
An Overview of Recent Advances of the Catalytic Selective Oxidation of Ethane to Oxygenates



Abstract
:
1. Introduction
2. Current Industrial Approaches to Ethane Upgrading
3. Partial Oxidation of Ethane
3.1. High Temperature Approaches
3.2. Low Temperature Approaches
3.2.1. Homogeneous Catalytic Approaches
3.2.2. Enzymatic Approaches
3.2.3. Biomimetic Approaches
3.2.4. Heterogeneous Approaches
3.2.5. Summary of Catalyst Performance
5. Conclusions and Outlook
Conflicts of Interest
References
- Degirmenci, V.; Uner, D.; Yilmaz, A. Methane to higher hydrocarbons via halogenation. Catal. Today 2005, 106, 252–255. [Google Scholar] [CrossRef]
- What Is Natural Gas? Available online: http://www.naturalgas.org/overview/background.asp (accessed on 20 November 2013).
- Jones, J.H. The cativa™ process for the manufacture of acetic acid. Platin. Met. Rev. 2000, 44, 94–105. [Google Scholar]
- Olah, G.A. Beyond oil and gas: The methanol economy. Angew. Chem. Int. Ed. 2005, 44, 2636–2639. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, N.; Karimzadeh, R. Catalytic cracking of hydrocarbons over modified zsm-5 zeolites to produce light olefins: A review. Appl. Catal. A Gen. 2011, 398, 1–17. [Google Scholar] [CrossRef]
- Kosaric, N.; Duvnjak, Z.; Farkas, A.; Sahm, H.; Bringer-Meyer, S.; Goebel, O.; Mayer, D. Ethanol, Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Hoboken, NJ, USA, 2000. [Google Scholar]
- Forde, M.M.; Armstrong, R.D.; Hammond, C.; He, Q.; Jenkins, R.L.; Kondrat, S.A.; Dimitratos, N.; Lopez-Sanchez, J.A.; Taylor, S.H.; Willock, D.; et al. Partial Oxidation of Ethane to Oxygenates Using Fe- and Cu-Containing ZSM-5. J. Am. Chem. Soc. 2013, 135, 11087–11099. [Google Scholar] [CrossRef] [PubMed]
- Pacala, S.; Socolow, R. Stabilization wedges: Solving the climate problem for the next 50 years with current technologies. Science 2004, 305, 968–972. [Google Scholar] [CrossRef] [PubMed]
- Blanksby, S.J.; Ellison, G.B. Bond dissociation energies of organic molecules. Acc. Chem. Res. 2003, 36, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Cossee, P. Ziegler-natta catalysis i. Mechanism of polymerization of α-olefins with ziegler-natta catalysts. J. Catal. 1964, 3, 80–88. [Google Scholar] [CrossRef]
- Fan, D.; Dai, D.J.; Wu, H.S. Ethylene formation by catalytic dehydration of ethanol with industrial considerations. Materials 2013, 6, 101–115. [Google Scholar] [CrossRef]
- Östman, M. Information on Substances: Acetic Acid. Available online: http://apps.kemi.se/flodessok/floden/kemamne_eng/attiksyra_eng.htm (accessed on 15 November 2013).
- Yoneda, N.; Kusano, S.; Yasui, M.; Pujado, P.; Wilcher, S. Recent advances in processes and catalysts for the production of acetic acid. Appl. Catal. A Gen. 2001, 221, 253–265. [Google Scholar] [CrossRef]
- Sunley, G.J.; Watson, D.J. High productivity methanol carbonylation catalysis using iridium: The cativa™ process for the manufacture of acetic acid. Catal. Today 2000, 58, 293–307. [Google Scholar] [CrossRef]
- Schultz, R.G.; Montgome, P.D. Vapour phase carbonylation of methanol to acetic acid. J. Catal. 1969, 13, 105–106. [Google Scholar] [CrossRef]
- Krzywicki, A.; Marczewski, M. Formation and evolution of the active site for methanol carbonylation on oxide catalysts containing rhcl3. J. Mol. Catal. 1979, 6, 431–440. [Google Scholar] [CrossRef]
- Scurrell, M.S.; Howe, R.F. Highly active rhodium-zeolite catalyst for methanol carbonylation. J. Mol. Catal. 1980, 7, 535–537. [Google Scholar]
- Takeshi, M.; Kazuhiko, H.; Kenji, S.; Yoshinmi, S. Process for Preparing Organic Carboxylic Acid; Chiyoda Corporation: Yokohama, Japan, 1995. [Google Scholar]
- Yoneda, N.; Tokyo, Y.; Shiroto, T.; Kazuhiko, H.; Kawasaki, S.; Sachio Asaoka, Y.; Testsuo Maejima, K. Process for the Production of Acetic Acid from Methanol and Carbon Monoxide Using Supported Rhodium Catalyst; Chiyoda Corporation: Yokohama, Japan, 1993. [Google Scholar]
- Takeshi, M.; Kenji, S.; Kazuhiko, H.; Kawasaki, S.; Yoshimi Shiroto, Y.; Yoneda, N. Supported Rhodium Catalyst, Method for Preparing Same and Process of Producing Acetic Acid by Methanol Cabonylation Using Same; Chiyoda Corporation: Yokohama, Japan, 1993. [Google Scholar]
- Boronat, M.; Martínez-Sánchez, C.; Law, D.; Corma, A. Enzyme-like specificity in zeolites: A unique site position in mordenite for selective carbonylation of methanol and dimethyl ether with co. J. Am. Chem. Soc. 2008, 130, 16316–16323. [Google Scholar] [CrossRef] [PubMed]
- Cheung, P.; Bhan, A.; Sunley, G.J.; Iglesia, E. Selective carbonylation of dimethyl ether to methyl acetate catalyzed by acidic zeolites. Angew. Chem. Int. Ed. 2006, 45, 1617–1620. [Google Scholar] [CrossRef] [PubMed]
- Cheung, P.; Bhan, A.; Sunley, G.J.; Law, D.J.; Iglesia, E. Site requirements and elementary steps in dimethyl ether carbonylation catalyzed by acidic zeolites. J. Catal. 2007, 245, 110–123. [Google Scholar] [CrossRef]
- Park, S.Y.; Shin, C.-H.; Bae, J.W. Selective carbonylation of dimethyl ether to methyl acetate on ferrierite. Catal. Commun. 2016, 75, 28–31. [Google Scholar] [CrossRef]
- Bañares, M.A. Supported metal oxide and other catalysts for ethane conversion: A review. Catal. Today 1999, 51, 319–348. [Google Scholar] [CrossRef]
- Gärtner, C.A.; van Veen, A.C.; Lercher, J.A. Oxidative dehydrogenation of ethane: Common principles and mechanistic aspects. ChemCatChem 2013, 5, 3196–3217. [Google Scholar] [CrossRef]
- Thorsteinson, E.M.; Wilson, T.P.; Young, F.G.; Kasai, P.H. The oxidative dehydrogenation of ethane over catalysts containing mixed oxides of molybdenum and vanadium. J. Catal. 1978, 52, 116–132. [Google Scholar] [CrossRef]
- Oyama, S.T.; Somorjai, G.A. Effect of structure in selective oxide catalysis—Oxidation reactions of ethanol and ethane on vanadium- oxide. J. Phys. Chem. 1990, 94, 5022–5028. [Google Scholar] [CrossRef]
- Roy, M.; GubelmannBonneau, M.; Ponceblanc, H.; Volta, J.C. Vanadium-molybdenum phosphates supported by tio2-anatase as new catalysts for selective oxidation of ethane to acetic acid. Catal. Lett. 1996, 42, 93–97. [Google Scholar] [CrossRef]
- Karim, K.; Al-Hazmi, M.; Khan, A. Catalysts for the Oxidation of Ethane to Acetic Acid, Methods of Making and Using the Same; Saudi Basic Industries Corporation: Riyadh, Saudi Arabia, 2000. [Google Scholar]
- Roy, M.; Ponceblanc, H.; Volta, J.C. Vanadium-molybdenum phosphates supported by TiO2 for ethane oxidation to acetic acid: A correlation between the local environment of vanadium and the reactivity. Top. Catal. 2000, 11, 101–109. [Google Scholar] [CrossRef]
- Linke, D.; Wolf, D.; Baerns, M.; Dingerdissen, U.; Zeyß, S. Catalytic partial oxidation of ethane to acetic acid over Mo1V0.25Nb0.12Pd0.0005Ox—Catalyst performance, reaction mechanism, kinetics and reactor operation. In Studies in Surface Science and Catalysis; E. Iglesia, J.J.S., Fleisch, T.H., Eds.; Elsevier: Amsterdam, The Netherlands, 2001; Volume 136, pp. 477–482. [Google Scholar]
- Rosen, B.I. Oxidation Catalyst Supported on Alumina and Its Preparation; BP Chemicals Limited: Hull Site, UK, 2004; p. 25. [Google Scholar]
- Brazdil, J.F.; George, R.J.; Rosen, B. Catalyst composition and use thereof in ethane oxidation. BP Chemicals Limited: Sunbury-on-Thames, UK, 2004. [Google Scholar]
- Galownia, J.M.; Wight, A.P.; Blanc, A.; Labinger, J.A.; Davis, M.E. Partially reduced heteropolyanions for the oxidative dehydrogenation of ethane to ethylene and acetic acid at atmospheric pressure. J. Catal. 2005, 236, 356–365. [Google Scholar] [CrossRef]
- Roussel, M.; Bouchard, M.; Bordes-Richard, E.; Karim, K.; Al-Sayari, S. Oxidation of ethane to ethylene and acetic acid by movnbo catalysts. Catal. Today 2005, 99, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Iglesia, E. Kinetics and mechanism of ethane oxidation to acetic acid on catalysts based on Mo-V-Nb oxides. J. Phys. Chem. C 2008, 112, 15001–15008. [Google Scholar] [CrossRef]
- Li, X.; Iglesia, E. Support and promoter effects in the selective oxidation of ethane to acetic acid catalyzed by Mo-V-Nb oxides. Appl. Catal. A Gen. 2008, 334, 339–347. [Google Scholar] [CrossRef]
- Karim, K.; Al-Hazmi, M.H.; Mamedov, E. Oxidation of Ethane to Acetic Acid and Ethylene Using Molybdenum and Vanadium Based Catalysts. U.S. Patent 6531631 B1, 7 January 2003. [Google Scholar]
- Ueda, W.; Chen, N.F.; Oshihara, K. Hydrothermal synthesis of Mo-V-M-O complex metal oxide catalysts active for partial oxidation of ethane. Chem. Commun. 1999, 6, 517–518. [Google Scholar]
- Smejkal, Q.; Linke, D.; Baerns, M. Energetic and economic evaluation of the production of acetic acid via ethane oxidation. Chem. Eng. Process. Process Intensif. 2005, 44, 421–428. [Google Scholar] [CrossRef]
- Cook, J.; Ellis, B.; Howard, P.; Jones, M.D.; Kitchen, S.J. Process for the production of acetic acid; Nixon & Vanderhye: Arlington, VA, USA, 2001. [Google Scholar]
- McCain, J.H.; Kaiser, S.W.; O’connor, G.L. Acetic Acid from Ethane, Ethylene and Oxygen; Union Carbide Corporation: Houston, TX, USA, 1988. [Google Scholar]
- Mizuno, N.; Han, W.C.; Kudo, T. Selective oxidation of ethane, propane, and isobutane catalyzed by copper-containing Cs2.5H1.5PVMo11O40 under oxygen-poor conditions. J. Catal. 1998, 178, 391–394. [Google Scholar] [CrossRef]
- Moffat, J.B. Conversion of C-2-C-5 alkanes on heteropoly oxometalates. Appl. Catal. A Gen. 1996, 146, 65–86. [Google Scholar] [CrossRef]
- Sopa, A.; Waclaw-Held, A.; Grossy, M.; Pijanka, J.; Nowinska, K. Ethane to acetic acid oxidation over supported heteropoly acids. Appl. Catal. A Gen. 2005, 285, 119–125. [Google Scholar] [CrossRef]
- Erdohelyi, A.; Solymosi, F. Oxidation of ethane over silica- supported alkali-metal vanadate catalysts. J. Catal. 1991, 129, 497–510. [Google Scholar] [CrossRef]
- Erdohelyi, A.; Mate, F.; Solymosi, F. Partial oxidation of ethane over silica- supported alkali-metal molybdate catalysts. J. Catal. 1992, 135, 563–575. [Google Scholar] [CrossRef]
- Bodke, A.S.; Olschki, D.A.; Schmidt, L.D.; Ranzi, E. High selectivities to ethylene by partial oxidation of ethane. Science 1999, 285, 712–715. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, A.B.; Kudrik, E.V.; Alvarez, L.X.; Afanasiev, P.; Millet, J.M.M.; Bouchu, D. Oxidation of methane and ethylene in water at ambient conditions. Catal. Today 2010, 157, 149–154. [Google Scholar] [CrossRef]
- Sorokin, A.B.; Kudrik, E.V.; Bouchu, D. Bio-inspired oxidation of methane in water catalyzed by n-bridged diiron phthalocyanine complex. Chem. Commun. 2008, 22, 2562–2564. [Google Scholar] [CrossRef] [PubMed]
- Osako, T.; Watson, E.J.; Dehestani, A.; Bales, B.C.; Mayer, J.M. Methane oxidation by aqueous osmium tetroxide and sodium periodate: Inhibition of methanol oxidation by methane. Angew. Chem. Int. Ed. 2006, 45, 7433–7436. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.J.; Taube, D.; Ziatdinov, V.R.; Periana, R.A.; Nielsen, R.J.; Oxgaard, J.; Goddard, W.A. Selective oxidation of methane to methanol catalyzed, with C–H activation, by homogeneous, cationic gold. Angew. Chem. Int. Ed. 2004, 43, 4626–4629. [Google Scholar] [CrossRef] [PubMed]
- Bar-Nahum, I.; Khenkin, A.M.; Neumann, R. Mild, aqueous, aerobic, catalytic oxidation of methane to methanol and acetaldehyde catalyzed by a supported bipyrimidinylplatinum−polyoxometalate hybrid compound. J. Am. Chem.Soc. 2004, 126, 10236–10237. [Google Scholar] [CrossRef] [PubMed]
- Seki, Y.; Min, J.S.; Misono, M.; Mizuno, N. Reaction mechanism of oxidation of methane with hydrogen peroxide catalyzed by 11-molybdo-1-vanadophosphoric acid catalyst precursor. J. Phys. Chem. B 2000, 104, 5940–5944. [Google Scholar] [CrossRef]
- Lunsford, J.H. Catalytic conversion of methane to more useful chemicals and fuels: A challenge for the 21st century. Catal. Today 2000, 63, 165–174. [Google Scholar] [CrossRef]
- Wolf, D. High yields of methanol from methane by C–H bond activation at low temperatures. Angew. Chem. Int. Ed. 1998, 37, 3351–3353. [Google Scholar] [CrossRef]
- Sen, A. Catalytic functionalization of carbon−hydrogen and carbon-carbon bonds in protic media. Acc. Chem. Res. 1998, 31, 550–557. [Google Scholar] [CrossRef]
- Periana, R.A.; Taube, D.J.; Gamble, S.; Taube, H.; Satoh, T.; Fujii, H. Platinum catalysts for the high-yield oxidation of methane to a methanol derivative. Science 1998, 280, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Nizova, G.V.; Suss-Fink, G.; Shul’pin, G.B. Catalytic oxidation of methane to methyl hydroperoxide and other oxygenates under mild conditions. Chem. Commun. 1997, 397–398. [Google Scholar] [CrossRef]
- Raja, R.; Ratnasamy, P. Direct conversion of methane to methanol. Appl. Catal. A Gen. 1997, 158, L7–L15. [Google Scholar] [CrossRef]
- Labinger, J.A. Methane activation in homogeneous systems. Fuel Process. Technol. 1995, 42, 325–338. [Google Scholar] [CrossRef]
- Lin, M.; Sen, A. Direct catalytic conversion of methane to acetic acid in aqueous medium. Nature 1994, 368, 613–615. [Google Scholar] [CrossRef]
- Periana, R.A.; Taube, D.J.; Evitt, E.R.; Löffler, D.G.; Wentrcek, P.R.; Voss, G.; Masuda, T. A mercury-catalyzed, high-yield system for the oxidation of methane to methanol. Science 1993, 259, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Shilov, A.E.; Shul’pin, G.B. Activation and catalytic reactions of alkanes in solutions of metal complexes. Russ. Chem. Rev. 1987, 56, 442–464. [Google Scholar] [CrossRef]
- Jia, C.; Kitamura, T.; Fujiwara, Y. Catalytic functionalization of arenes and alkanes via C–H bond activation. Acc. Chem. Res. 2001, 34, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Suss-Fink, G.; Gonzalez, L.; Shul’pin, G.B. Alkane oxidation with hydrogen peroxide catalyzed homogeneously by vanadium-containing polyphosphomolybdates. Appl. Catal. A Gen. 2001, 217, 111–117. [Google Scholar] [CrossRef]
- Shul’pin, G.B.; Süss-Fink, G.; Shul’pina, L.S. Oxidations by the system “hydrogen peroxide-manganese(IV) complex-carboxylic acid”: Part 3. Oxygenation of ethane, higher alkanes, alcohols, olefins and sulfides. J. Mol. Catal. A Chem. 2001, 170, 17–34. [Google Scholar] [CrossRef]
- Rahman, A.K.M.L.; Indo, R.; Hagiwara, H.; Ishihara, T. Direct conversion of ethane to acetic acid over H-ZSM-5 using H2O2 in aqueous phase. Appl. Catal. A Gen. 2013, 456, 82–87. [Google Scholar] [CrossRef]
- Shul’pin, G.B.; Süss-Fink, G.; Shul’pina, L.S. Oxidative functionalisation of ethane with hydrogen peroxide catalysed by chromic acid. J. Chem. Res. Part S 2000, 12, 576–577. [Google Scholar]
- Shul’pina, L.S.; Kirillova, M.V.; Pombeiro, A.J.L.; Shul’pin, G.B. Alkane oxidation by the H2O2-NaVO3-H2SO4 system in acetonitrile and water. Tetrahedron 2009, 65, 2424–2429. [Google Scholar] [CrossRef]
- Shul’pin, G.B.; Nizova, G.V.; Kozlov, Y.N.; Gonzalez Cuervo, L.; Süss-Fink, G. Hydrogen peroxide oxygenation of alkanes including methane and ethane catalyzed by iron complexes in acetonitrile. Adv. Synth. Catal. 2004, 346, 317–332. [Google Scholar] [CrossRef]
- Ensing, B.; Buda, F.; Blochl, P.E.; Baerends, E.J. A car-parrinello study of the formation of oxidizing intermediates from fenton’s reagent in aqueous solution. Phys. Chem. Chem. Phys. 2002, 4, 3619–3627. [Google Scholar] [CrossRef]
- Yuan, Q.; Deng, W.; Zhang, Q.; Wang, Y. Osmium-catalyzed selective oxidations of methane and ethane with hydrogen peroxide in aqueous medium. Adv. Synth. Catal. 2007, 349, 1199–1209. [Google Scholar] [CrossRef]
- Tse, C.-W.; Chow, T.W.-S.; Guo, Z.; Lee, H.K.; Huang, J.-S.; Che, C.-M. Nonheme iron mediated oxidation of light alkanes with oxone: Characterization of reactive oxoiron(IV) ligand cation radical intermediates by spectroscopic studies and dft calculations. Angew. Chem. Int. Ed. 2014, 53, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Hashiguchi, B.G.; Konnick, M.M.; Bischof, S.M.; Gustafson, S.J.; Devarajan, D.; Gunsalus, N.; Ess, D.H.; Periana, R.A. Main-group compounds selectively oxidize mixtures of methane, ethane, and propane to alcohol esters. Science 2014, 343, 1232–1237. [Google Scholar] [CrossRef] [PubMed]
- Konnick, M.M.; Hashiguchi, B.G.; Devarajan, D.; Boaz, N.C.; Gunnoe, T.B.; Groves, J.T.; Gunsalus, N.; Ess, D.H.; Periana, R.A. Selective ch functionalization of methane, ethane, and propane by a perfluoroarene iodine(III) complex. Angew. Chem. Int. Ed. 2014, 53, 10490–10494. [Google Scholar] [CrossRef] [PubMed]
- Periana, R.; Hashiguchi, B.G.; Konnick, M.M.; Bischof, S.M. Oxidation of Alkanes to Alcohols; The Scrips Research Institute: San Diego, CA, USA, 2014. [Google Scholar]
- Colby, J.; Stirling, D.I.; Dalton, H. Soluble methane mono-oxygenase of methylococcus-capsulatus-(bath)—Ability to oxygenate normal-alkanes, normal-alkenes, ethers, and alicyclic, aromatic and heterocyclic-compounds. Biochem. J. 1977, 165, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Tonge, G.M.; Harrison, D.E.F.; Higgins, I.J. Purification and properties of methane mono-oxygenase enzyme-system from methylosinus-trichosporium ob3b. Biochem. J. 1977, 161, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Shteinman, A.A. The role of metal-oxygen intermediates in biological and chemical monooxygenation of alkanes. Russ. Chem. Bull. 2001, 50, 1795–1810. [Google Scholar] [CrossRef]
- Meinhold, P.; Peters, M.W.; Chen, M.M.Y.; Takahashi, K.; Arnold, F.H. Direct conversion of ethane to ethanol by engineered cytochrome P450 BM3. ChemBioChem 2005, 6, 1765–1768. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Bell, S.G.; Lednik, J.; Insley, A.; Rao, Z.; Wong, L.-L. The heme monooxygenase cytochrome P450CAM can be engineered to oxidize ethane to ethanol. Angew. Chem. Int. Ed. 2005, 44, 4029–4032. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, N.; Shoji, O.; Watanabe, Y. Direct hydroxylation of primary carbons in small alkanes by wild-type cytochrome P450BM3 containing perfluorocarboxylic acids as decoy molecules. Chem. Sci. 2013, 4, 2344–2348. [Google Scholar] [CrossRef]
- Chen, M.M.; Coelho, P.S.; Arnold, F.H. Utilizing terminal oxidants to achieve p450-catalyzed oxidation of methane. Adv. Synth. Catal. 2012, 354, 964–968. [Google Scholar] [CrossRef]
- Wang, Y.; Otsuka, K. Partial oxidation of ethane by reductively activated oxygen over iron phosphate catalyst. J. Catal. 1997, 171, 106–114. [Google Scholar] [CrossRef]
- Wang, Y.; Otsuka, K. Catalytic oxidation of methane to methanol with H2-O2 gas mixture at atmospheric pressure. J. Catal. 1995, 155, 256–267. [Google Scholar] [CrossRef]
- Nizova, G.V.; Krebs, B.; Suss-Fink, G.; Schindler, S.; Westerheide, L.; Gonzalez Cuervo, L.; Shul’pin, G.B. Hydroperoxidation of methane and other alkanes with H2O2 catalyzed by a dinuclear iron complex and an amino acid. Tetrahedron 2002, 58, 9231–9237. [Google Scholar] [CrossRef]
- Nagababu, P.; Yu, S.S.F.; Maji, S.; Ramu, R.; Chan, S.I. Developing an efficient catalyst for controlled oxidation of small alkanes under ambient conditions. Catal. Sci. Technol. 2014, 4, 930–935. [Google Scholar] [CrossRef]
- Xiao, D.J.; Bloch, E.D.; Mason, J.A.; Queen, W.L.; Hudson, M.R.; Planas, N.; Borycz, J.; Dzubak, A.L.; Verma, P.; Lee, K.; et al. Oxidation of ethane to ethanol by N2O in a metal-organic framework with coordinatively unsaturated iron(II) sites. Nat. Chem. 2014, 6, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Sen, A. A highly catalytic system for the direct oxidation of lower alkanes by dioxygen in aqueous medium. A formal heterogeneous analog of alkane monooxygenases. J. Am. Chem. Soc. 1992, 114, 7307–7308. [Google Scholar] [CrossRef]
- Shul’pin, G.B.; Sooknoi, T.; Romakh, V.B.; Süss-Fink, G.; Shul’pina, L.S. Regioselective alkane oxygenation with H2O2 catalyzed by titanosilicalite Ts-1. Tetrahedron Lett. 2006, 47, 3071–3075. [Google Scholar] [CrossRef]
- Kudrik, E.V.; Afanasiev, P.; Alvarez, L.X.; Dubourdeaux, P.; Clemancey, M.; Latour, J.-M.; Blondin, G.; Bouchu, D.; Albrieux, F.; Nefedov, S.E.; et al. An n-bridged high-valent diiron-oxo species on a porphyrin platform that can oxidize methane. Nat. Chem. 2012, 4, 1024–1029. [Google Scholar] [CrossRef] [PubMed]
- Forde, M.M.; Grazia, B.C.; Armstrong, R.; Jenkins, R.L.; Rahim, M.H.A.; Carley, A.F.; Dimitratos, N.; Lopez-Sanchez, J.A.; Taylor, S.H.; McKeown, N.B.; et al. Methane oxidation using silica-supported n-bridged di-iron phthalocyanine catalyst. J. Catal. 2012, 290, 177–185. [Google Scholar] [CrossRef]
- Alvarez, L.X.; Sorokin, A.B. Mild oxidation of ethane to acetic acid by H2O2 catalyzed by supported μ-nitrido diiron phthalocyanines. J. Organomet. Chem. 2015, 793, 139–144. [Google Scholar] [CrossRef]
- Armstrong, R.D.; Freakley, S.J.; Forde, M.M.; Peneau, V.; Jenkins, R.L.; Taylor, S.H.; Moulijn, J.A.; Morgan, D.J.; Hutchings, G.J. Low temperature catalytic partial oxidation of ethane to oxygenates by Fe- and Cu-zsm-5 in a continuous flow reactor. J. Catal. 2015, 330, 84–92. [Google Scholar] [CrossRef]
- Forde, M.M.; Armstrong, R.D.; McVicker, R.; Wells, P.P.; Dimitratos, N.; He, Q.; Lu, L.; Jenkins, R.L.; Hammond, C.; Lopez-Sanchez, J.A.; et al. Light alkane oxidation using catalysts prepared by chemical vapour impregnation: Tuning alcohol selectivity through catalyst pre-treatment. Chem. Sci. 2014, 5, 3603–3616. [Google Scholar] [CrossRef]
- Hammond, C.; Forde, M.M.; Ab Rahim, M.H.; Thetford, A.; He, Q.; Jenkins, R.L.; Dimitratos, N.; Lopez-Sanchez, J.A.; Dummer, N.F.; Murphy, D.M.; et al. Direct catalytic conversion of methane to methanol in an aqueous medium by using copper-promoted Fe-ZSM-5. Angew. Chem. Int. Ed. 2012, 51, 5129–5133. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
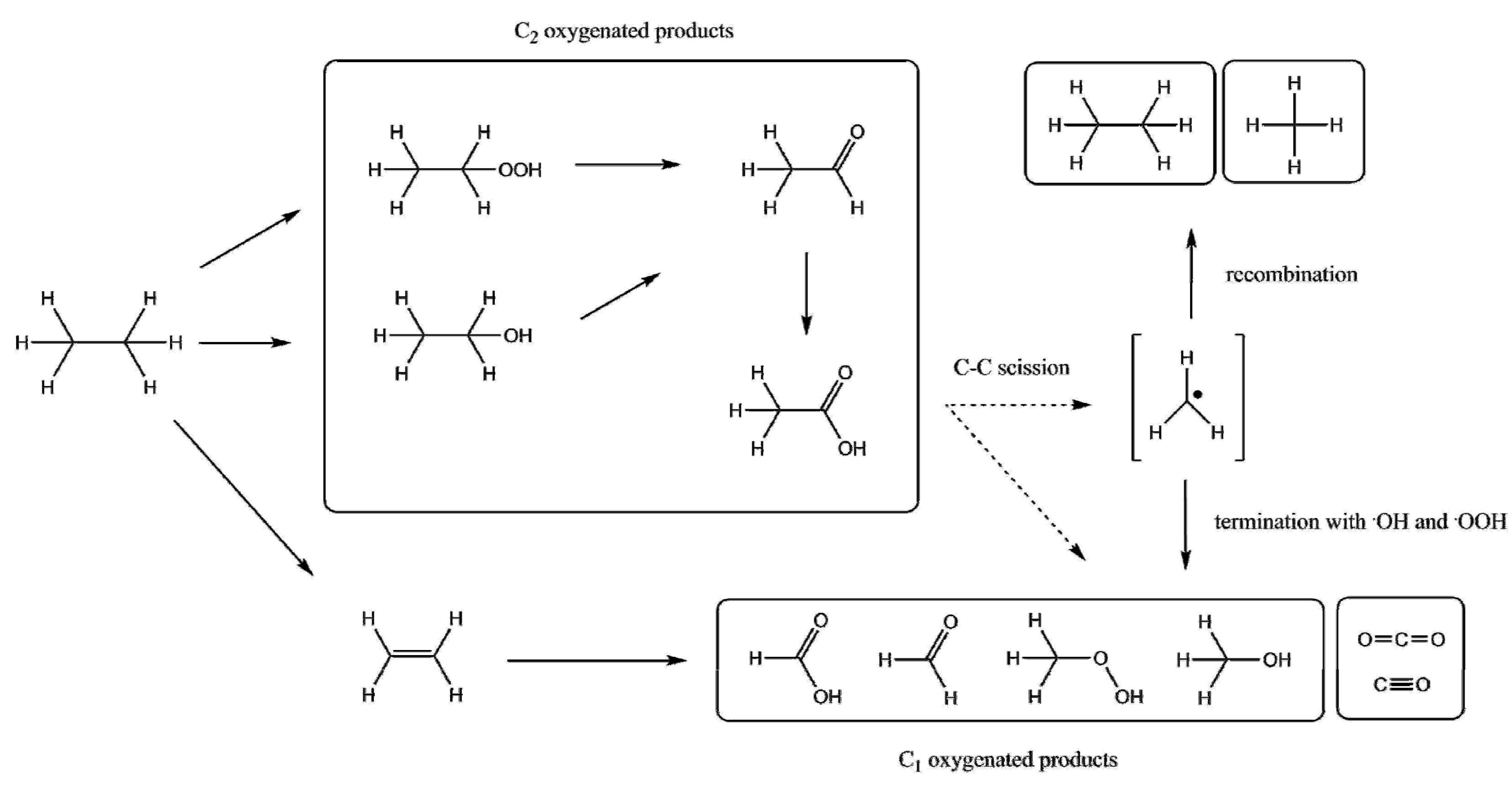{kind=link}
| Entry | Catalyst | Regime | Oxidant | Solvent(s) | P (C2H6)/bar | T/°C | Time/h | Major Product Selectivities/% | Mass Normalised Conversion Rate a | TOF b | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | [FeIII(Tp)2]ClO4 c | Batch. L/G | KHSO5 | MeCN/H2O | 6.9 | RT | 0.08 | CH3COOH (80), EtOH (20) | 20.64 | 12.0 | [75] |
| 2 | [FeIII(L)-(acac)Cl]ClO4 d | Batch. L/G | KHSO5 | MeCN/H2O | 6.9 | RT | 0.5 | CH3COOH (83), EtOH (17) | 13.01 | 6.0 | [75] |
| 3 | [FeIII(L)-(3-Cl-acac)Cl]ClO4 d | Batch. L/G | KHSO5 | MeCN/H2O | 6.9 | RT | 0.5 | CH3COOH (85), EtOH (15) | 13.72 | 6.8 | [75] |
| 4 | H2CrO4 | Batch. L/G | H2O2 | MeCN | 30 | 60 | 1 | CH3CHO (52), EtOOH (23), EtOH (19) | 5253.71 | 620 | [70] |
| 5 | NaVO3 + H2SO4 | Batch. L/G | H2O2 | MeCN | 30 | 50 | 4 | EtOH (51), CH3CHO (32), CH3COOH (17) | 385.47 | 47.5 | [71] |
| 6 | [PMo11VO40](Bu4N)4 | Batch. L/G | H2O2 | MeCN | 30 | 60 | 10 | CH3CHO (44), EtOOH (34), EtOH (22) | 0.50 | 1.4 | [67] |
| 7 | [L2Mn2O3](PF6)2 d | Batch. L/G | H2O2 | MeCN | 20 | 25 | 2 | EtOOH (39), CH3CHO (33), EtOH (28) | 227.74 | 180 | [68] |
| 8 | Fe(ClO4)3 | Batch. L/G | H2O2 | MeCN | 27 | 25 | 3 | EtOOH (88), CH3CHO (9), EtOH (3) | 64.00 | 22.7 | [72] |
| 9 | Fe(OAC)2(OH) + PCA e | Batch. L/G | H2O2 | MeCN | 27 | 25 | 2 | EtOH (68), CH3CHO (29), EtOOH (3) | 31.43 | 6.0 | [72] |
| 10 | FeCl3 | Batch. L/G | H2O2 | H2O | 30 | 90 | 1 | CH3CHO (66), EtOH (18), CO2 (16) | 189.27 | 30.7 | [74] |
| 11 | PdCl2 | Batch. L/G | H2O2 | H2O | 30 | 90 | 1 | CH3CHO (56), CO2 (31), EtOH (13) | 170.87 | 30.3 | [74] |
| 12 | OsCl3 | Batch. L/G | H2O2 | H2O | 30 | 90 | 1 | CH3CHO (56), CO2 (26) EtOH (18) | 161.17 | 47.8 | [74] |
| 13 | H2PtCl6 | Batch. L/G | H2O2 | H2O | 30 | 90 | 1 | CH3CHO (67), EtOH (33) | 13.18 | 5.4 | [74] |
| 14 | HAuCl4 | Batch. L/G | H2O2 | H2O | 30 | 90 | 1 | CH3CHO (62), CO2 (22) EtOH (15) | 94.32 | 32.1 | [74] |
| 15 | - | Batch. L/G | Tl(TFA)3 | HTFA | 34.4 | 180 | 3 | EtTFA (67), EG(TFA)2 (33) | 0.46 f | - | [76] |
| 16 | - | Batch L/G | Pb(TFA)4 | HTFA | 34.4 | 180 | 3 | EtTFA (70), EG(TFA)2 (30) | 0.46 f | - | [76] |
| 17 | - | Batch. L/G | C6F5IIII(TFA)2 | TFAA/HTFA | 34.5 | 150 | 3 | EtTFA (91), 1,2-Et(TFA)2 (8%) | 0.51 f | - | [77] |
| 18 | Methylococcus capsulatus (sMMO) | Batch. L/G | O2/NADH | H2O | - | 45 | 0.2 | EtOH (100) | 4.10 | - | [79] |
| 19 | Cytochrome P450 BM3 | Batch. L/G | O2/NADPH | H2O | 5 | 20 | 2 | EtOH (100) | - | 40.0 | [84] |
| 20 | Cytochrome P450 BM3 mutant | Batch. L/G | O2/NADPH | H2O | 1.38 | 25 | 0.5 | EtOH (100) | - | 24.0 | [82] |
| 21 | Cytochrome P450cam mutant | Batch. L/G | O2/NADH | H2O | - | 30 | - | EtOH (100) | - | 4700 | [83] |
| 22 | Cytochrome 450 PMO A6 | Batch. L/G | PhIO | H2O | 1.38 | 25 | 0.17 | EtOH (100) | - | 15.0 | [85] |
| 23 | Cytochrome 450 PMO A6 | Batch. L/G | MCPBA | H2O | 1.38 | 25 | 0.17 | EtOH (100) | - | 2.0 | [85] |
| 24 | Cytochrome 450 PMO A6 | Batch. L/G | H2O2 | H2O | 1.38 | 25 | 0.17 | EtOH (100) | - | 1.4 | [85] |
| 25 | FePO4 | Flow, G/S | O2/H2 | - | 0.34 | 400 | - | CH3CHO (24), HCHO (18), EtOH (12) g | 4.20 | 1.3 | [86] |
| 26 | [CuICuICuI(7-N-Etppz)]1+ h | Batch. L/G | H2O2 | MeCN | 1.79 | RT | 1 | EtOH (100) | 19.06 | 11.0 | [89] |
| 27 | Fe0.1Mg1.9(dobdc) | Batch. G/S | N2O | - | 7.5 | 75 | 24 | EtOH (96), CH3CHO (4) | - | 0.07 | [90] |
| 28 | [Fe2(HPTB)(μ-OH)(NO3)2](NO3)2·CH3OH·2H2O + PCA e | Batch. L/G | H2O2 | MeCN | 30 | 25 | 6 | EtOOH (82), CH3CHO (17), CH3COOH (1) | 3.28 | 3.5 | [88] |
| 29 | 5% Pd/C | Batch. L/G/S | H2O2 i | DCl/D2O | 34.5 | 70 | 24 | CH3COOH (85), HCOOH (10), EtOH (6) | 0.65 | 1.4 | [91] |
| 30 | 5% Pd/C | Batch. L/G/S | H2O2 i | DCl/D2O | 34.5 | 85 | 24 | CH3COOH (78), HCOOH (22) | 3.40 | 7.2 | [91] |
| 31 | 5% Pt/C | Batch. L/G/S | H2O2 i | DCl/D2O | 34.5 | 95 | 24 | CH3OOH (100) | 0.14 | 0.5 | [91] |
| 32 | TS-1 | Batch. L/G/S | H2O2 | H2O | 30 | 60 | 12 | CH3CHO (94), EtOH (6) | 0.25 | - | [92] |
| 33 | (FePc)2N/SiO2 | Batch. L/G/S | H2O2 | H2O | 32 | 60 | 20 | CH3COOH (69), HCOOH (31) | 0.054 | 2.7 | [95] |
| 33 | (FePctBu)2N/SiO2 | Batch. L/G/S | H2O2 | H2O | 32 | 60 | 20 | CH3COOH (71), HCOOH (29) | 0.047 | 2.3 | [95] |
| 34 | TS-1 | Batch. L/G/S | H2O2 | PPh3/H2O | 30 | 120 | 2 | CH3COOH (84), CO2 (9), HCOOH (4) | 9.53 | - | [69] |
| 35 | H-β | Batch. L/G/S | H2O2 | PPh3/H2O | 30 | 120 | 2 | CH3COOH (65), HCOOH (20), CO2 (11) | 14.09 | - | [69] |
| 36 | 5% W/H-ZSM-5 | Batch. L/G/S | H2O2 | PPh3/H2O | 30 | 120 | 2 | CH3COOH (44), HCOOH (38), CO2 (16) | 13.41 | - | [69] |
| 37 | H4PVMoO40 | Batch. L/G/S | H2O2 | PPh3/H2O | 30 | 120 | 2 | CH3COOH (61), HCOOH (19), CO2 (12) | 15.37 | - | [69] |
| 38 | H-ZSM-5 | Batch. L/G/S | H2O2 | PPh3/H2O | 30 | 120 | 2 | CH3COOH (48), HCOOH (36), CO2 (12) | 17.24 | - | [69] |
| 39 | H-ZSM-5 | Batch. L/G/S | H2O2 | H2O | 20 | 50 | 0.5 | CH3COOH (37), EtOH (26) HCOOH (17) | 3.00 | 1211.4 | [7] |
| 40 | 0.4% Fe/ZSM-5 | Batch. L/G/S | H2O2 | H2O | 20 | 50 | 0.5 | CH3COOH (49), EtOH (19) HCOOH (14) | 16.50 | 233.2 | [7] |
| 41 | 1.1% Fe/ZSM-5 j | Batch. L/G/S | H2O2 | H2O | 20 | 50 | 0.5 | CH3COOH (55), EtOH (23) HCOOH (16) | 49.50 | 251.3 | [7] |
| 42 | 1.25% Fe 1.25% Cu/ZSM-5 | Batch. L/G/S | H2O2 | H2O | 20 | 50 | 0.5 | C2H4 (34), CH3COOH (31), EtOH (26) | 24.00 | 32.6 | [7] |
| 43 | 2.5% Fe/SiO2 | Batch. L/G/S | H2O2 | H2O | 20 | 50 | 0.5 | CH3CH3OOH (34), CH3CHO (33), CH3COOH (13) | 4.50 | 12.9 | [7] |
| 44 | 0.5% Fe-Silicalite-1 | Batch. L/G/S | H2O2 | H2O | 20 | 50 | 0.5 | EtOH (40), CH3COOH (30), HCOOH (14) | 6.00 | 66.5 | [7] |
| 45 | 1.1% Fe/ZSM-5 k | Batch. L/G/S | H2O2 | H2O | 20 | 50 | 0.5 | EtOH (33), CH3COOH (44), HCOOH (13) | 56.00 | 284.3 | [97] |
| 46 | 0.4% Fe/ZSM-5 | Flow. L/G/S | H2O2 | H2O | 2 | 50 | 0.06 l | CH3COOH (73), HCOOH (19), CH3CHO (3) | 0.26 | 3.6 | [96] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Armstrong, R.D.; Hutchings, G.J.; Taylor, S.H. An Overview of Recent Advances of the Catalytic Selective Oxidation of Ethane to Oxygenates. Catalysts 2016, 6, 71. https://doi.org/10.3390/catal6050071
Armstrong RD, Hutchings GJ, Taylor SH. An Overview of Recent Advances of the Catalytic Selective Oxidation of Ethane to Oxygenates. Catalysts. 2016; 6(5):71. https://doi.org/10.3390/catal6050071
Chicago/Turabian StyleArmstrong, Robert D., Graham J. Hutchings, and Stuart H. Taylor. 2016. "An Overview of Recent Advances of the Catalytic Selective Oxidation of Ethane to Oxygenates" Catalysts 6, no. 5: 71. https://doi.org/10.3390/catal6050071
APA StyleArmstrong, R. D., Hutchings, G. J., & Taylor, S. H. (2016). An Overview of Recent Advances of the Catalytic Selective Oxidation of Ethane to Oxygenates. Catalysts, 6(5), 71. https://doi.org/10.3390/catal6050071