to multi-pulse sequences of CO, H2, and CO/H2 mixtures. This is independent of the CO/H2 ratio. Hence, also under PROX reaction conditions CO and H2 compete for TiO2 surface lattice oxygen close to the Au nanoparticles/at the perimeter of the Au-TiO2 interface as active oxygen species.
- (2)
- The selectivity of Au/TiO2 catalysts for CO oxidation in CO/H2 containing gas mixtures observed in kinetic measurements is proposed to mainly result from the much higher efficiency of CO for active oxygen removal compared to hydrogen under present reaction conditions, in the simultaneous presence of H2 and CO in the reaction atmosphere. The latter is illustrated also by the much higher amount of H2 pulses required for complete removal of active oxygen from the catalyst compared to CO pulsing.
- (3)
- Since we can rule out the presence of weakly-bound adsorbed water under present reaction conditions, in multi-pulse sequences, the much higher efficiency of CO for reaction with active oxygen species or the preference for this reaction pathway cannot result from effects caused by weakly-bound water species. On the other hand, stable adsorbed (strongly bound) water species, which are known to be present on the surface upon H2 pulsing, leave the dominant reaction pathway for CO oxidation under present reaction conditions, namely reaction of COad with surface lattice oxygen at the perimeter of the Au NP-support interface, unchanged, although the reaction rate may change.
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Oact | Active Oxygen |
| PROX | Preferential CO Oxidation |
| TAP | Temporal Analysis of Products |
| PEM | Polymer Electrolyte Membrane |
| TEM | Transmission Electron Microscopy |
| TPD | Temperature Programmed Desorption |
References
- Hashmi, A.S.K.; Hutchings, G.J. Gold Catalysis. Angew. Chem. Int. Ed. 2006, 45, 7896–7936. [Google Scholar] [CrossRef] [PubMed]
- Min, B.K.; Friend, C.M. Heterogeneous Gold-Based Catalysis for Green Chemistry: Low-Temperature CO Oxidation and Propene Oxidation. Chem. Rev. 2007, 107, 2709–2724. [Google Scholar] [CrossRef] [PubMed]
- Freakley, S.J.; He, Q.; Kiely, C.J.; Hutchings, G.J. Gold Catalysis: A Reflection on Where We are Now. Catal. Lett. 2015, 145, 71–79. [Google Scholar] [CrossRef]
- Haruta, M. Role of perimeter interfaces in catalysis by gold nanoparticles. Faraday Discuss. 2011, 152, 11–32. [Google Scholar] [CrossRef] [PubMed]
- Freund, H.J.; Meijer, G.; Scheffler, M.; Schlögl, R.; Wolf, M. CO Oxidation as a Prototypical Reaction for Heterogeneous Processes. Angew. Chem. Int. Ed. 2011, 50, 10064–10094. [Google Scholar] [CrossRef] [PubMed]
- Trimm, D.L.; Önsan, Z.I. On-board fuel conversion for hydrogen-fuel-cell-driven vehicles. Catal. Rev. 2001, 43, 31–84. [Google Scholar] [CrossRef]
- Ghenciu, A.F. Review of fuel processing catalysts for hydrogen production in PEM fuel cell systems. Curr. Opin. Solid State Mater. Sci. 2002, 6, 389–399. [Google Scholar] [CrossRef]
- Corti, C.W.; Holliday, R.J.; Thompson, D.T. Progress towards the commercial application of gold catalysts. Top. Catal. 2007, 44, 331–343. [Google Scholar] [CrossRef]
- Park, E.D.; Lee, D.; Lee, H.C. Recent progress in selective CO removal in a H2-rich stream. Catal. Today 2009, 139, 280–290. [Google Scholar] [CrossRef]
- Rossignol, C.; Arrii, S.; Morfin, F.; Piccolo, L.; Caps, V.; Rousset, J.-L. Selective oxidation of CO over model gold-based catalysts in the presence of H2. J. Catal. 2005, 230, 476–483. [Google Scholar] [CrossRef]
- Bion, N.; Epron, F.; Moreno, M.; Marino, F.; Duprez, D. Preferential Oxidation of Carbon Monoxide in the Presence of Hydrogen (PROX) over Noble Metals and Transition Metal Oxides: Advantages and Drawbacks. Top. Catal. 2008, 51, 76–88. [Google Scholar] [CrossRef]
- Quinet, E.; Piccolo, L.; Morfin, F.; Avenier, P.; Diehl, F.; Caps, V.; Rousset, J.L. On the mechanism of hydrogen-promoted gold-catalyzed CO oxidation. J. Catal. 2009, 268, 384–389. [Google Scholar] [CrossRef]
- Kahlich, M.J.; Gasteiger, H.A.; Behm, R.J. Kinetics of the Selective Low-Temperature Oxidation of CO in H2-rich Gas over Au/Fe2O3. J. Catal. 1999, 182, 430–440. [Google Scholar] [CrossRef]
- Schumacher, B.; Denkwitz, Y.; Plzak, V.; Kinne, M.; Behm, R.J. Kinetics, mechanism and the influence of H2 on the CO oxidation reaction on a Au/TiO2 catalyst. J. Catal. 2004, 224, 449–462. [Google Scholar] [CrossRef]
- Denkwitz, Y.; Schumacher, B.; Kucèrová, G.; Behm, R.J. Activity, stability and deactivation behavior of supported Au/TiO2 catalysts in the CO oxidation and preferential CO oxidation reaction at elevated temperatures. J. Catal. 2009, 267, 78–88. [Google Scholar] [CrossRef]
- Grzybowska, B. Nano-Au/oxide support catalysts in oxidation reactions: Provenance of active oxygen species. Catal. Today 2006, 112, 3–7. [Google Scholar] [CrossRef]
- Bollinger, M.A.; Vannice, M.A. A kinetic and DRIFTS study of low-temperature carbon monoxide oxidation over Au-TiO2 catalysts. Appl. Catal. B 1996, 8, 417–443. [Google Scholar] [CrossRef]
- Boccuzzi, F.; Chiorino, A. FTIR study on Au/TiO2 at 90K and room temperature. An insight into the nature of the reaction centers. J. Phys. Chem. B 2000, 104, 5414–5416. [Google Scholar] [CrossRef]
- Green, I.X.; Tang, W.; Neurock, M.; Yates, J.T. Spectroscopic Observation of Dual Catalytic Sites During Oxidation of CO on a Au/TiO2 Catalyst. Science 2011, 333, 736–739. [Google Scholar] [CrossRef] [PubMed]
- Boronat, M.; Francesc, I.; Corma, A. Active Sites for H2 Adsorption and Activation in Au/TiO2 and the Role of the Support. J. Phys. Chem. A 2009, 113, 3750–3757. [Google Scholar] [CrossRef] [PubMed]
- Panayotov, D.A.; Burrows, V.A.; Yates, J.T., Jr.; Morris, L. Mechanistic Studies of Hydrogen Dissociation and Spillover on Au/TiO2: IR Spectroscopy of Coadsorbed CO and H-Donated Electrons. J. Phys. Chem. C 2011, 115, 22400–22408. [Google Scholar] [CrossRef]
- Fujitani, T.; Nakamura, I.; Akita, T.; Okumura, M.; Haruta, M. Hydrogen Dissociation by Gold Clusters. Angew. Chem. 2009, 121, 9679–9682. [Google Scholar] [CrossRef]
- Nakamura, I.; Mantoku, H.; Furukawa, T.; Fujitani, T. Active Sites for Hydrogen Dissociation over TiOx/Au(111) Surfaces. J. Phys. Chem. C 2011, 115, 16074–16080. [Google Scholar] [CrossRef]
- Bus, E.; Miller, J.T.; van Bokhoven, J.A. Hydrogen Chemisorption on Al2O3-Supported Gold Catalysts. J. Phys. Chem. B 2005, 109, 14581–14587. [Google Scholar] [CrossRef] [PubMed]
- Kotobuki, M.; Leppelt, R.; Hansgen, D.; Widmann, D.; Behm, R.J. Reactive oxygen on a Au/TiO2 supported catalyst. J. Catal. 2009, 264, 67–76. [Google Scholar] [CrossRef]
- Vilhelmsen, L.B.; Hammer, B. Indentification of the Catalytic Site at the Interface Perimeter of Au clusters on Rutile TiO2(110). ACS Catal. 2014, 4, 1626–1631. [Google Scholar] [CrossRef]
- Widmann, D.; Behm, R.J. Active oxygen on a Au/TiO2 catalyst: Formation, stability and CO oxidation activity. Angew. Chem. Int. Ed. 2011, 50, 10241–10245. [Google Scholar] [CrossRef] [PubMed]
- Maeda, Y.; Iizuka, Y.; Kohyama, M. Generation of Oxygen Vacancies at a Au/TiO2 Perimeter Interface during CO Oxidation Detected by in Situ Electrical Conductance Measurement. J. Am. Chem. Soc. 2013, 135, 906–909. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, A.; Qiao, B.; Lin, J.; Huang, Y.; Wang, X.; Zhang, T. Origin of the high activity of Au/FeOx for low-temperature CO oxidation: Direct evidence for a redox mechanism. J. Catal. 2013, 299, 90–100. [Google Scholar] [CrossRef]
- Kim, H.Y.; Henkelman, G. CO Oxidation at the Interface of Au Nanoclusters and the Stepped-CeO2(111) Surface by the Mars-van Krevelen Mechanism. J. Phys. Chem. Lett. 2013, 4, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Widmann, D.; Behm, R.J. Activation of Molecular Oxygen and the Nature of the Active Oxygen Species for CO Oxidation on Oxide Supported Au Catalysts. Acc. Chem. Res. 2014, 47, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.; Henkelman, G. CO Oxidation at the Au/TiO2 Boundary: The Role of the Au/Ti5c Site. ACS Catal. 2015, 5, 1589–1595. [Google Scholar] [CrossRef]
- Saqlain, M.A.; Hussain, A.; Siddiq, M.; Ferreira, A.R.; Leitao, A.A. Thermally activated surface oxygen defects at the perimeter of Au/TiO2: A DFT + U study. Phys. Chem. Chem. Phys. 2015, 17, 25403–25410. [Google Scholar] [CrossRef] [PubMed]
- Green, I.X.; Tang, W.; Neurock, M.; Yates, J.T. Insights into Catalytic Oxidation at the Au/TiO2 Dual Perimeter Sites. Acc. Chem. Res. 2013, 47, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Widmann, D.; Hocking, E.; Behm, R.J. On the origin of the selectivity in the preferential CO oxidation on Au/TiO2—Nature of the active oxygen species for H2 oxidation. J. Catal. 2014, 317, 272–276. [Google Scholar] [CrossRef]
- Sun, K.; Kohyama, M.; Tanaka, S.; Takeda, S. A Study on the Mechanism for H2 Dissociation on Au/TiO2 Catalysts. J. Phys. Chem. C 2014, 118, 1611–1617. [Google Scholar] [CrossRef]
- Daté, M.; Haruta, M. Moisture effect on CO oxidation over Au/TiO2 catalyst. J. Catal. 2001, 201, 221–224. [Google Scholar] [CrossRef]
- Diemant, T.; Bansmann, J.; Behm, R.J. CO oxidation on planar Au/TiO2 model catalysts: Deactivation and the influence of water. Vacuum 2009, 84, 193–196. [Google Scholar] [CrossRef]
- Gao, F.; Wood, T.E.; Goodman, D.W. The Effects of Water on CO Oxidation over TiO2 Supported Au Catalysts. Catal. Lett. 2010, 134, 9–12. [Google Scholar] [CrossRef]
- Saavedra, J.; Doan, H.A.; Pursell, C.J.; Grabow, L.C.; Chandler, B.D. The critical role of water at the gold-titania interface in catalytic CO oxidation. Science 2014, 345, 1599–1602. [Google Scholar] [CrossRef] [PubMed]
- Widmann, D.; Liu, Y.; Schüth, F.; Behm, R.J. Support effects in the Au catalyzed CO oxidation—Correlation between activity, oxygen storage capacity and support reducibility. J. Catal. 2010, 276, 292–305. [Google Scholar] [CrossRef]
- Wang, L.-C.; Widmann, D.; Behm, R.J. Reactive removal of surface oxygen by H2, CO and CO/H2 on a Au/CeO2 catalyst and its relevance to the preferential CO oxidation (PROX) and reverse water gas shift (RWGS) reaction. Catal. Sci. Technol. 2015, 5, 925–941. [Google Scholar] [CrossRef]
- Schubert, M.M.; Plzak, V.; Garche, J.; Behm, R.J. Activity, Selectivity, and Long-Term Stability of Different Metal Oxide Supported Gold Catalysts for the Preferential CO Oxidation in H2-rich Gas. Catal. Lett. 2001, 76, 143. [Google Scholar] [CrossRef]
- Arrii, S.; Morfin, F.; Renouprez, A.J.; Rousset, J.L. Oxidation of CO on Gold Supported Catalysts Prepared by Laser Vaporization: Direct Evidence of Support Contribution. J. Am. Chem. Soc. 2004, 126, 1199–1205. [Google Scholar] [CrossRef] [PubMed]
- Lakshmanan, P.; Park, J.E.; Park, E.D. Recent Advances in Preferential Oxidation of CO in H2 Over Gold Catalysts. Catal. Surv. Asia 2014, 18, 75–88. [Google Scholar] [CrossRef]
- Leppelt, R.; Hansgen, D.; Widmann, D.; Häring, T.; Bräth, G.; Behm, R.J. Design and characterization of a temporal analysis of products reactor. Rev. Sci. Instrum. 2007, 78, 104103. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hartadi, Y.; Behm, R.J.; Widmann, D. Competition of CO and H2 for Active Oxygen Species during the Preferential CO Oxidation (PROX) on Au/TiO2 Catalysts. Catalysts 2016, 6, 21. https://doi.org/10.3390/catal6020021
Hartadi Y, Behm RJ, Widmann D. Competition of CO and H2 for Active Oxygen Species during the Preferential CO Oxidation (PROX) on Au/TiO2 Catalysts. Catalysts. 2016; 6(2):21. https://doi.org/10.3390/catal6020021
Chicago/Turabian StyleHartadi, Yeusy, R. Jürgen Behm, and Daniel Widmann. 2016. "Competition of CO and H2 for Active Oxygen Species during the Preferential CO Oxidation (PROX) on Au/TiO2 Catalysts" Catalysts 6, no. 2: 21. https://doi.org/10.3390/catal6020021
APA StyleHartadi, Y., Behm, R. J., & Widmann, D. (2016). Competition of CO and H2 for Active Oxygen Species during the Preferential CO Oxidation (PROX) on Au/TiO2 Catalysts. Catalysts, 6(2), 21. https://doi.org/10.3390/catal6020021