
Methanol Reforming over Cobalt Catalysts Prepared from Fumarate Precursors: TPD Investigation



Abstract
:
1. Introduction
2. Results and Discussion
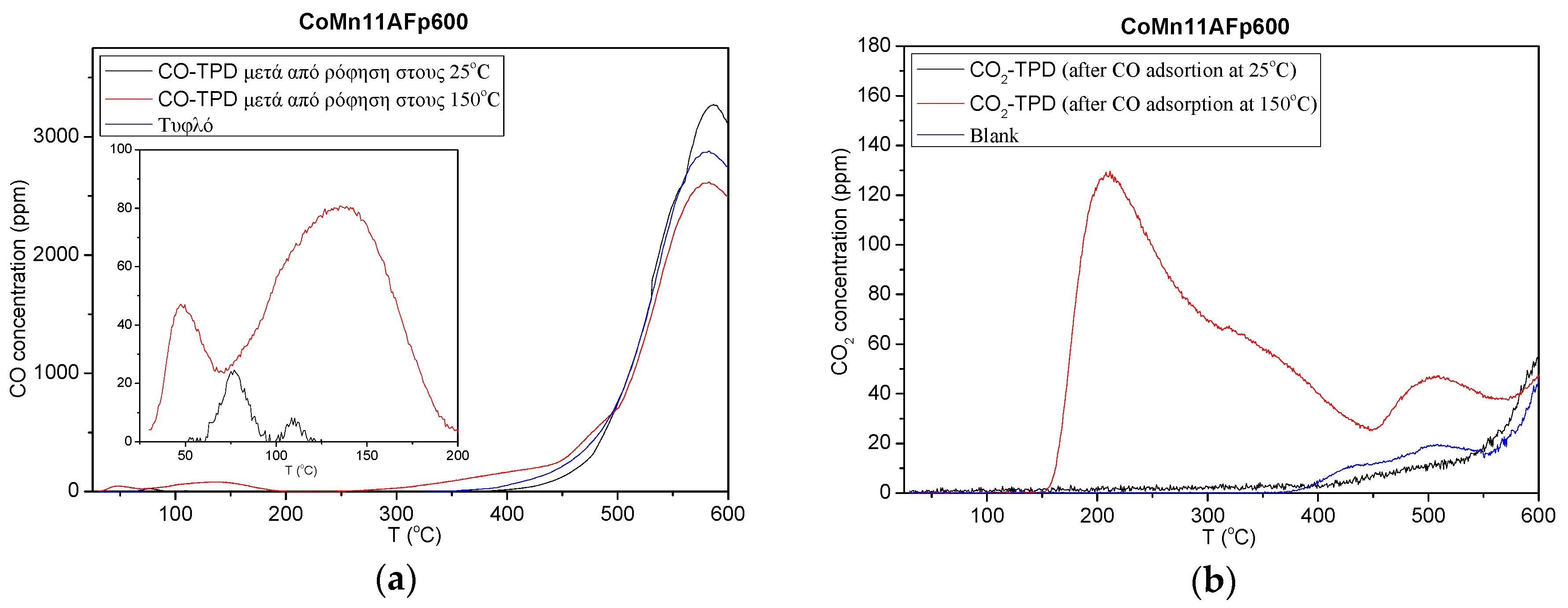
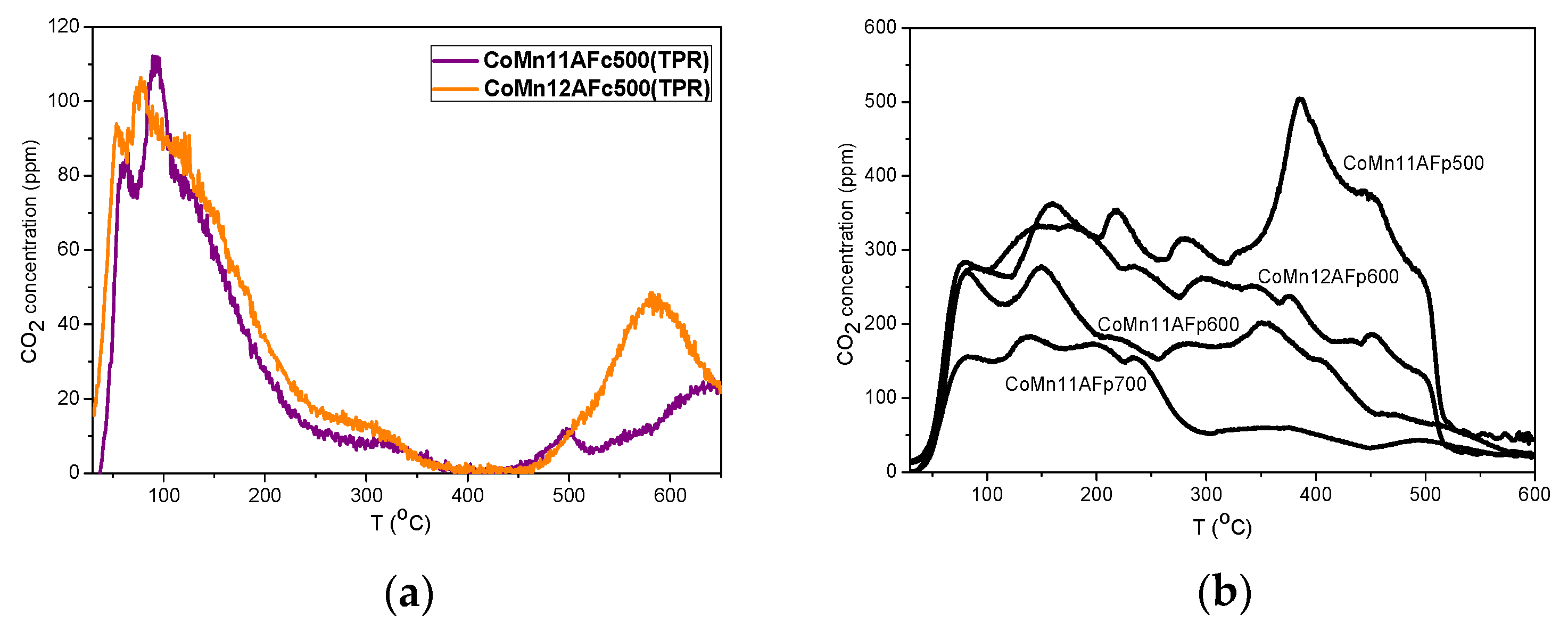
2.1. TPD of Adsorbed CO or CO2
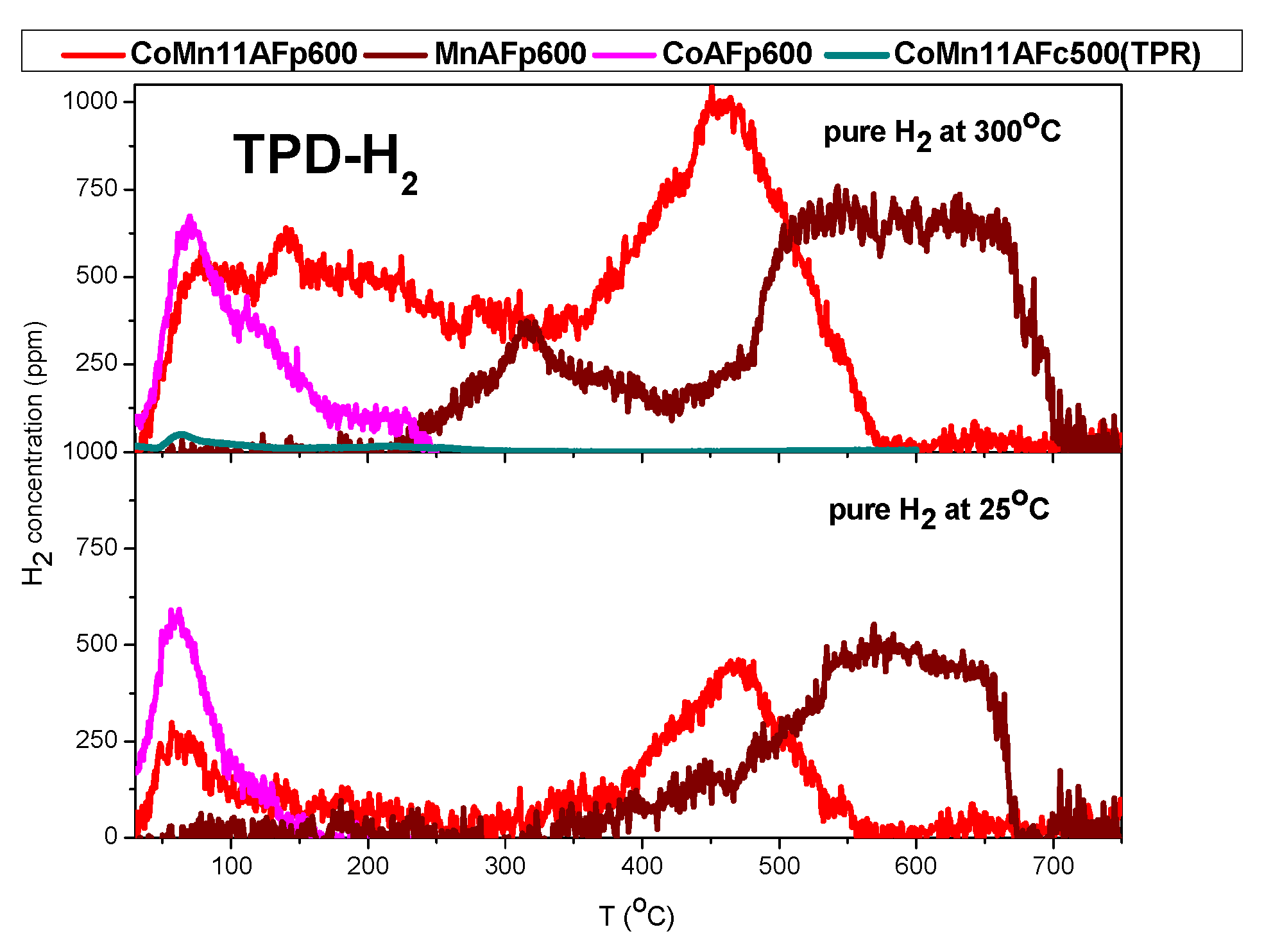
2.2. TPD of Adsorbed H2
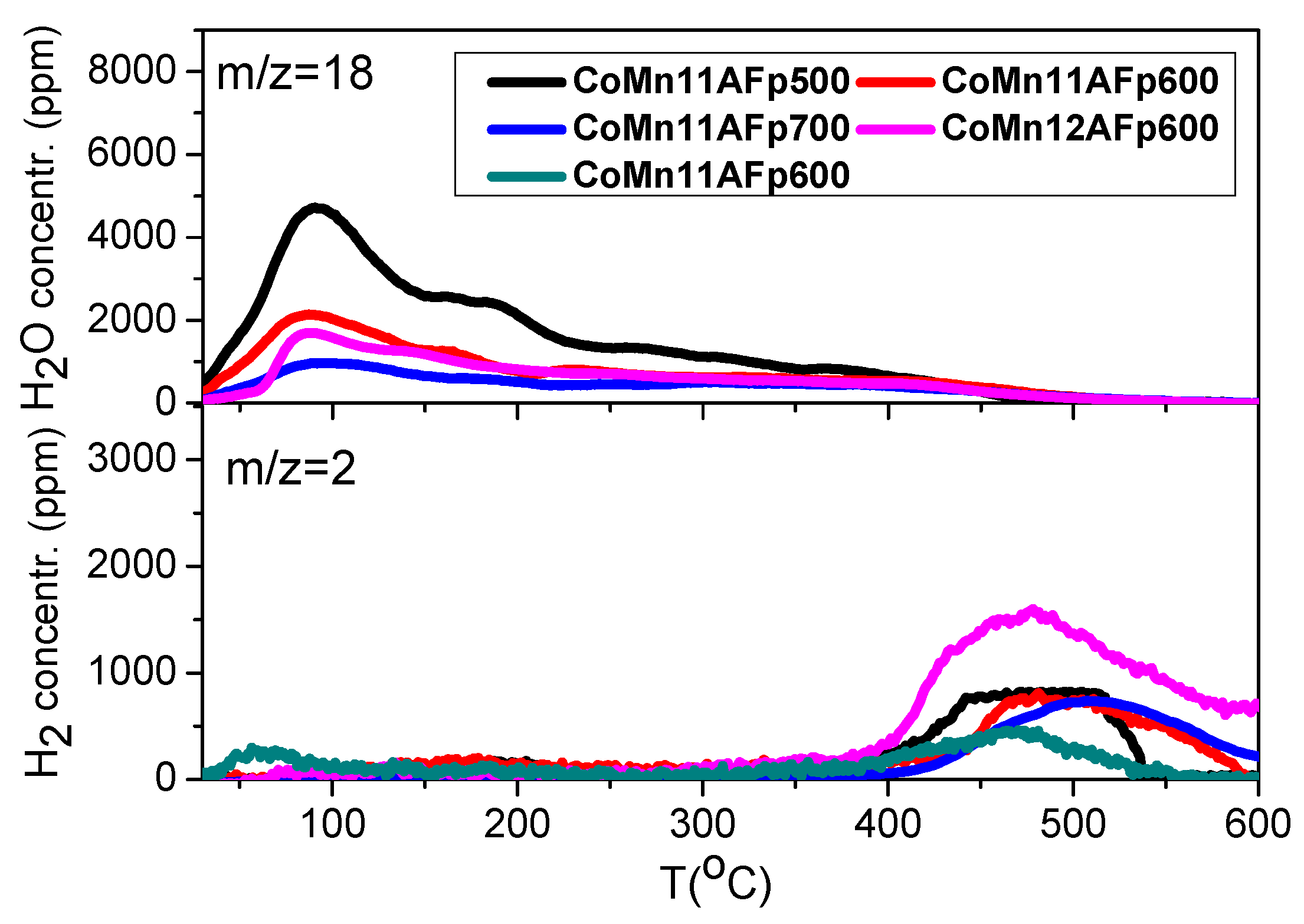
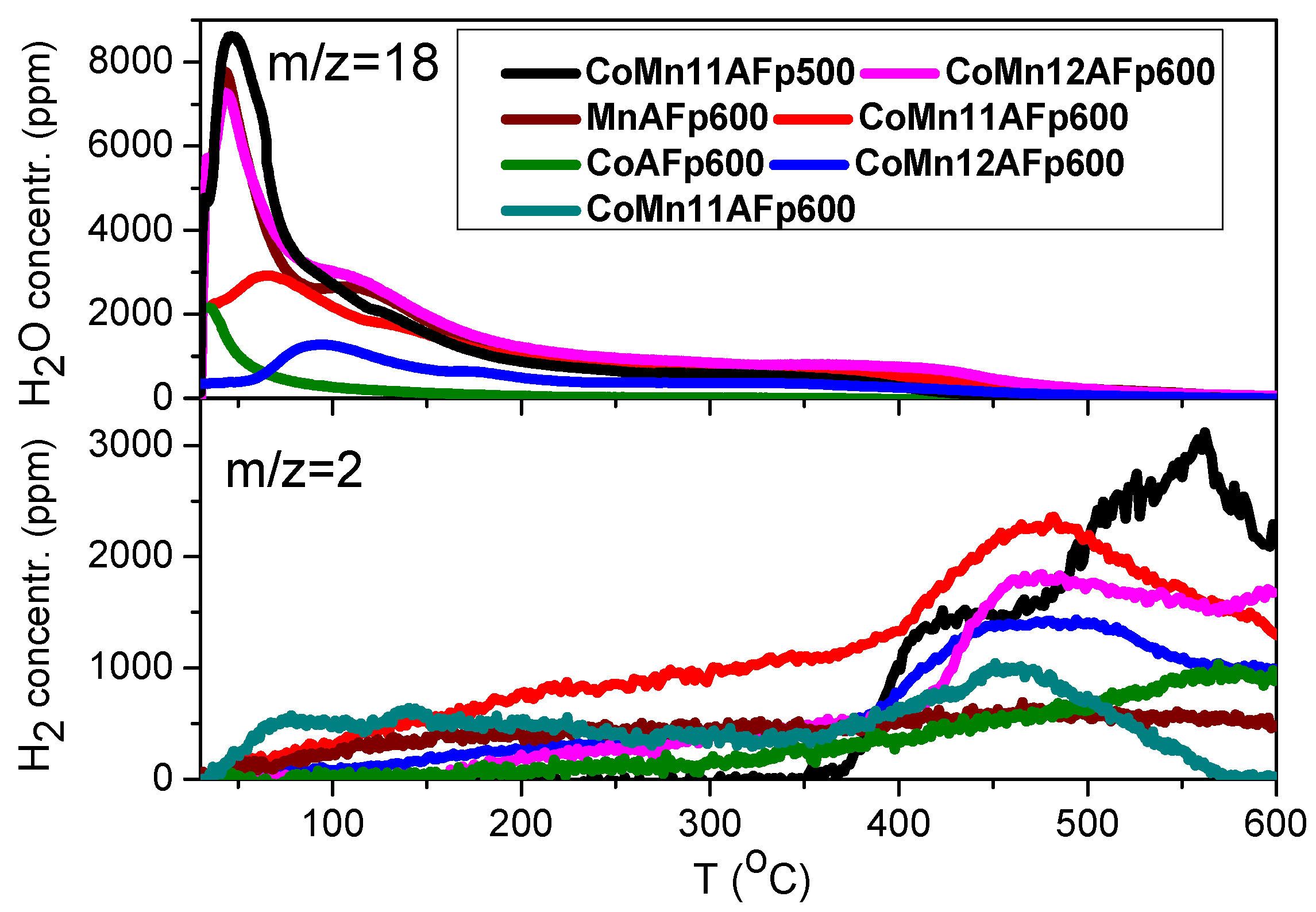
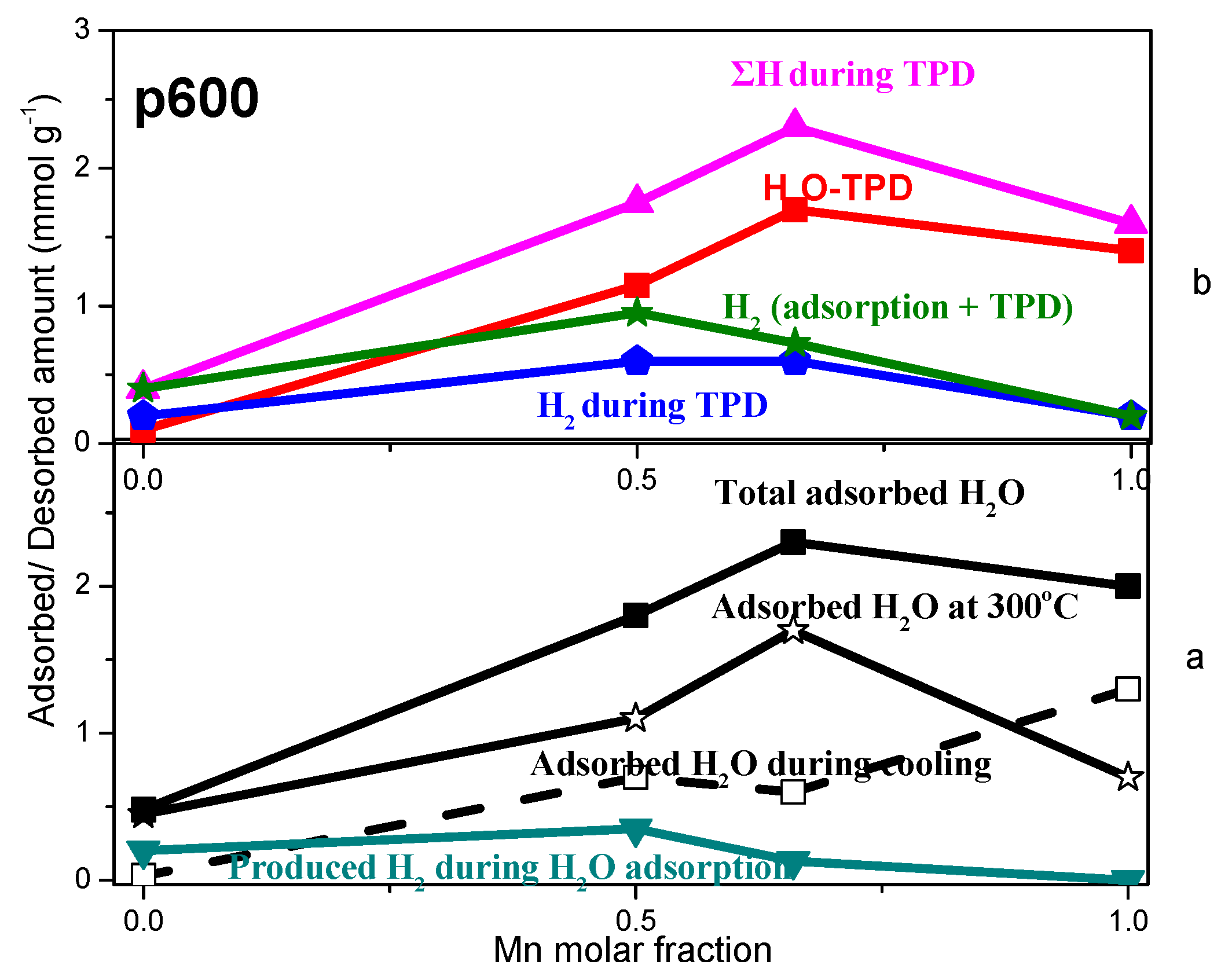
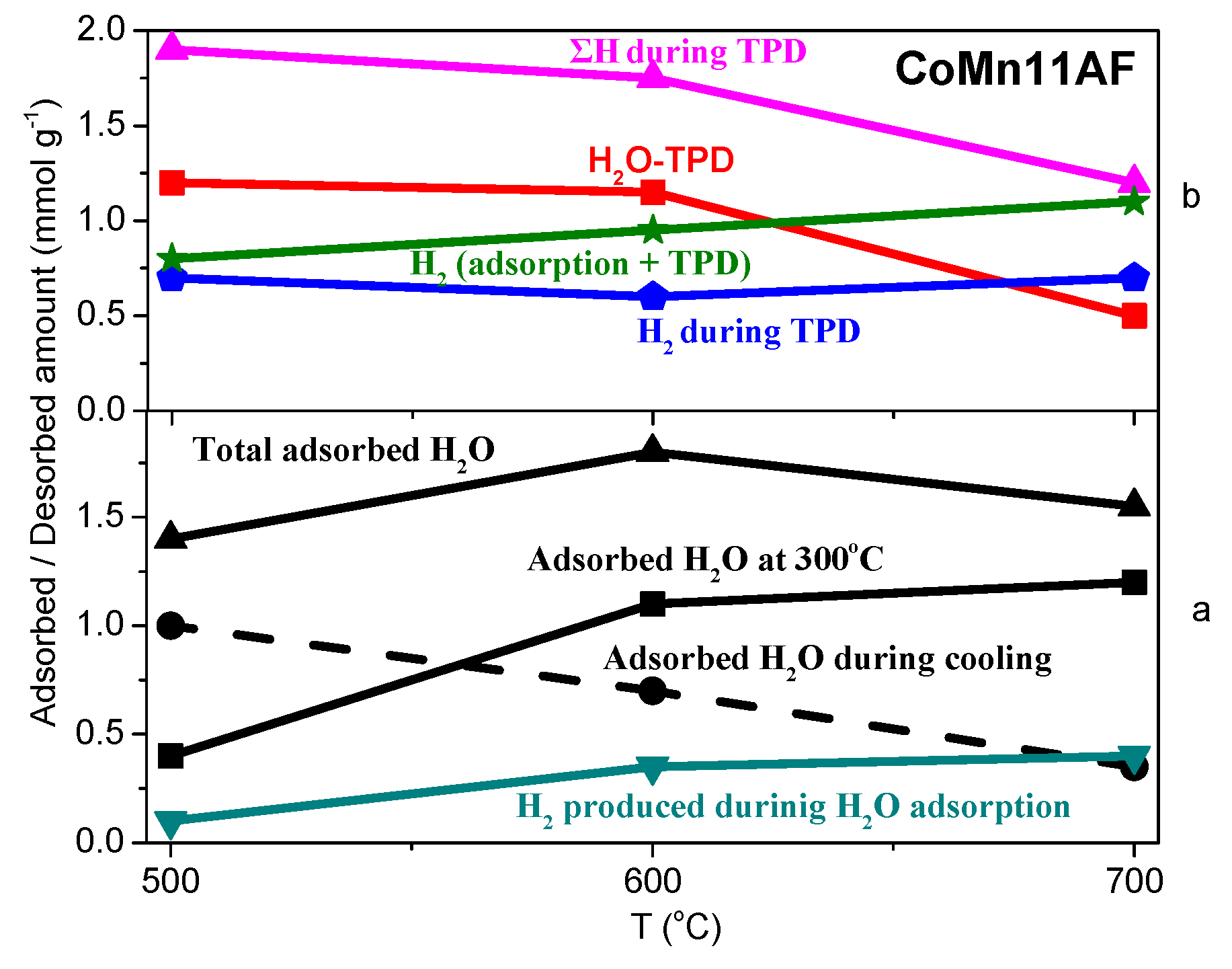
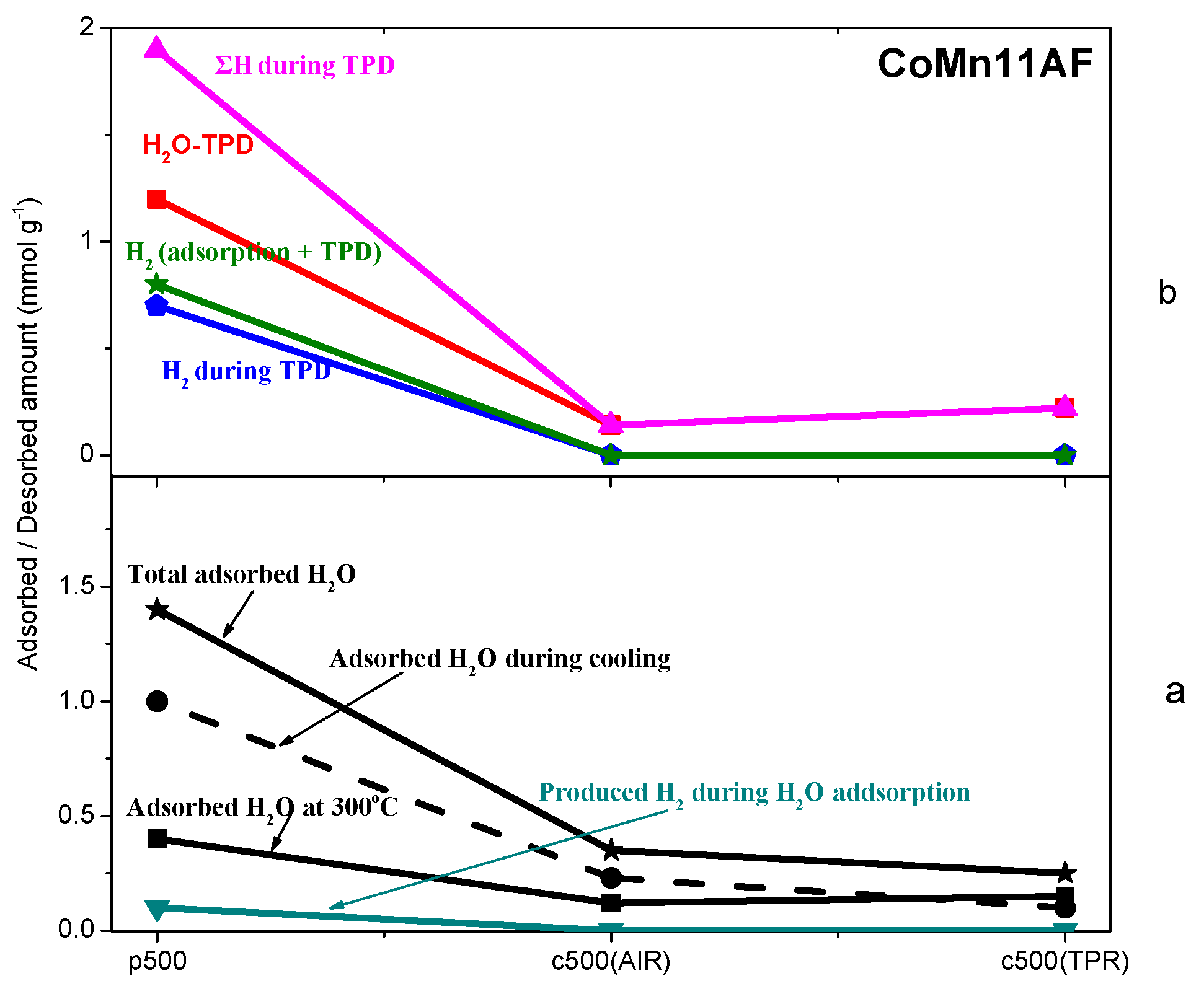
2.3. TPD of Adsorbed H2O
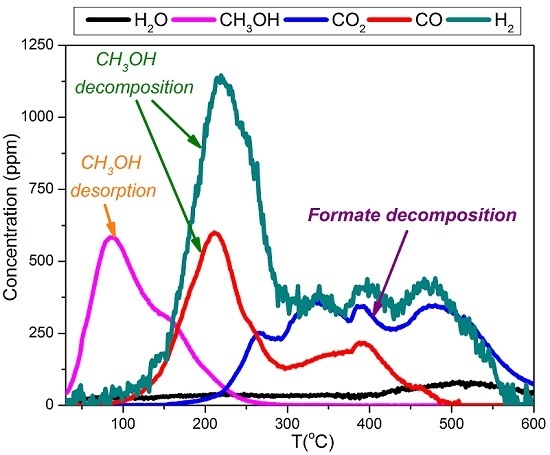
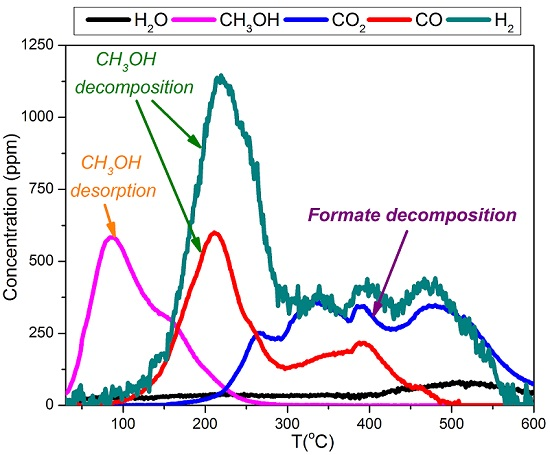
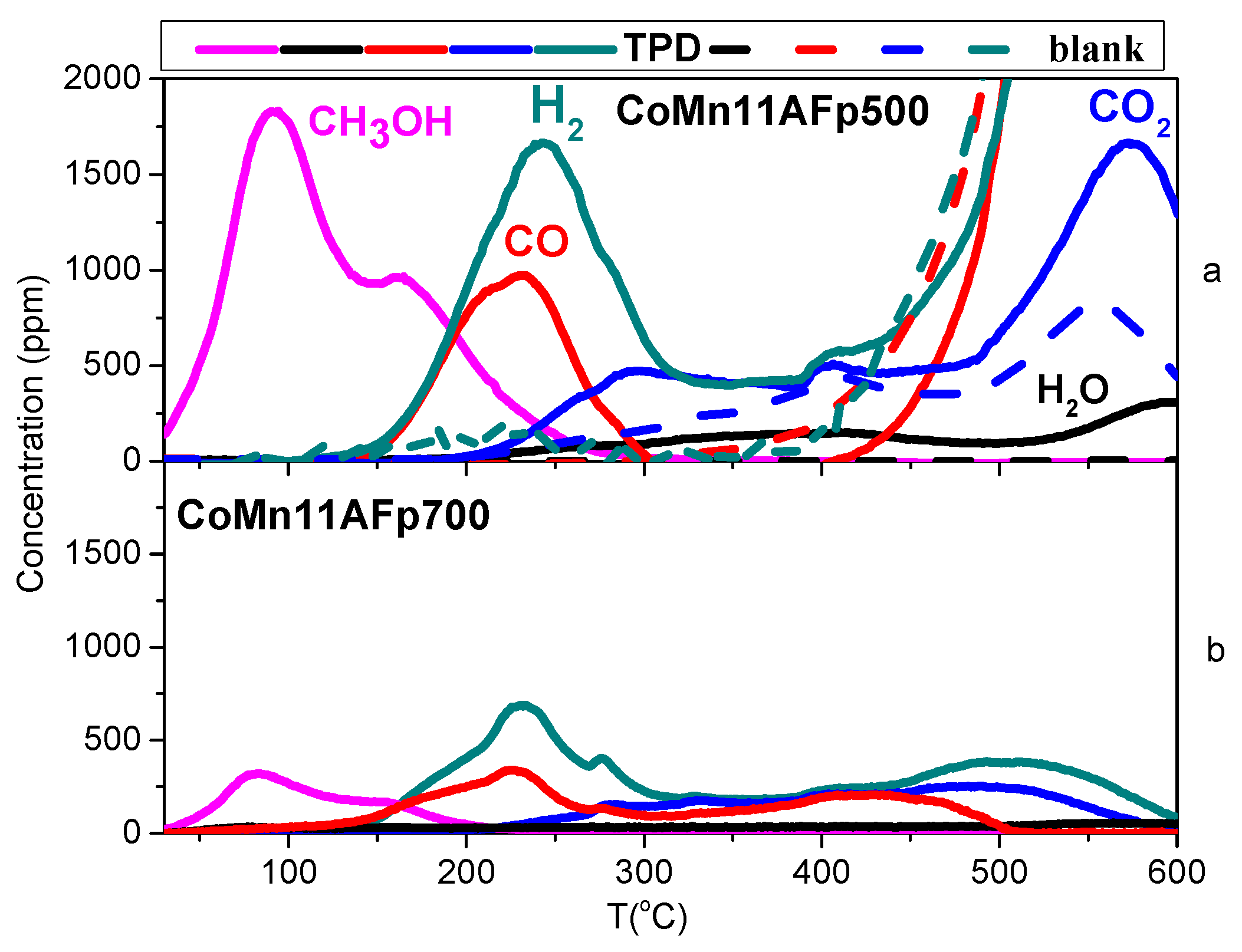
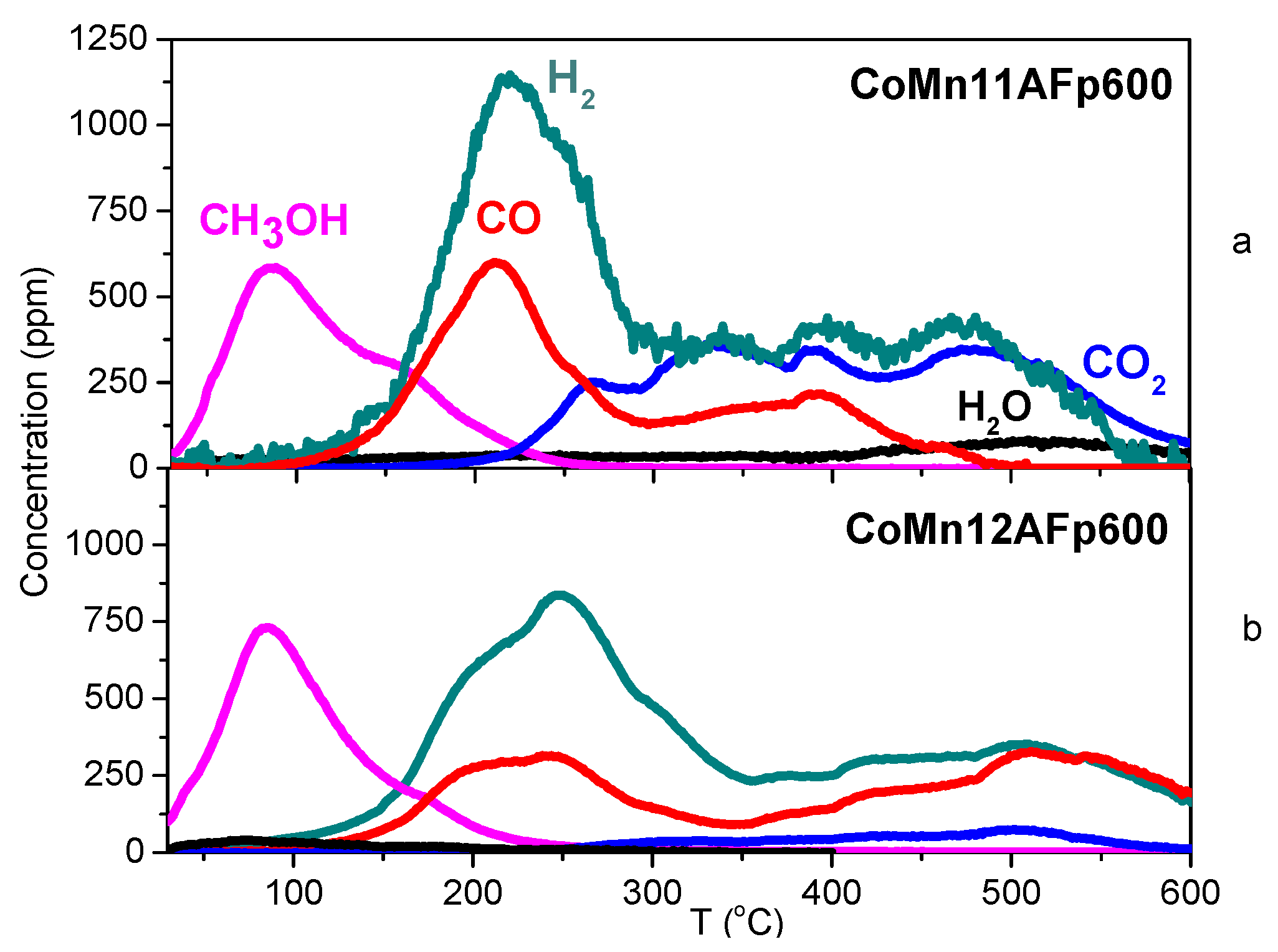
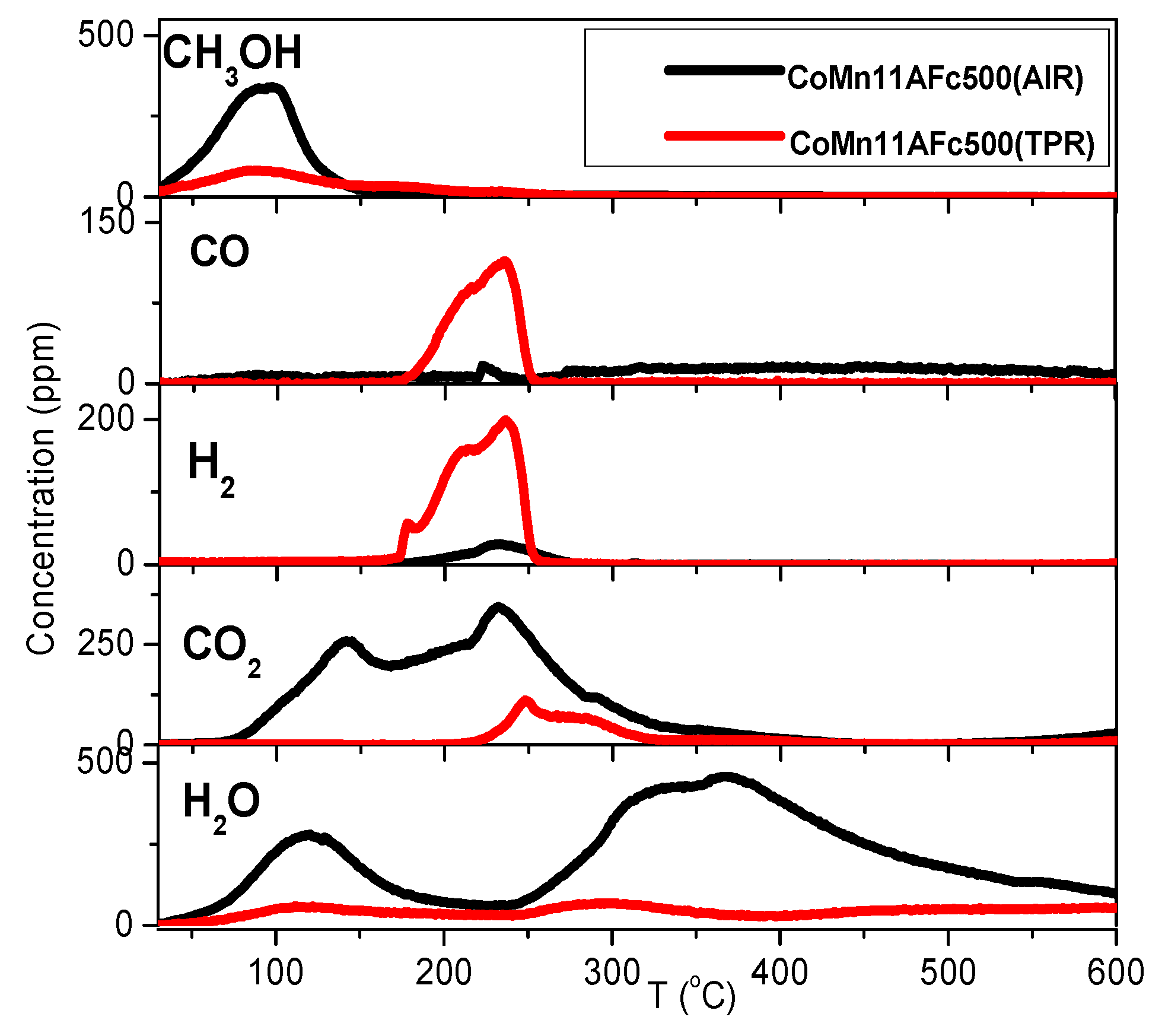
2.4. TPD of Adsorbed CH3OH
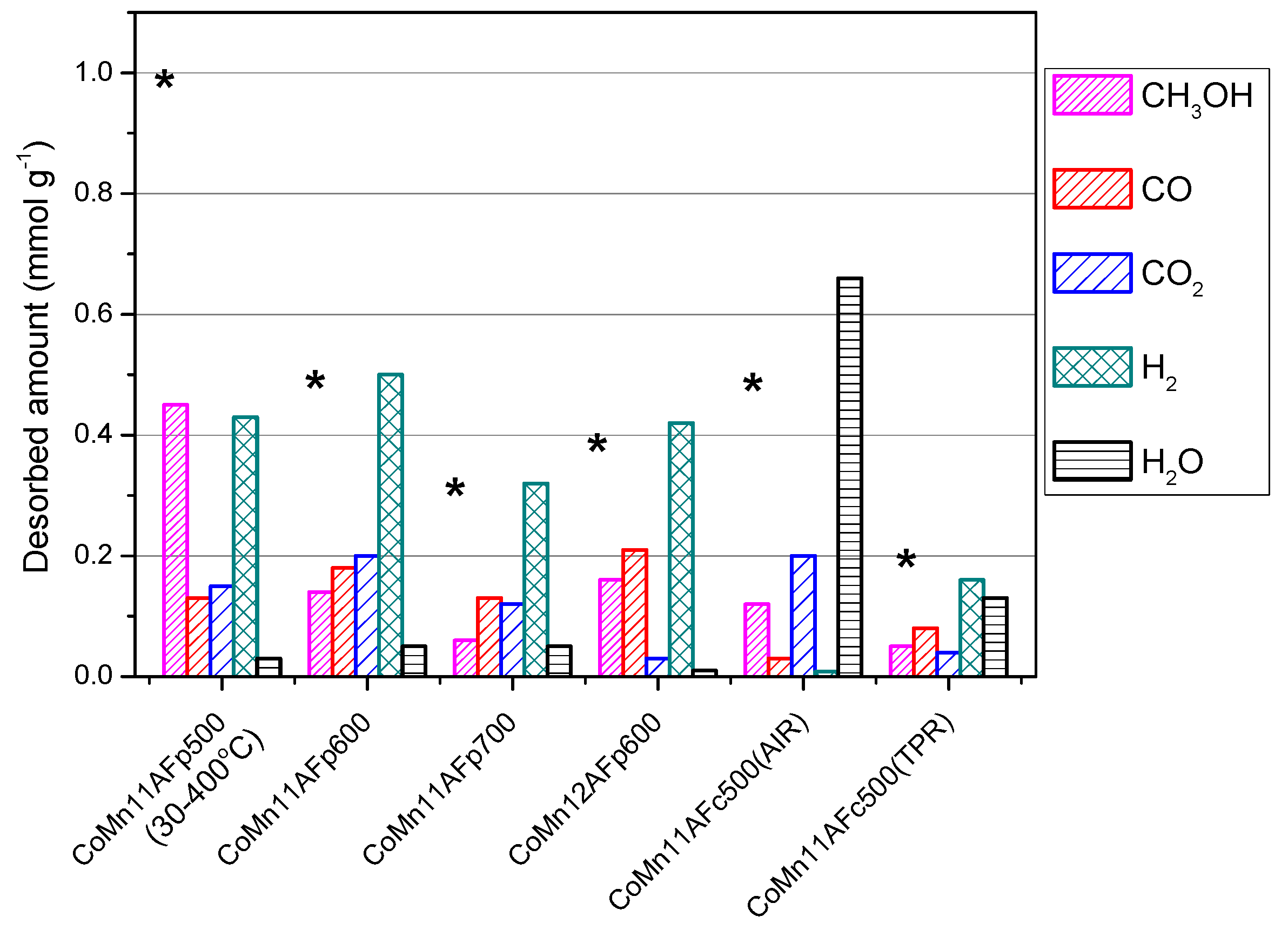
- With the exception of catalyst CoMn11AFp500, which adsorbs 1 mmol·g−1, all catalysts adsorb methanol in the range of 0.2–0.5 mmol·g−1. The lowest quantity is found over the oxidized/reduced sample.
- Less than half of adsorbed methanol desorbs molecularly.
- Increase of pyrolysis temperature and decrease of cobalt content lead to decrease of adsorbed methanol.
- The amounts of CO and CO2 produced during methanol TPD are 2–4 times higher over the pyrolyzed catalysts compared to those prepared via oxidation or oxidation/reduction.
- The oxygen mass balance between output and input shows a surplus indicating that adsorbed methanol acts as a reducing agent scavenging lattice oxygen from the catalysts. The amount of adsorbed methanol that gets oxidized towards CO2 and H2O depends on the oxidation state of the catalyst surface.
- The carbon mass balance is overall satisfied (error <10%) with the exception of the CoMn11AFp500 catalyst, because its reported values correspond to temperatures below 400 °C (at higher temperatures interference from residual pyrolysis does not allow for reliable measurement).
2.5. Discussion
3. Experimental Section
3.1. Catalyst Preparation
3.2. Catalyst Characterization
3.3. TPD Experiments
4. Conclusions
- Adsorption of CO and H2 is activated. Although activated hydrogen adsorption on cobalt is well established, activated adsorption of CO has not been reported previously.
- Hydrogen appears to adsorb both on cobalt and MnO components. Taking into account literature results concerning cobalt and MnO and results of the present work concerning cobalt, MnO and Co-MnO samples, it is inferred that hydrogen desorbing below 250 °C originates from cobalt crystallites, hydrogen desorbing above 500 °C originates from MnO, while hydrogen desorbing in the intermediate temperature range (250–500 °C) probably originates from sites created at the interface of Co and MnO or from a mixed reduced oxide phase.
- Water adsorption is dissociative at an adsorption temperature of 300 °C, but not at 25 °C, leading to surface oxidation of the catalyst. Hydrogen produced from water dissociation remains partially adsorbed on the catalyst surface confirming that part of hydrogen is quite strongly bound on the catalysts.
- Reaction paths of adsorbed methanol during TPD include decomposition to CO and H2, as well as creation of rather stable surface formates, which decompose at higher temperatures to CO2 and H2. Adsorbed methanol acts as a reducing agent during TPD leading to catalyst reduction.
- Differences of the pyrolyzed and oxidized/reduced catalysts appear to be mainly in the number density of active sites, which, however, is not directly analogous to differences in specific surface area.
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Papadopoulou, E.; Ioannides, T. Steam reforming of methanol over cobalt catalysts: effect of cobalt oxidation state. Int. J. Hydrogen Energy 2015, 40, 5251–5255. [Google Scholar] [CrossRef]
- Papadopoulou, E.; Delimaris, D.; Denis, A.; Machocki, A.; Ioannides, T. Alcohol reforming on cobalt-based catalysts prepared from organic salt precursors. Int. J. Hydrogen Energy 2012, 37, 16375–16381. [Google Scholar] [CrossRef]
- Yong, S.T.; Ooi, C.W.; Chai, S.P.; Wu, X.S. Review of methanol reforming-Cu-based catalysts, surface reaction mechanisms, and reaction schemes. Int. J. Hydrogen Energy 2013, 38, 9541–9552. [Google Scholar] [CrossRef]
- Sá, S.; Silva, H.; Brandão, L.; Sousa, J.M.; Mendes, A. Catalysts for methanol steam reforming-A review. Appl. Catal. B 2010, 99, 43–57. [Google Scholar] [CrossRef]
- Palo, D.R.; Dagle, R.A.; Holladay, J.D. Methanol Steam Reforming for Hydrogen Production. Chem. Rev. 2007, 107, 3992–4021. [Google Scholar] [CrossRef] [PubMed]
- Agrell, J.; Birgersson, H.; Boutonnet, M.; Melián-Cabrera, I.; Navarro, R.M.; Fierro, J.L.G. Production of hydrogen from methanol over Cu/ZnO catalysts promoted by ZrO2 and Al2O3. J. Catal. 2003, 219, 389–403. [Google Scholar] [CrossRef]
- De Wild, P.J.; Verhaak, M.J.F.M. Catalytic production of hydrogen from methanol. Catal. Today 2000, 60, 3–10. [Google Scholar] [CrossRef]
- Matter, P.H.; Braden, D.J.; Ozkan, U.S. Steam reforming of methanol to H2 over nonreduced Zr-containing CuO/ZnO catalysts. J. Catal. 2004, 223, 340–351. [Google Scholar] [CrossRef]
- Tartakovsky, L.; Baibikov, V.; Veinblat, M. Comparative Performance Analysis of SI Engine Fed by Ethanol and Methanol Reforming Products. SAE Tech. Pap. 2013. [Google Scholar] [CrossRef]
- Habermehl-Cwirzen, K. An Insight: Studies of Atomic and Molecular Adsorption on Co(0001); Helsinki University of Technology: Helsinki, Finland, 2006. [Google Scholar]
- Blyholder, G.; Wyatt, W.V. Infrared spectra and structures of some C,H,O compounds adsorbed on silica-supported iron, cobalt, and nickel. J. Phys. Chem. 1966, 70, 1745–1750. [Google Scholar] [CrossRef]
- Natile, M.M.; Glisenti, A. Study of surface reactivity of cobalt oxides: interaction with methanol. Chem. Mater. 2002, 14, 3090–3099. [Google Scholar] [CrossRef]
- Grellner, F.; Klingenberg, B.; Borgmann, D.; Wedler, G. Electron spectroscopic study of the interaction of oxygen with Co(1120) and of coadsorption with water. J. Electron Spectrosc. 1995, 71, 107–115. [Google Scholar] [CrossRef]
- Heras, J.M.; Papp, H.; Spiess, W. Face specificity of the H2O adsorption and decomposition on Co surfaces: A LEED, UPS, sp and TPD study. Surf. Sci. 1982, 117, 590–604. [Google Scholar] [CrossRef]
- Reuel, R.C.; Bartholomew, C.H. The stoichiometries of H2 and CO adsorptions on cobalt: Effects of support and preparation. J. Catal. 1984, 85, 63–77. [Google Scholar] [CrossRef]
- Bartholomew, C.H.; Reuel, R.C. Cobalt-support interactions: Their effects on adsorption and CO hydrogenation activity and selectivity properties. Ind. Eng. Chem. Prod. Res. Dev. 1985, 24, 56–61. [Google Scholar] [CrossRef]
- Zowtiak, J.M.; Bartholomew, C.H. The kinetics of H2 adsorption on and desorption from cobalt and the effects of support thereon. J. Catal. 1983, 83, 107–120. [Google Scholar] [CrossRef]
- Tejuca, L.G.; Bell, A.T.; Fierro, J.L.G.; Pena, M.A. Surface behavior of reduced LaCoO3 as studied by TPD of CO, CO2 and H2 probes and by XPS. Appl. Surf. Sci. 1988, 31, 301–316. [Google Scholar] [CrossRef]
- Narayanan, S.; Unnikrishnan, R.P. Comparison of hydrogen adsorption and aniline hydrogenation over co-precipitated Co/Al2O3 and Ni/Al2O3 catalysts. J. Chem. Soc. Faraday Trans. 1997, 93, 2009–2013. [Google Scholar] [CrossRef]
- Sakka, Y.; Ohno, S. Hydrogen desorption characteristics of composite Co-TiN nanoparticles. Appl. Surf. Sci. 1996, 100/101, 232–237. [Google Scholar] [CrossRef]
- Soonq, Y.; Rao, V.U.S.; Zarochak, M.F.; Gormley, R.J.; Zhang, B. Temperature-programmed desorption study on manganese-iron catalysts. Appl. Catal. 1991, 78, 97–108. [Google Scholar] [CrossRef]
- Gauthier, Y.; Schmidt, M.; Padovani, S.; Lundgren, E.; Bus, V.; Kresse, G.; Redinger, J.; Vagra, P. Adsorption sites and ligand effect for CO on an alloy surface: A direct view. Phys. Rev. Lett. 2001, 87, 036103. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Koizumi, N.; Ozaki, T.; Yamada, M. Adsorption properties of cobalt and cobalt-manganese catalysts studied by in situ diffuse reflectance FTIR using CO and CO+H2 as probes. Appl. Catal. A 2001, 209, 59–70. [Google Scholar] [CrossRef]
- Mohana Rao, K.; Scarano, D.; Spoto, G.; Zecchina, A. CO adsorption on cobalt particles supported on MgO: An IR investigation. Surf. Sci. 1988, 204, 319–330. [Google Scholar] [CrossRef]
- Rostrup-Nielsen, J.R. Catalytic Steam Reforming. Catalysis 1984, 5, 1–117. [Google Scholar]
- Baker, R.T.K.; Kim, M.S.; Chambers, A.; Park, C.; Rodriguez, N.M. The relationship between metal particle morphology and the structural characteristics of carbon deposits. Stud. Surf. Sci. Catal. 1997, 111, 99–109. [Google Scholar]
- Ōya, A.; Ōtani, S. Catalytic graphitization of carbons by various metals. Carbon 1979, 17, 131–137. [Google Scholar] [CrossRef]
- Budiman, A.W.; Song, S.A.; Chang, T.S.; Shin, C.H.; Choi, M.J. Dry Reforming of Methane Over Cobalt Catalysts: A Literature Review of Catalyst Development. Catal. Surv. Asia 2012, 16, 183–197. [Google Scholar] [CrossRef]
- Tsakoumis, M.; Ronning, M.; Qyvind, B. Deactivation of cobalt based Fischer-Tropsch catalysts: A review. Catal. Today 2010, 154, 162–182. [Google Scholar] [CrossRef]
- Bartholomew, C.H. Mechanisms of catalyst deactivation. Appl. Catal. A 2001, 21, 17–60. [Google Scholar] [CrossRef]
- Bartholomew, C.H. Carbon Deposition in Steam Reforming and Methanation. Catal. Rev. Sci. Eng. 1982, 24, 67–112. [Google Scholar] [CrossRef]
- Nakamura, J.; Tanaka, K.; Toyoshima, I. Reactivity of deposited carbon on Co-Al2O3 catalyst. J. Catal. 1987, 108, 55–62. [Google Scholar] [CrossRef]
- Tagawa, T.; Pleizier, G.; Amenomiya, Y. Methanol synthesis from CO2+H2: Characterization of catalysts by TPD. Appl. Catal. 1985, 18, 285–293. [Google Scholar] [CrossRef]
- Meunier, F.C.; Reid, D.; Goguet, A.; Shekhtman, S.; Hardacre, C.; Burch, R.; Deng, W.; Flytzani-Stephanopoulos, M. Quantitative analysis of the reactivity of formate species seen by DRIFTS over a Au/Ce(La)O2 water-gas shift catalyst: First unambiguous evidence of the minority role of formates as reaction intermediates. J. Catal. 2007, 247, 269–279. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | H2-TPD Adsorption at 25 °C | H2-TPD Adsorption at 300 °C | ||
|---|---|---|---|---|
| μmol H2 g−1 | H/Co | μmol H2 g−1 | H/Co | |
| CoMn11AFp500 | 174 | 0.058 | 492 | 0.164 |
| CoMn11AFp600 | 189 | 0.060 | 661 | 0.210 |
| CoMn11AFp700 | 328 | 0.104 | 371 | 0.118 |
| CoMn12AFp600 | 195 | 0.096 | 478 | 0.236 |
| CoMn11AFc500(TPR) | 0 | 0 | 10 | 0.0034 |
| CoAFp600 | 34 | 0.00116 | 61 | 0.0212 |
| MnAFp600 | 100 | 0.034 * | 195 | 0.066 * |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papadopoulou, E.; Ioannides, T. Methanol Reforming over Cobalt Catalysts Prepared from Fumarate Precursors: TPD Investigation. Catalysts 2016, 6, 33. https://doi.org/10.3390/catal6030033
Papadopoulou E, Ioannides T. Methanol Reforming over Cobalt Catalysts Prepared from Fumarate Precursors: TPD Investigation. Catalysts. 2016; 6(3):33. https://doi.org/10.3390/catal6030033
Chicago/Turabian StylePapadopoulou, Eftichia, and Theophilos Ioannides. 2016. "Methanol Reforming over Cobalt Catalysts Prepared from Fumarate Precursors: TPD Investigation" Catalysts 6, no. 3: 33. https://doi.org/10.3390/catal6030033
APA StylePapadopoulou, E., & Ioannides, T. (2016). Methanol Reforming over Cobalt Catalysts Prepared from Fumarate Precursors: TPD Investigation. Catalysts, 6(3), 33. https://doi.org/10.3390/catal6030033