2.1. FTIR Analysis of CO Adsorption on Catalysts
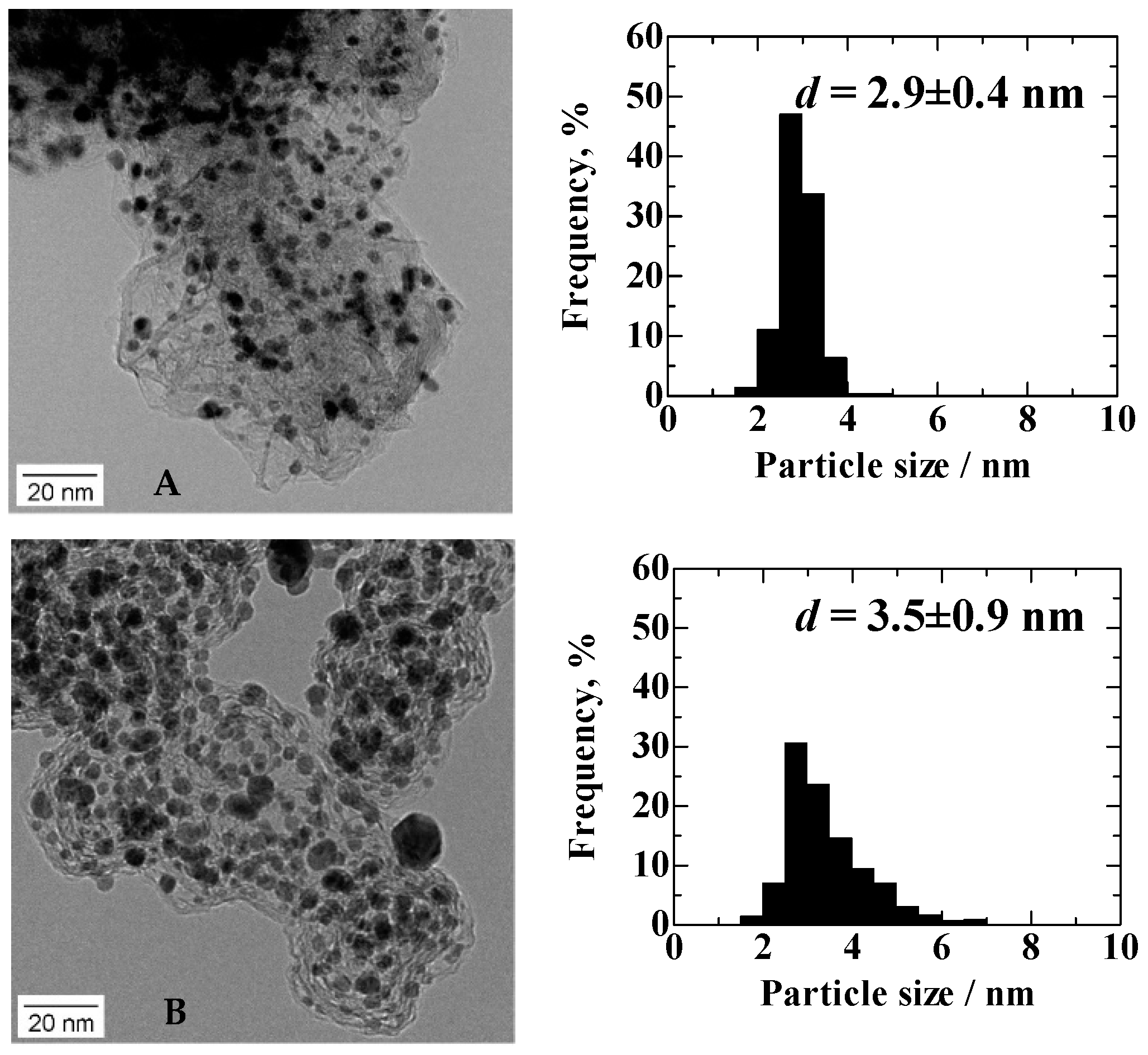
TEM images and particle size distribution histograms of Pt
2AL–PtFe/C and c-Pt
2Ru
3/C (
Figure 1) show that the average particle sizes and the standard deviations (
σd) of the Pt
2AL–PtFe/C and c-Pt
2Ru
3/C, which were determined from 500 particles in several TEM images, were 2.9 ± 0.4 nm and 3.5 ± 0.9 nm, respectively. As reported previously [
11], the Pt
2AL–PtFe nanoparticles were more uniform in size (smaller
σd) and highly dispersed on the carbon black support, compared with c-Pt
2Ru
3/C.
By the use of an in situ FTIR cell with the attenuated total reflection configuration (ATR-FTIR) [
7,
12], the IR spectra on both catalysts at 0.02 V (practical operating potential in PEFCs) versus an RHE and 60 °C by bubbling 1% CO (H
2-balance) continuously in 0.1 M HClO
4 solution were measured together with the HOR current. The electrolyte solution was first saturated with pure H
2 to measure the initial HOR current and the reference IR spectrum at 0.02 V, followed by changing the gas to 1% CO/H
2.
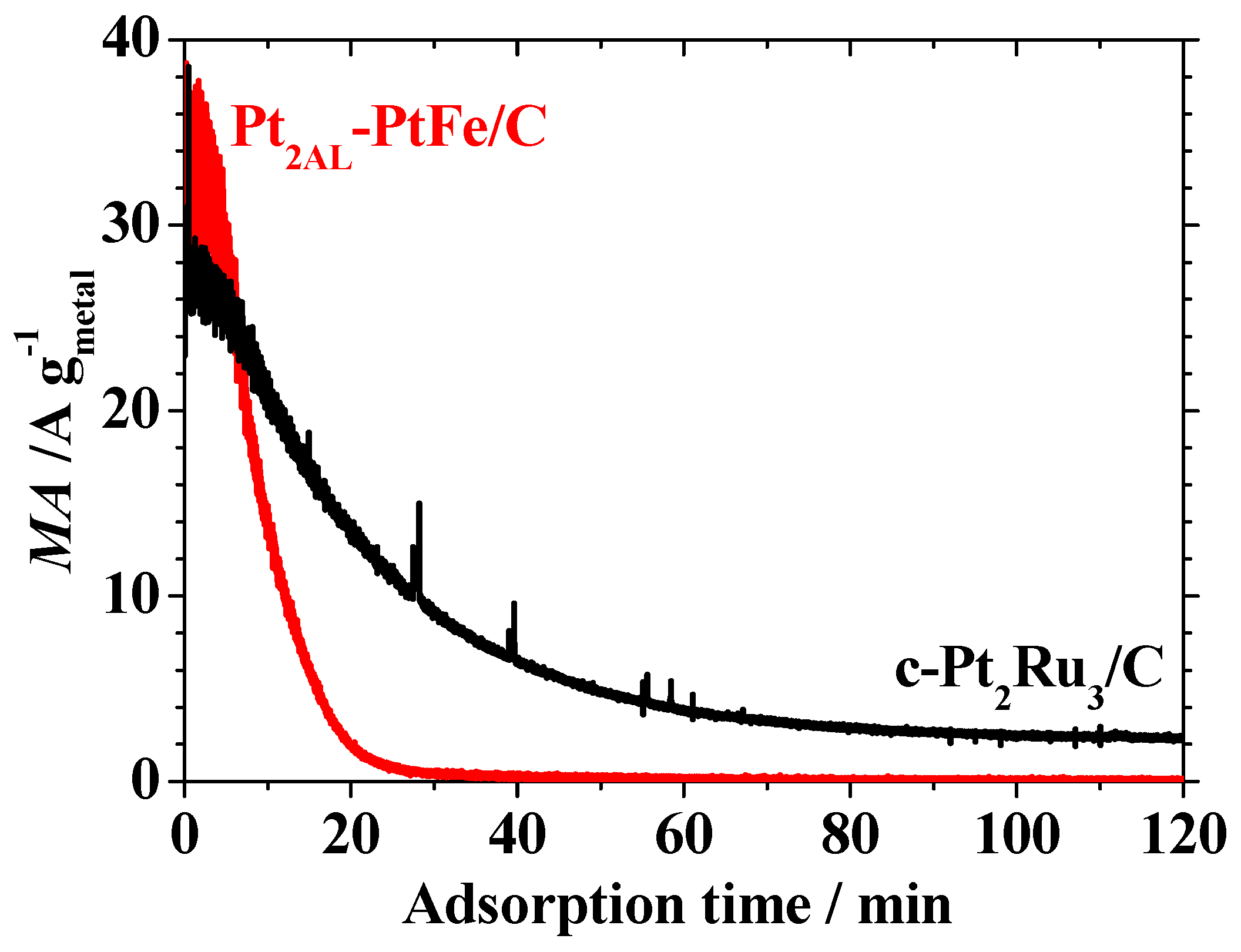
Figure 2 shows change in the HOR mass activity (
MA, HOR current per unit metal mass) at 0.02 V vs. RHE during the in situ ATR-FTIR measurement of CO adsorption. The initial
MA value (measured in pure H
2-saturated solution) on the Pt
2AL–PtFe/C catalyst (35 A g
metal−1) was ca. 1.3 times larger than that on the c-Pt
2Ru
3/C. Such an enhancement factor in the
MA for the HOR of pure H
2 on the Pt
2AL–PtFe/C was smaller than that (ca. 2.5 times) measured in the CFE at 70 and 90 °C [
10]. This is ascribed with certainty to the fact that it is difficult for all of the catalyst particles to work effectively for the HOR, because the thickness of the catalyst layer loaded on the ATR prism corresponded to about 6.5 monolayers of the carbon black particles (to increase the signal/noise ratio of the IR spectrum), whereas the amount of the catalyst loaded in the CFE cell was ca. two monolayers of carbon black particles to obtain the HOR activity in the ideal electrochemical condition. Focusing on the changes in the
MA values during CO adsorption on both catalysts in
Figure 2, the CO-poisoning rate at the Pt
2AL–PtFe/C was rapid, losing 99% of
MA after 30 min, whereas that at the c-Pt
2Ru
3/C was slower, so that about 10% of the initial
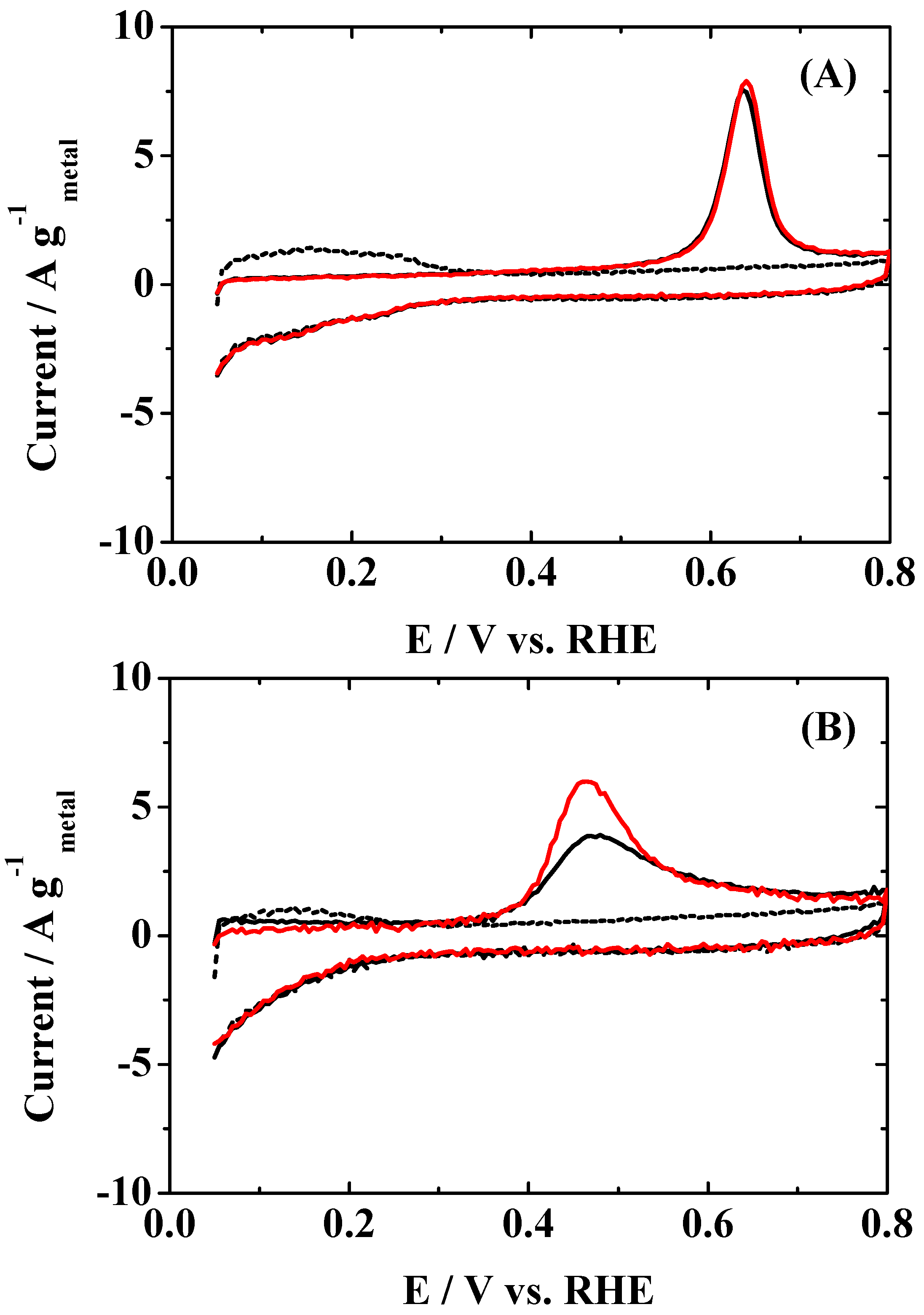
MA was still maintained after 2 h. Then, the CO coverage θ
CO on the surface just after the in situ ATR-FTIR measurement was evaluated from the CO-stripping voltammogram in N
2-purged solution, as shown in
Figure 3. The loss of
MA on the Pt
2AL–PtFe/C catalyst is reasonably explained by the θ
CO value of 0.99. The value of θ
CO on the c-Pt
2Ru
3/C catalyst was also 0.99, but, as stated above, the HOR activity was still maintained. It is clear that the adsorbed CO (CO
ad) cannot be oxidized during the HOR at 0.02 V, even on c-Pt
2Ru
3/C, because the values of onset potential for the CO
ad oxidation on Pt
2AL–PtFe/C and c-P
2tRu
3/C were ca. 0.50 V and 0.33 V, respectively. The difference in the CO-tolerance will be discussed in more detail below.
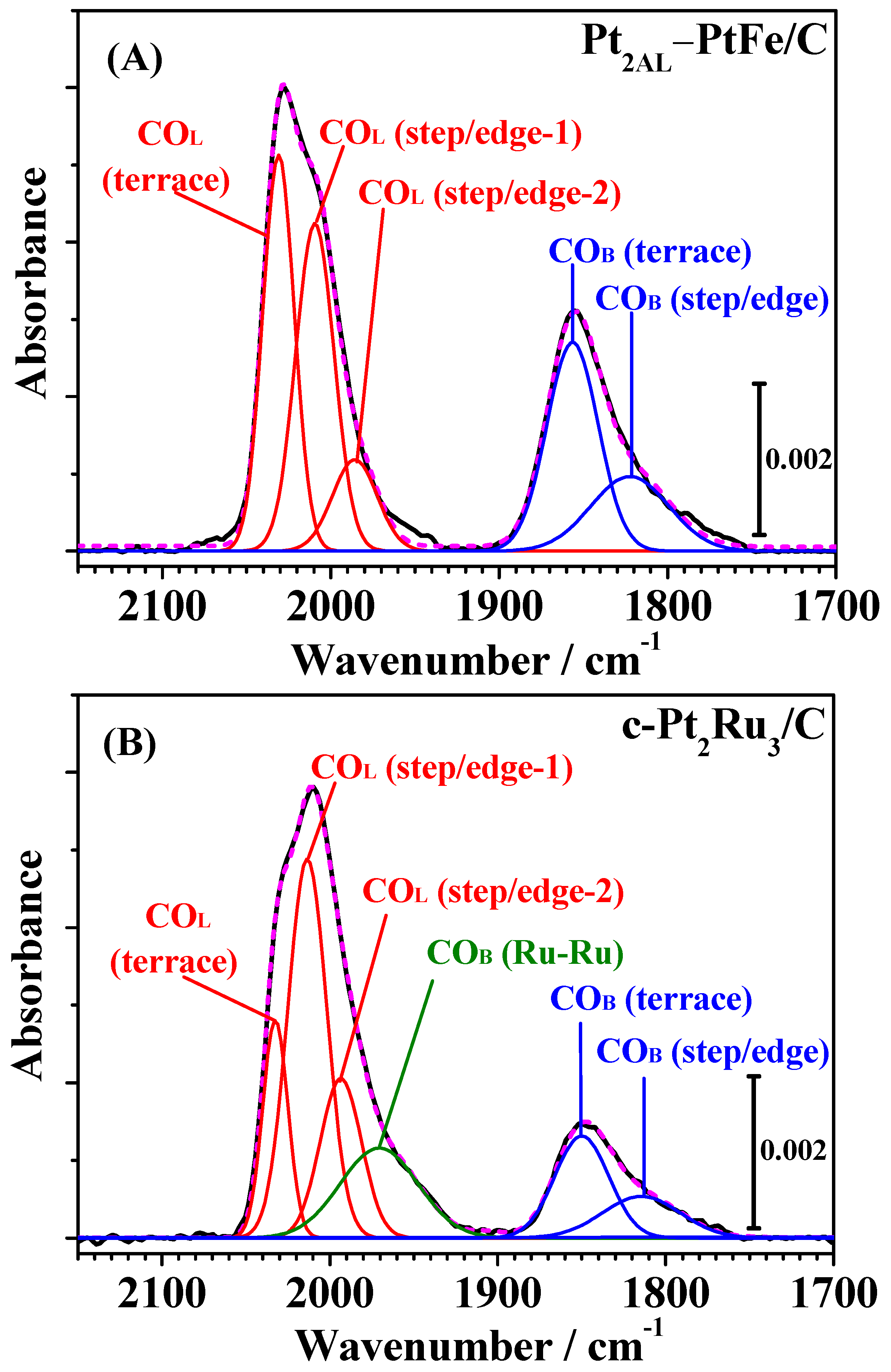
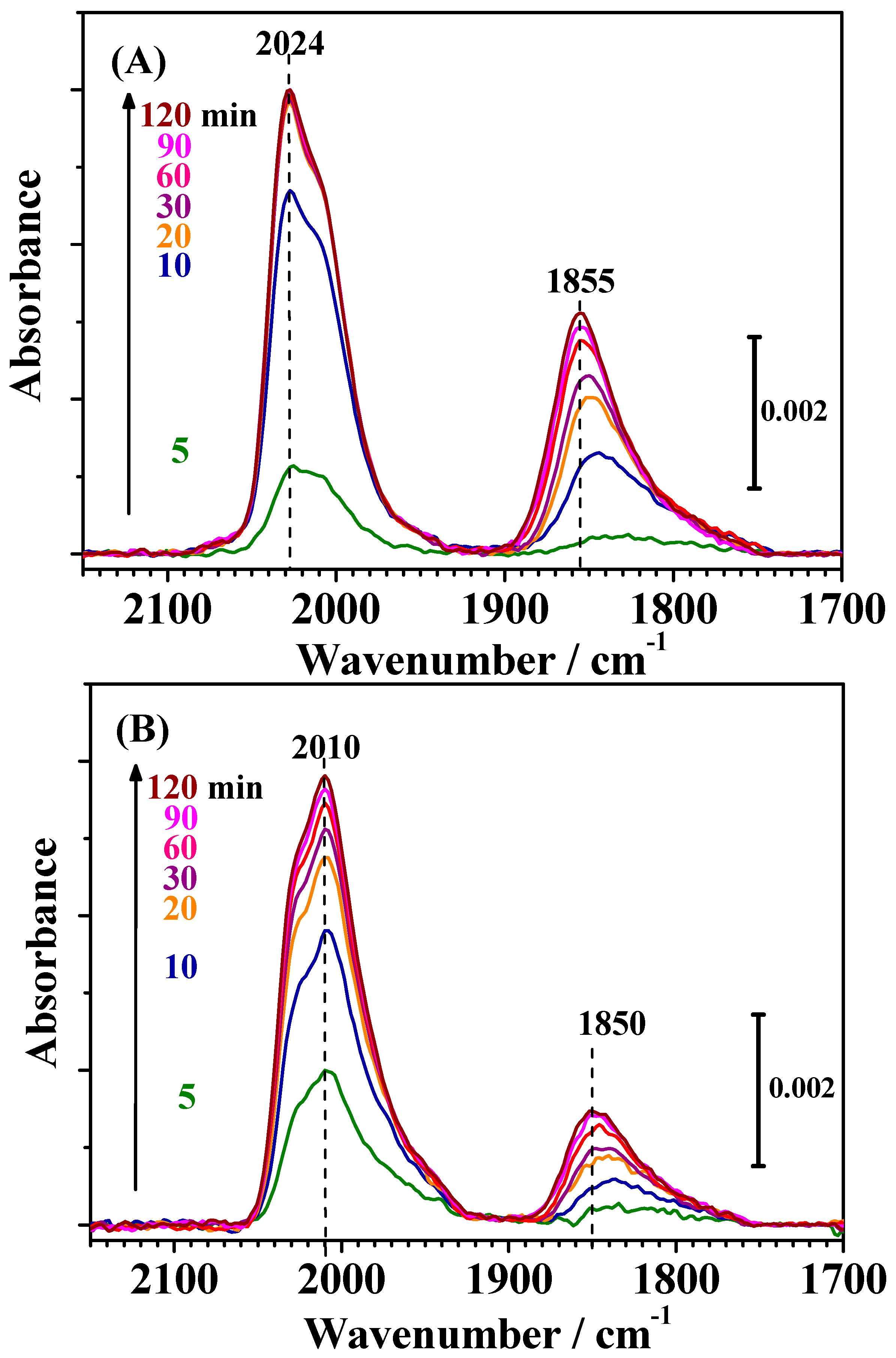
Figure 4 shows changes in the IR spectra observed on both catalysts during CO adsorption measured simultaneously with the HOR current shown in
Figure 2. The bands observed for both catalysts around 2050–1950 cm
−1 and 1900–1750 cm
−1 were assigned to linearly adsorbed (on-top) CO on Pt [CO
L] and bridged CO on Pt-Pt pair sites [CO
B(Pt-Pt)], respectively [
7,
12,
13,
14]. A small band around 1950 cm
−1 on c-Pt
2Ru
3/C (see
Figure 5) was assigned to CO
ad in a bridged configuration on Pt-Ru and/or Ru-Ru sites [CO-Ru, CO
B(Pt-Ru) or CO
B(Ru-Ru)] [
13].
A nearly identical CO-Ru band around 1950 to 2020 cm
−1 at high CO coverage was reported, but it was previously assigned to on-top CO on Ru sites for Ru-decorated Pt [
15]. However, based on FTIR experiments carried out by the use of size-controlled Pt, Ru, and Pt-Ru particles, Baranova et al. clearly assigned such a band to CO
ad bridged on Pt-Ru and/or Ru-Ru sites [
16]. As has been observed for the CO adsorption process on various catalysts, the changes in the IR spectra in
Figure 4 indicate that the bands consists of multiple components, which can be ascribed to CO
ad in slightly different configurations or environments, specifically, terraces or step/edge sites [
12,
13].
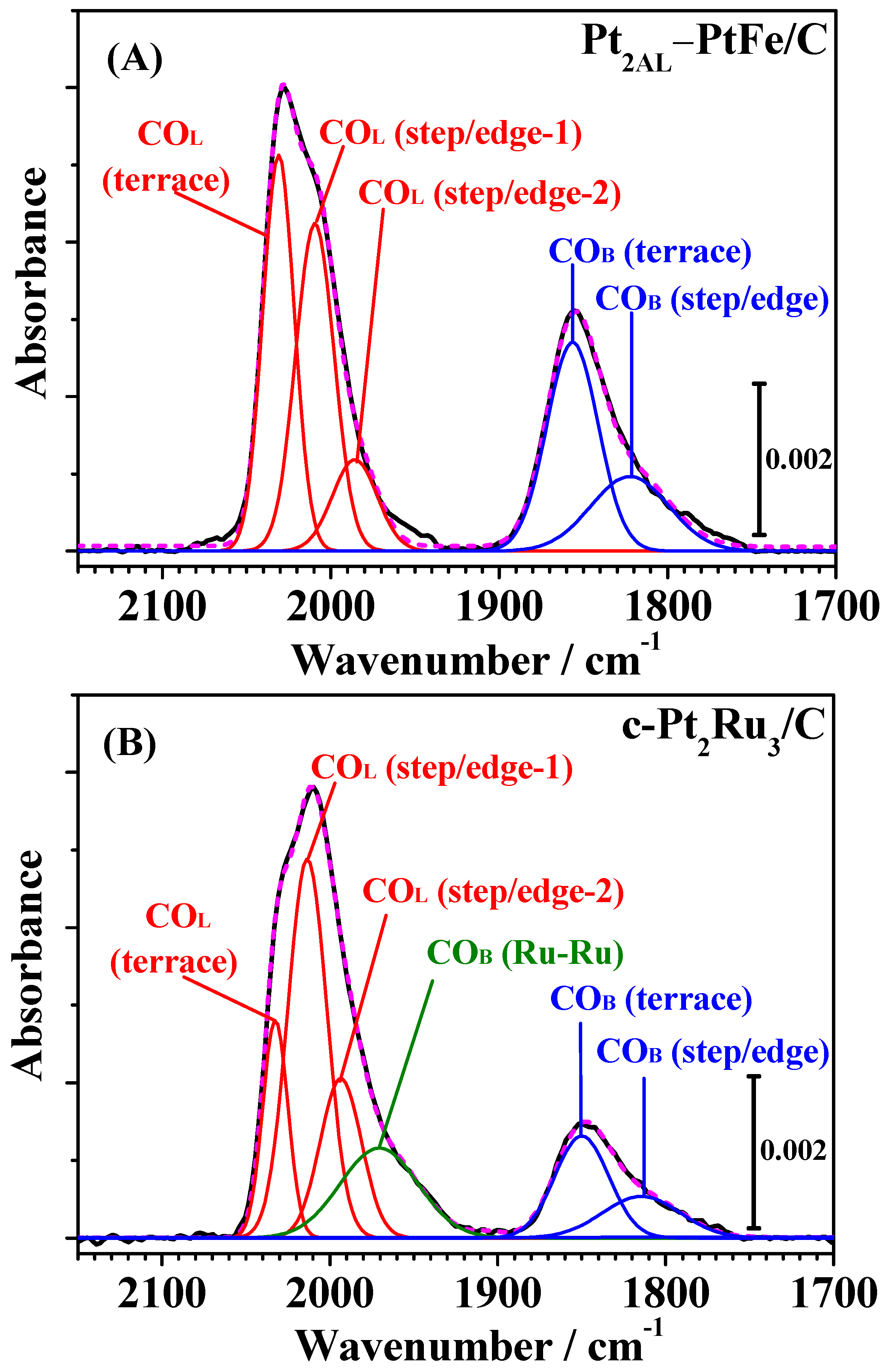
Then, these bands were deconvoluted into several symmetric Gaussian peaks, and curve fitting was carried out for all spectra assuming the full width at half maximum (FWHM) to be constant and allowing the peak maxima (wavenumber) and areas to vary, as described our previous work [
7,
12,
13]. As shown in
Figure 5, the FTIR spectra on Pt
2AL–PtFe/C and c-Pt
2Ru
3/C were successfully deconvoluted into five and six components, respectively, with close correspondence between the fitted and measured spectra (see also
Figure S2 in the Supplementary Materials, as another example of deconvolution of FTIR spectra measured after 1 h). The values of peak wavenumber and FWHM of all peaks are summarized in
Table S1 in the Supplementary Materials. The CO
L band on c-Pt
2Ru
3/C was deconvoluted into three components, i.e., CO
L on Pt terrace sites (CO
L, terrace, 2031 cm
−1) and two types of CO
L, on Pt step/edge sites (CO
L, step/edge-1, 2011 cm
−1 and CO
L, step/edge-2, 1993 cm
−1). These peak wavenumbers were very close to those previously reported for c-Pt
2Ru
3/C (same composition, but different lot number) measured at 25 °C [
13]. The values of peak wavenumbers for the three types of CO
L on Pt
2AL–PtFe/C were similar to those of c-Pt
2Ru
3/C. However, the ratio of integrated intensities of CO
L components was quite different so that the intensity of CO
L, terrace,
I[CO
L, terrace], on c-Pt
2Ru
3/C was smaller than that for Pt
2AL–PtFe/C. With respect to the ratio
I[CO
L, step/edge] to
I[CO
L, terrace], the number of atoms on the terraces and step/edges was calculated, based on a cuboctahedral model, for simplicity, for the fcc nanoparticles Pt
2AL–PtFe with
d = 2.9 nm and Pt
2Ru
3 with
d = 3.5 nm. The calculation method [
17,
18] is shown in
Section S1 in the Supplementary Materials. The number ratio of atoms at the step/edge to those at the terrace for Pt
2AL–PtFe/C was calculated to be 57% (see in
Table S2), which is in accord with the percentage of
I[CO
L, step/edge]/
I[CO
L, terrace] of 57% after CO adsorption for 2 h. In contrast, the value of
I[CO
L, step/edge]/
I[CO
L, terrace] at c-Pt
2Ru
3/C was as large as 73%, although the number ratio of atoms at the step/edge to those at the terrace was estimated to be 45%. Hence, even at θ
CO ≈ 0.99, the adsorption of CO
L at the terrace sites was suppressed on c-Pt
2Ru
3/C. This effect of the suppression of CO
L, terrace will be discussed later.
The bridged CO
ad on Pt-Ru and/or Ru-Ru sites was approximated by a broad single band with a peak at 1967 cm
−1, which will be denoted as CO-Ru. The CO
B(Pt-Pt) band on both catalysts was deconvoluted into two components: CO
B on Pt-Pt pairs on terraces and step/edges. The integrated intensity of CO
B(Pt-Pt), specifically on terraces, on the Pt
2AL–PtFe/C was larger than that on c-Pt
2Ru
3/C, which is ascribed to the fact that two atomic layers of Pt-skin layers were formed on the PtFe alloy. However, it should be noted that the intensity ratio of
I[CO
B(Pt-Pt)s] to
I[CO
Ls] observed on c-Pt
2Ru
3/C in
Figure 3 and
Figure 4 was larger than that reported previously [
13], suggesting that a Pt-rich surface layer was formed on the present c-Pt
2Ru
3/C catalyst, while Ru sites were still present on the surface, because CO-Ru was observed. Thus, we found definite differences in the adsorption state of CO on Pt
2AL–PtFe/C and c-Pt
2Ru
3/C in the CO adsorption experiment using in situ ATR-FTIR.
2.2. FTIR Analysis of Recovery of HOR Activity on CO-Adsorbed Catalysts
It is important to note that the apparently inferior CO-tolerance of the Pt
2AL–PtFe/C catalyst is entirely inconsistent with that evaluated by the CFE [
10]. Specifically, there were three differences in experimental conditions, i.e., temperatures (60 °C in the present work vs. 70 and 90 °C for the CFE experiment), CO concentrations (1% vs. 0.1%), and protocols. In the CFE experiment, CO was adsorbed on the catalysts by flowing 0.1 M HClO
4 solution saturated with 0.1% CO/H
2 for various time intervals, and the HOR activity was evaluated by a hydrodynamic voltammogram in pure H
2-saturated solution, followed by the CO-stripping voltammogram in N
2-purged solution to evaluate θ
CO. This protocol was employed to determine the dependence of HOR activity on θ
CO with a minimum change in θ
CO during the HOR measurement with a slow potential scan rate (1 mV·s
−1). Then, we adopted a similar dynamic change of the atmosphere to the present in situ ATR-FTIR measurements at 60 °C. After CO adsorption by bubbling 1% CO/H
2 for 30 min, pure H
2 was introduced to saturate the electrolyte solution, to remove dissolved CO. Lastly, the θ
CO values on the catalyst were evaluated by measuring the CO-stripping voltammogram.
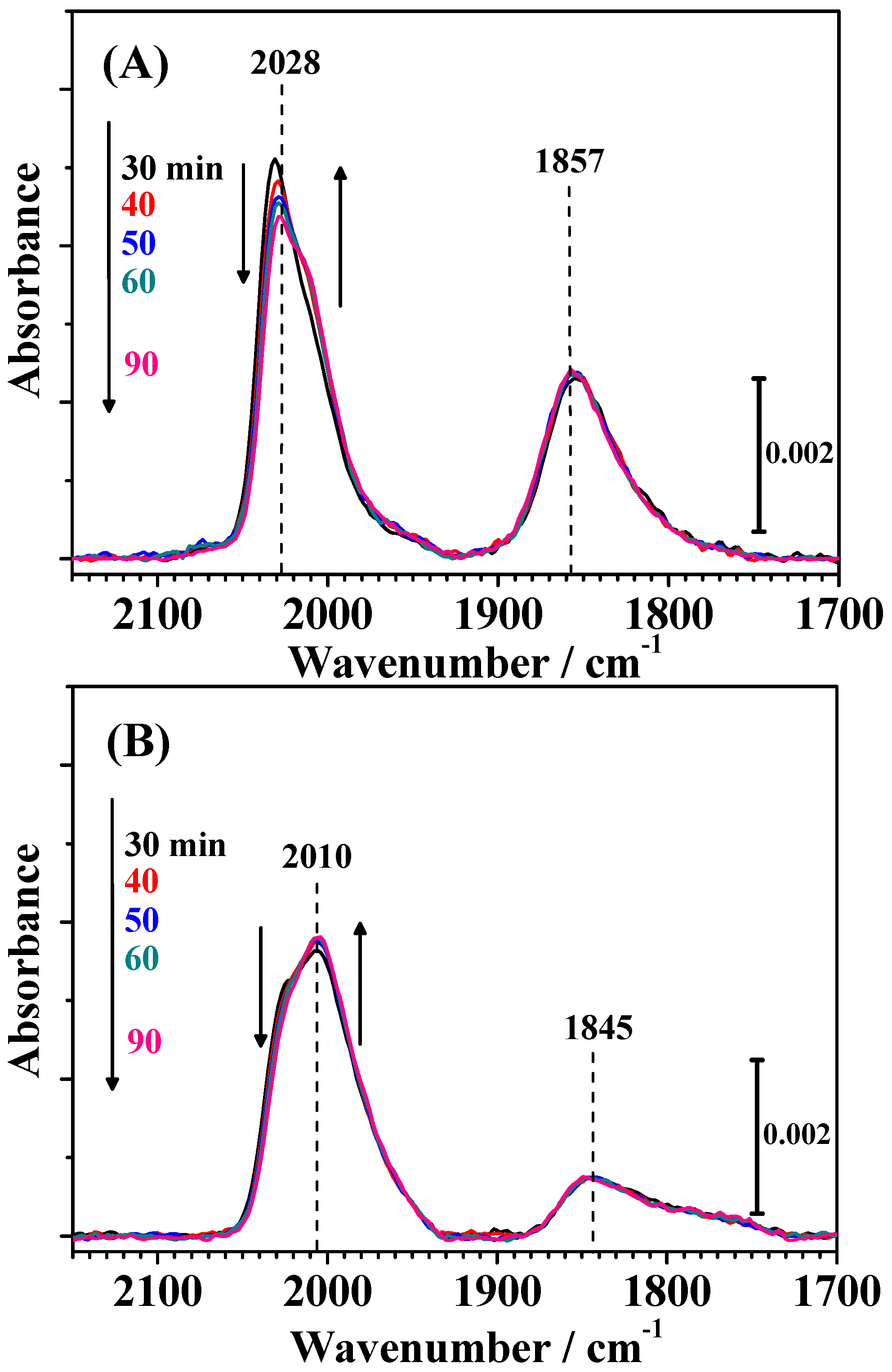
Changes in the IR spectra during CO adsorption on both catalysts for 30 min (shown in
Figure S1 in the Supplementary Materials) were, of course, nearly identical with the corresponding time intervals shown in
Figure 4. As shown in
Figure 6, small changes in the shape of the CO
L band were observed by the introduction of pure H
2 in the solution. The
MA for the HOR on both catalysts decreased by CO adsorption similarly to the case of
Figure 2, but the
MA recovered appreciably just after bubbling pure H
2, as shown in
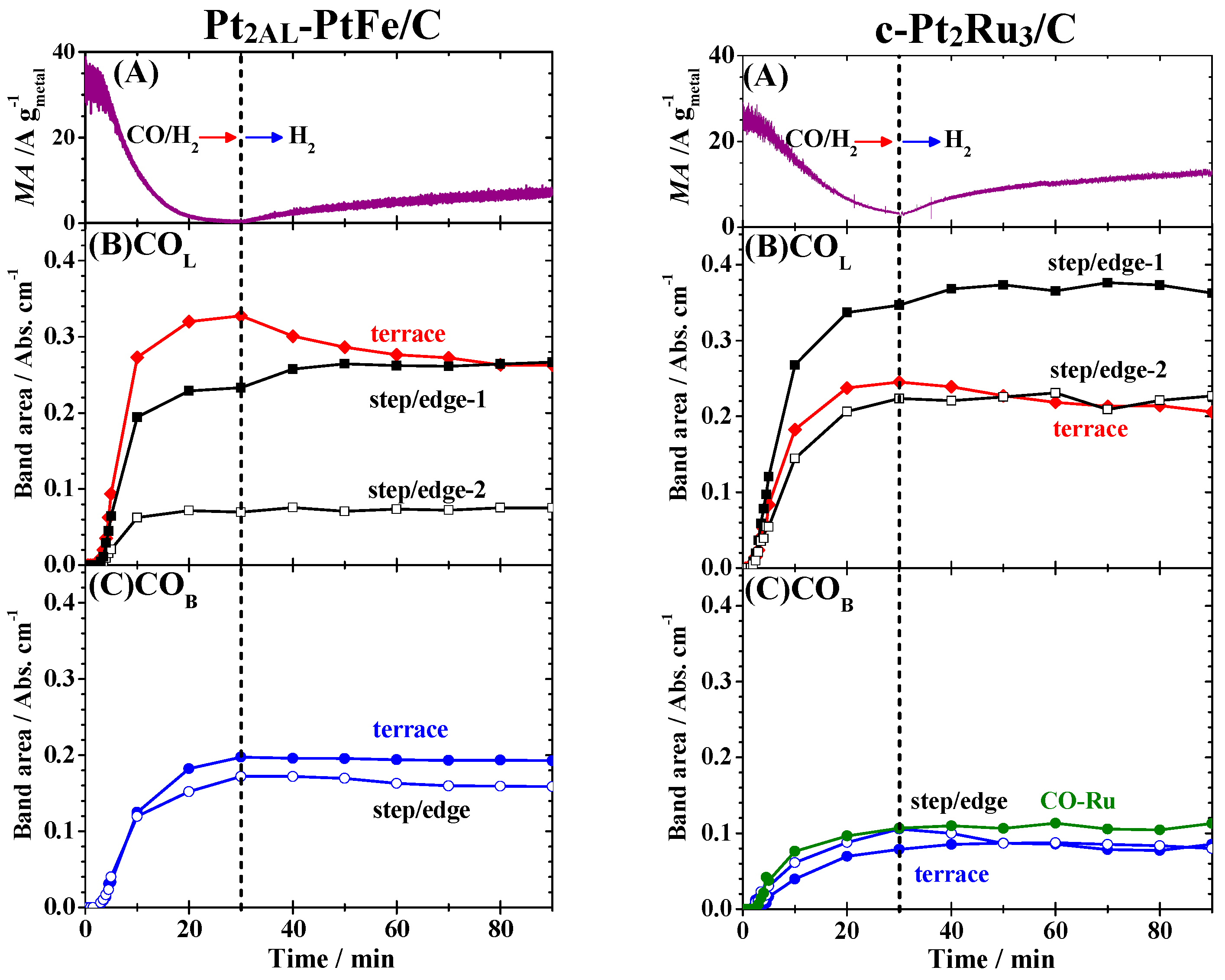
Figure 7A. For example, the
MA on the Pt
2AL–PtFe/C increased from only 1% of the initial value to as high as 22%. To examine the changes in the IR spectra, time courses of integrated intensities of all peaks were plotted in
Figure 7B,C. When the CO-adsorbed Pt
2AL–PtFe/C catalyst was contacted with CO-free pure H
2, the
I[CO
L, terrace] decreased by ca. 20% after 60 min of H
2 bubbling, accompanied by an increase in
I[CO
L, step/edge-1] by ca. 13%. Only a slight decrease in
I[CO
B, step/edge] was seen, while
I[CO
B, terrace] was nearly unchanged. It is striking that the θ
CO evaluated (solid black line in
Figure 3A) was still 0.99 even after H
2 bubbling for 60 min, suggesting that the recovery of the HOR activity can be ascribed to a transfer of CO
ad within the surface, not by a removal of CO
ad from the surface. For the case of c-Pt
2Ru
3/C, the
MA increased from 14% of the initial value to 52% after 60 min of H
2 bubbling, where
I[CO
L, terrace] decreased by ca. 12% with an increase in
I[CO
L, step/edge-1] by 8%. The
I[CO-Ru] was nearly unchanged during the recovery of the
MA. A decrease in
I[CO
B, step/edge] (about 10%) was larger than that for Pt
2AL–PtFe/C. Because the value of θ
CO evaluated was 0.74, in accord with the remaining
MA (24%) at 30 min of CO adsorption, the θ
CO on c-Pt
2Ru
3/C was, with certainty, unchanged even after H
2 bubbling for 60 min, similar to the case for Pt
2AL–PtFe/C.
Here, we discuss a mechanism for the recovery of the HOR activity on CO-adsorbed catalysts by contacting with CO-free pure H
2. It has been reported for Pt
2Ru
3/C catalysts that the presence of a Pt-rich surface overlaid upon Pt-Ru alloy was essential in providing the HOR active sites by weakening the CO adsorption on Pt [
13]. The Pt-rich surface of the present c-Pt
2Ru
3/C satisfies this criterion. In our previous work on the CO-tolerance of Pt
2Ru
3 nanoparticles dispersed on carbon black and Sb-SnO
2, we proposed a possible mechanism that the electronic modification (ligand effect) of Pt
2Ru
3 nanoparticles by the Sb-SnO
2 support gave rise to a weakening of the CO
L, terrace adsorption and suppression of CO
L, step/edge [
7]. In ref. [
19], the Pt(110) surface exhibited the highest catalytic activity for the HOR, in comparison to those for Pt(111) and Pt(100). Truncated octahedral or truncated cuboctohedral Pt-based (fcc) nanoparticles include (111) and (100) terraces, with (110)-like sites at the edges between two (111) facets. In addition, (110) steps exist on the (111) terraces. Both of these types of (110) sites can be HOR-active. Thus, we considered that, in order to improve CO-tolerant HOR activity on the Pt
2Ru
3/Sb-SnO
2 catalyst, lowering the coverage of CO
L, step/edge could be beneficial. However, very recently, we proposed a modified HOR mechanism [
10] in which, after H
2 dissociates at the step/edges, the dissociated H atoms can “spill over” to the (111) terraces, which can accommodate larger numbers of atoms, prior to oxidative desorption:
In this mechanism, H2 adsorbs and spontaneously dissociates at step sites due to stronger adsorption of H2 at these sites, since H2 cannot compete with water adsorption on the terraces. After dissociation, however, the H atoms can compete with water more successfully on the terraces, and, even though the adsorption strength would still be larger at the steps at the same coverage, at high coverage, decreased adsorption strength at the steps might allow H to move to the terraces.
This mechanism was derived to explain the orders of CO-tolerant HOR activities of various catalysts (Pt
2AL–PtFe > Pt
2AL–PtCo > Pt
2AL–PtNi > PtRu > Pt) in the CFE experiments [
10]. Increased HOR activity was considered to correlate with decreased H adsorption strength on (111) terraces. However, the results shown in
Figure 7 can also be well explained by this HOR mechanism. The decrease in the coverage of CO
L, terrace would result in an increased number of active sites for reaction (4) on the terrace, assuming that the dissociation rate of H
2 were maintained at the step/edge, for example, step/edge-2. A decrease in the coverage of CO
B, step/edge would also be beneficial for reactions (1) and (2), because two Pt active sites (Pt-Pt pair site) are formed by the desorption of one CO
B. Furthermore, the mobility of CO
ad would be enhanced with increasing temperature, consistent with our observation of excellent CO-tolerance for Pt
2AL–PtFe/C at 70 and 90 °C. According to our DFT calculations on nearly all of the surfaces studied, we found that at both step edges and terraces, CO adsorption is stronger by approximately 1 eV compared with the adsorption of atomic hydrogen. Hence, as is well known, the CO concentration must be maintained at very low levels in order for H to compete effectively with CO for adsorption sites.
Even though the use of 1% CO was a very challenging condition for both Pt2AL–PtFe/C and c-Pt2Ru3/C, we have, for the first time, observed the recovery of the HOR activity of these CO-adsorbed catalysts (without removal of COad) and clarified the mechanism correlated with the mobility of COad to create HOR active sites. Such an enhanced mobility of COad can be ascribed to a modification of the electronic structure of the Pt2AL skin on PtFe alloy and the Pt-rich layer on PtRu alloy.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




 ) experimental spectrum, (
) experimental spectrum, (  ) sum of all peaks, (
) sum of all peaks, (  ) COL peaks, (
) COL peaks, (  ) CO-Ru peaks, and (
) CO-Ru peaks, and (  ) COB peaks.
) COB peaks.