1. Introduction
The widely used
C-acylation, Friedel–Crafts acylation [
1,
2] and Fries rearrangement [
2] of aromatic compounds are efficient methods that result in satisfactory product yields. The fundamental feature of these reactions is the use of a homogeneous or heterogeneous Lewis or Brønsted acid that catalyzes the reaction of the acylating agent with the corresponding substrates. Conventionally, the Friedel–Crafts acylation of aromatic derivatives with acyl chloride or anhydrides requires at least 1 equiv. of AlCl
3 [
2,
3]. Up to now, an improvement on this reaction is continuing at the time of writing this review. The Fries rearrangement, a typical rearrangement of a phenyl ester to
o- or
p-hydroxyphenyl ketone, has been carried out with a number of esters that differ in their phenolic and carboxylic acid substituent [
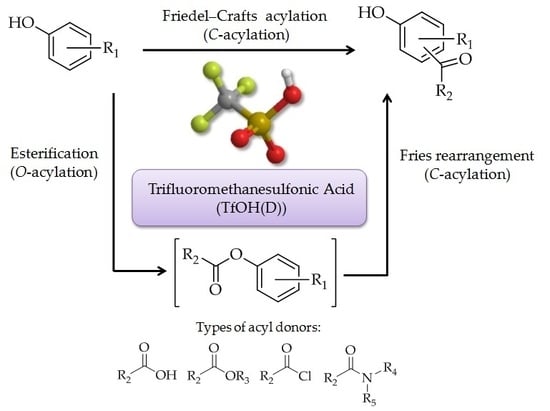
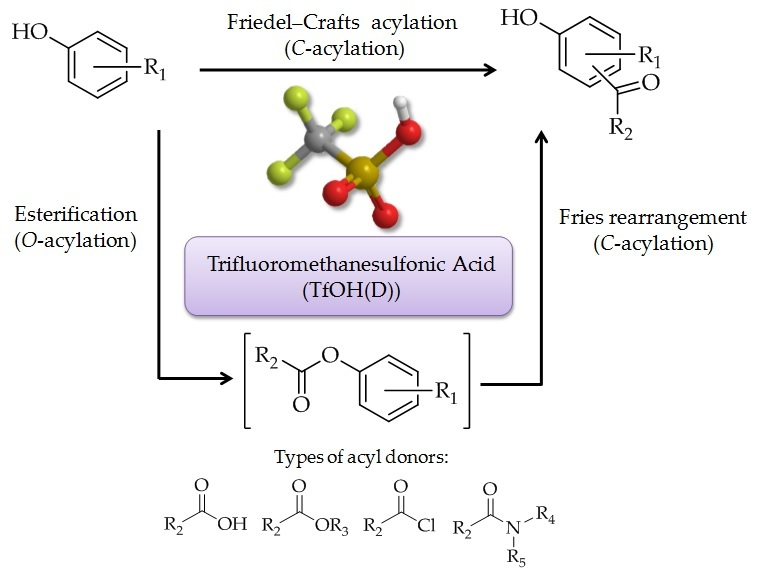
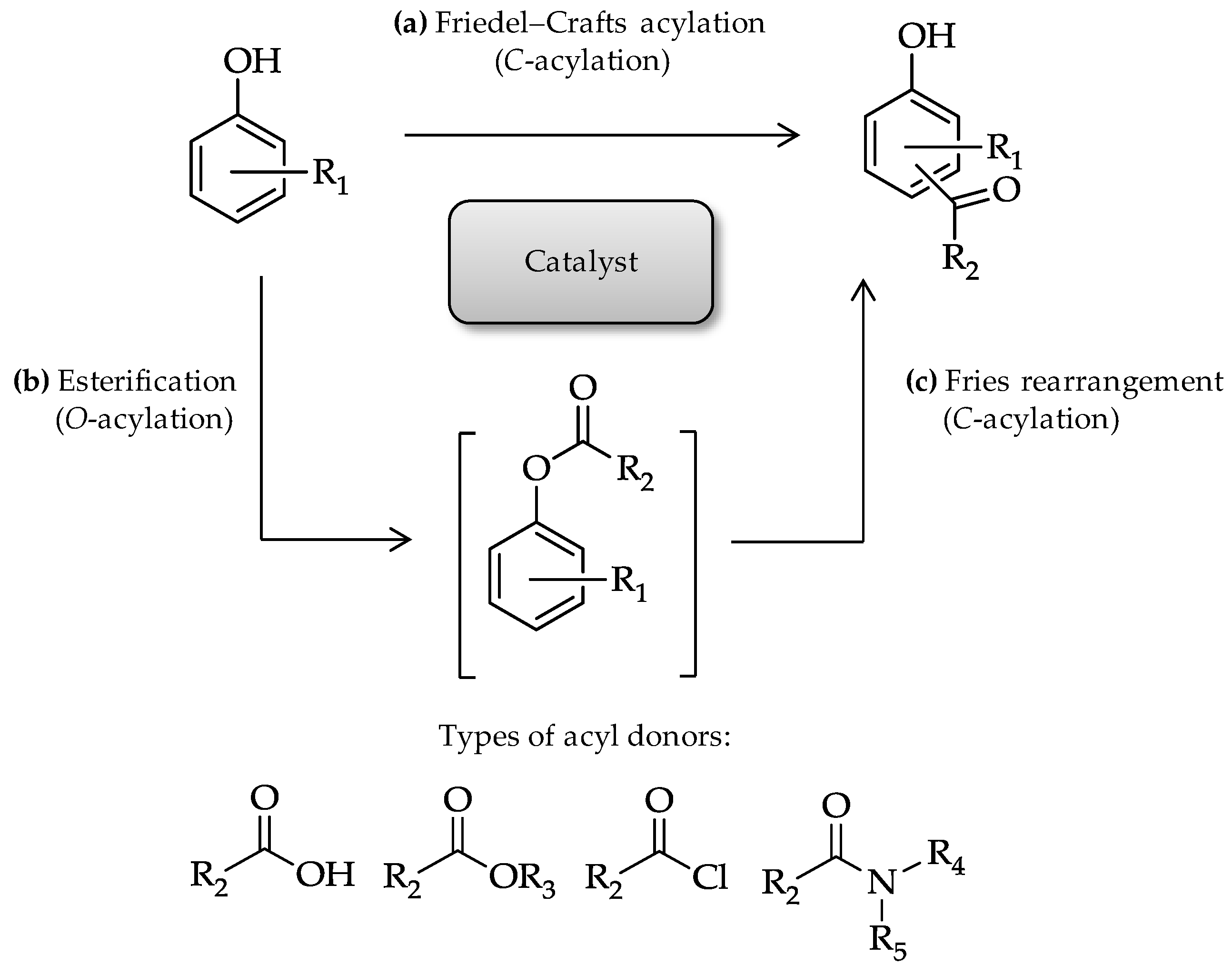
4]. When phenol and phenol derivatives react with acylating reagents in the presence of appropriate catalyst, they can form aryl ketones via
C-acylation of the aromatic ring (
Figure 1a), as well as form phenyl esters via
O-acylation (
Figure 1b). In the reaction, phenyl ester derivatives can also undergo Fries rearrangement (
Figure 1c). Direct phenol
C-acylation and Fries rearrangement usually compete with one another, and are difficult to distinguish from their reaction mechanisms [
5].
Many kinds of acid-promoted acylations have been developed in order to improve their reactivity and selectivity, and some of these activators have been applied to industrial processes [
6,
7]. A comparative study of the catalytic activities of various acids in acylation has demonstrated that trifluoromethanesulfonic acid (triflic acid, TfOH) is far superior to all other Lewis and Brønsted acids tested [
8]. TfOH is a perfluoroalkanesulfonic acid [
9] that is extremely thermostable and highly resistant to decomposition by aqueous bases [
10]. The synthesis of this acid was first reported in 1954 [
8,
10]. Since then, the novel properties of TfOH have been developed for modern chemical applications [
11]; thus, comprehensive discussions of its utilization in various reactions and applications have been published [
5,
8,
9,
11,
12,
13]. From the point of view of TfOH as a catalyst for
C- or
O-acylation, consideration of comprehensive substrates with the reaction condition are needed to achieve optimum conditions. Recently, the general trends for various substrates
C- or
O-acylation catalyzed by TfOH are carried out, not only by the simple arenes with ordinary acyl component utilization, but also with extraordinary substrates to achieve efficient reactions. Therefore, the use of TfOH as a catalyst has broadened. In this review, the catalytic activity of TfOH in
C- or
O-acylation is discussed. The recent progress of TfOH as a promising catalyst for both Friedel–Crafts acylation and the Fries rearrangement, where esterification (
O-acylation) plays an important role to connect these two reactions (from the point of view of phenol and phenol derivatives), is described. Furthermore, the controllable TfOH catalytic system for
C-acylation and/or
O-acylation of various substrates is also discussed in this review.
2. General Features of the TfOH Catalytic System for C-Acylation
Carbon–carbon bond formation in Friedel–Crafts acylation (
Figure 1a) is one of the most important routes for preparing various ketones; especially in the synthesis of aryl ketones. In the case of the acylation of phenol, it has been extensively studied for
o- or
p-hydroxyphenyl ketones synthesis and since it employs more readily available and less expensive raw materials, direct Friedel–Crafts
C-acylation might offer a convenient approach. Generally, the Friedel–Crafts acylation is carried out using acylating reagents such as acyl chlorides, carboxylic anhydrides or carboxylic acids in the presence of a Lewis or Brønsted acid. TfOH possesses a large negative
H0 value (−14.1) [
9,
14], which is stronger than other Lewis acids. Therefore, it is categorized as a superacid and expected to be an effective catalyst for Friedel–Crafts
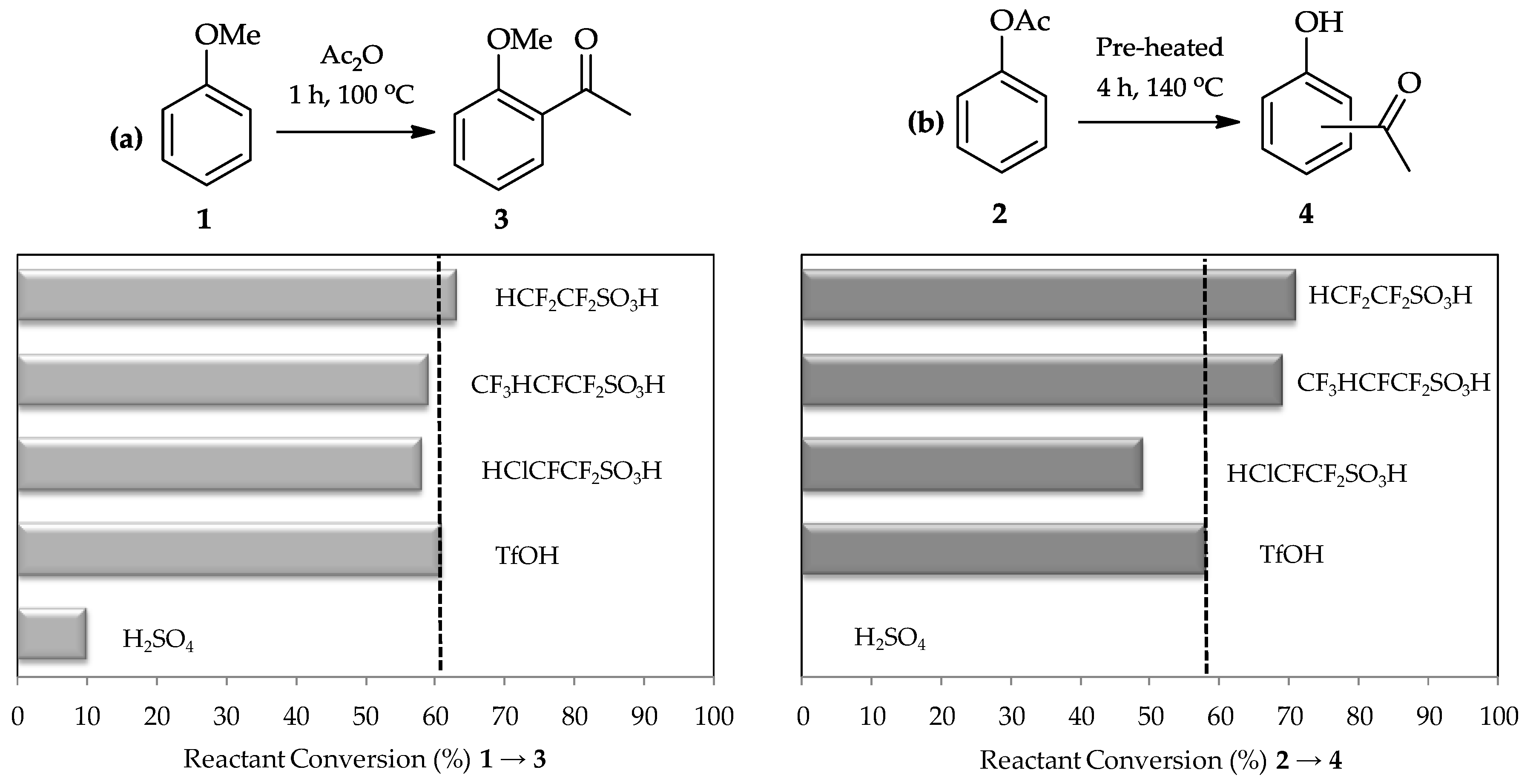
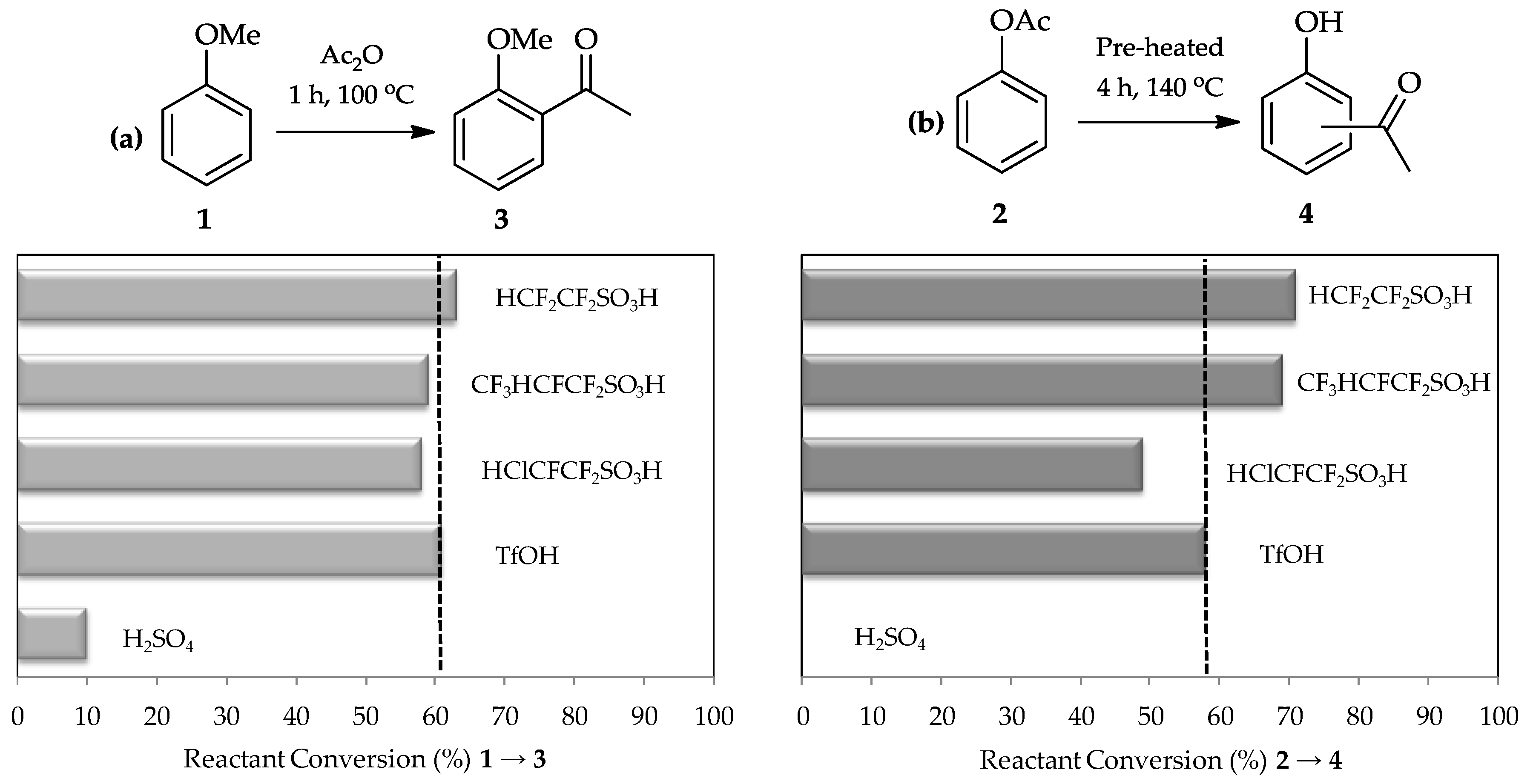
C-acylation. A comparative study using sulfuric acid, TfOH, and three other perfluorinated sulfonic acids synthesized by Harmer et al. [
15] in the acylation of anisole
1 (
Figure 2a) proved this expectation. Moreover, the Fries rearrangement of phenyl acetate
2 (
Figure 2b) also shows high efficiency of TfOH utilization as a catalyst as it is 100 times stronger than sulfuric acid [
13,
16] and is a commercially available reagent which can be readily used for these reactions.
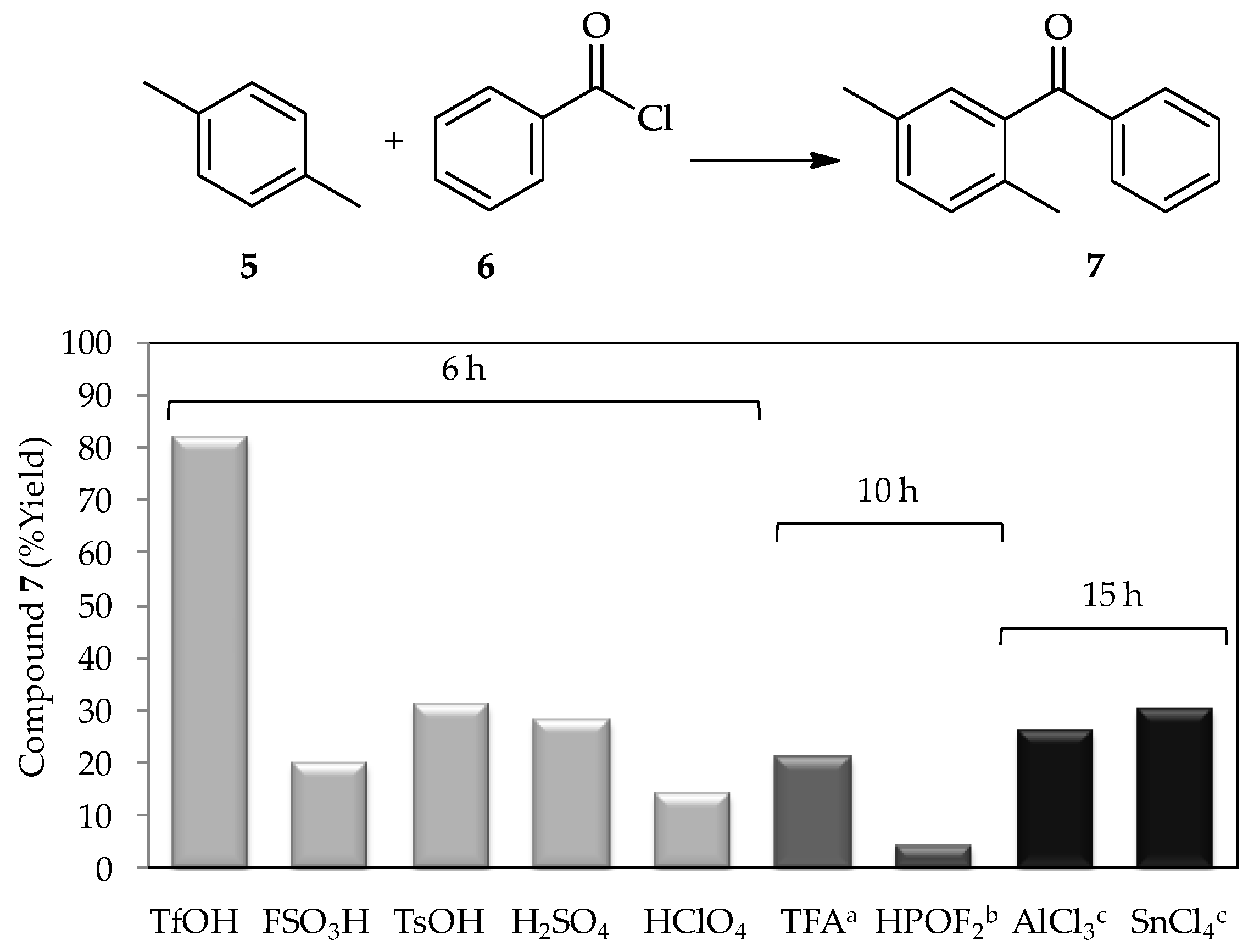
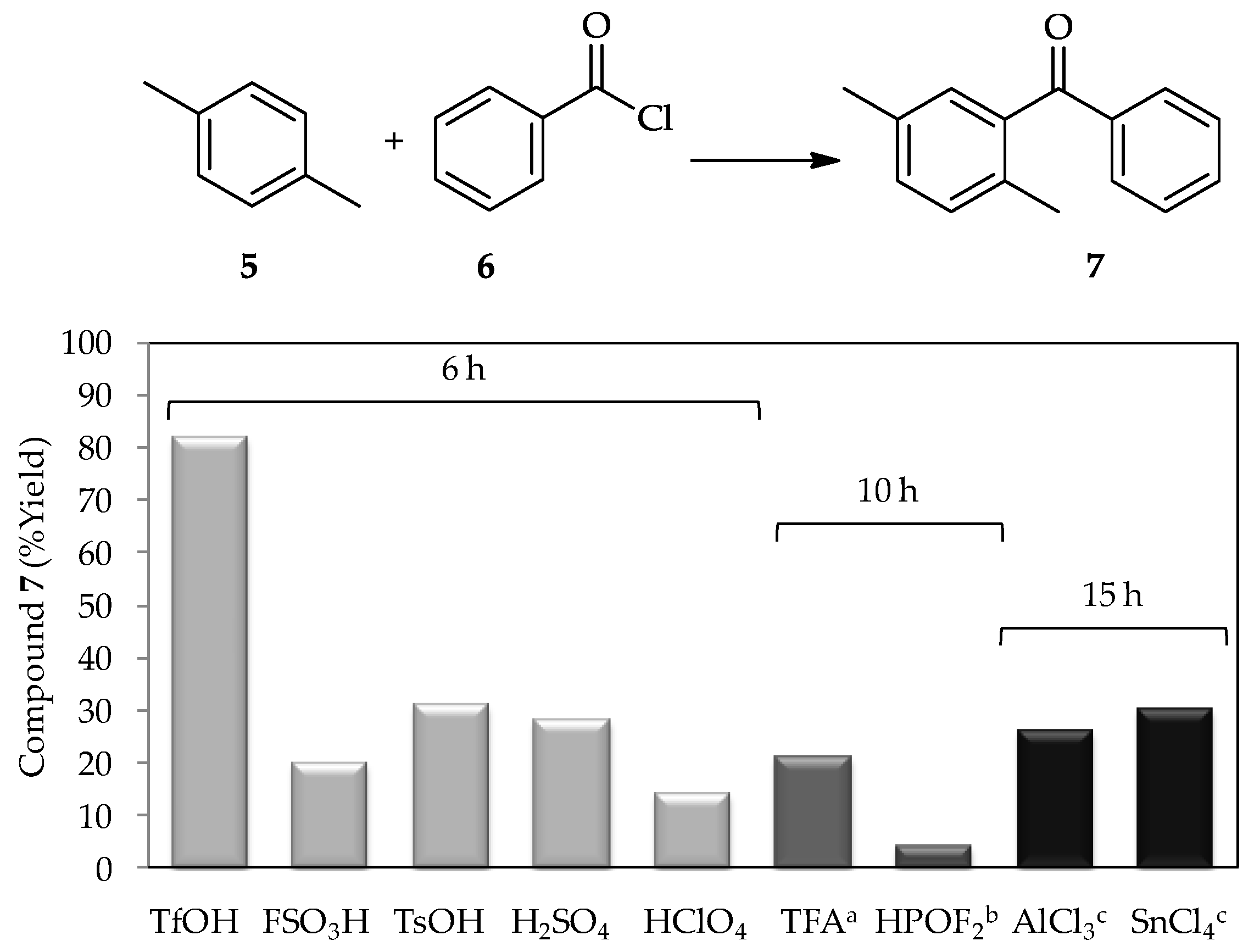
A study in 1972 using a low proportion of TfOH (~1 wt %) for the acylation of
p-xylene
5 with benzoyl chloride
6 [
3] found that this acid produced a higher yield of product
7 in comparison with other tested catalysts, including the conventional catalyst AlCl
3 (
Figure 3). Since then, the catalytic activity of TfOH has been extensively studied. In recent decades, research into TfOH not only focused on determining its optimal proportion for high efficiency acylations, but also in broadening the utilization of vast substrates, which contribute to new synthetic pathways, and the comprehensive study of suitable reaction conditions to optimize its application exploitation. For example, sterically hindered aromatic ketones and carboxylic acids can undergo either protodeacylation or decarboxylation, which are typically accompanied by side reactions in the presence of TfOH (at 110 °C) [
17]. As the direct mechanism of
C-acylation is identical to that of other reactions, there is a need to examine the selection of aromatic compounds used in these reactions [
18]. A summary of the general characteristics of the TfOH catalytic system for
C-acylation in this review is expected to discover excellent findings for TfOH catalytic activity in the future.
2.1. TfOH Catalytic Activity for Various Acyl Donors and Acyl Acceptors
A typical definition of the intermolecular
C-acylation is the addition of acyl groups to aromatic derivatives, which is conducted by an activator. Conventional acylation includes the use of simple arenes (both electron-rich and electron-poor) such as benzene, toluene, and xylene, which act as acyl acceptors, as well as typical acyl donors such as acyl chloride, carboxylic anhydride, esters, and carboxylic acid. The TfOH catalytic system has made the exploration of different acyl donors more feasible for
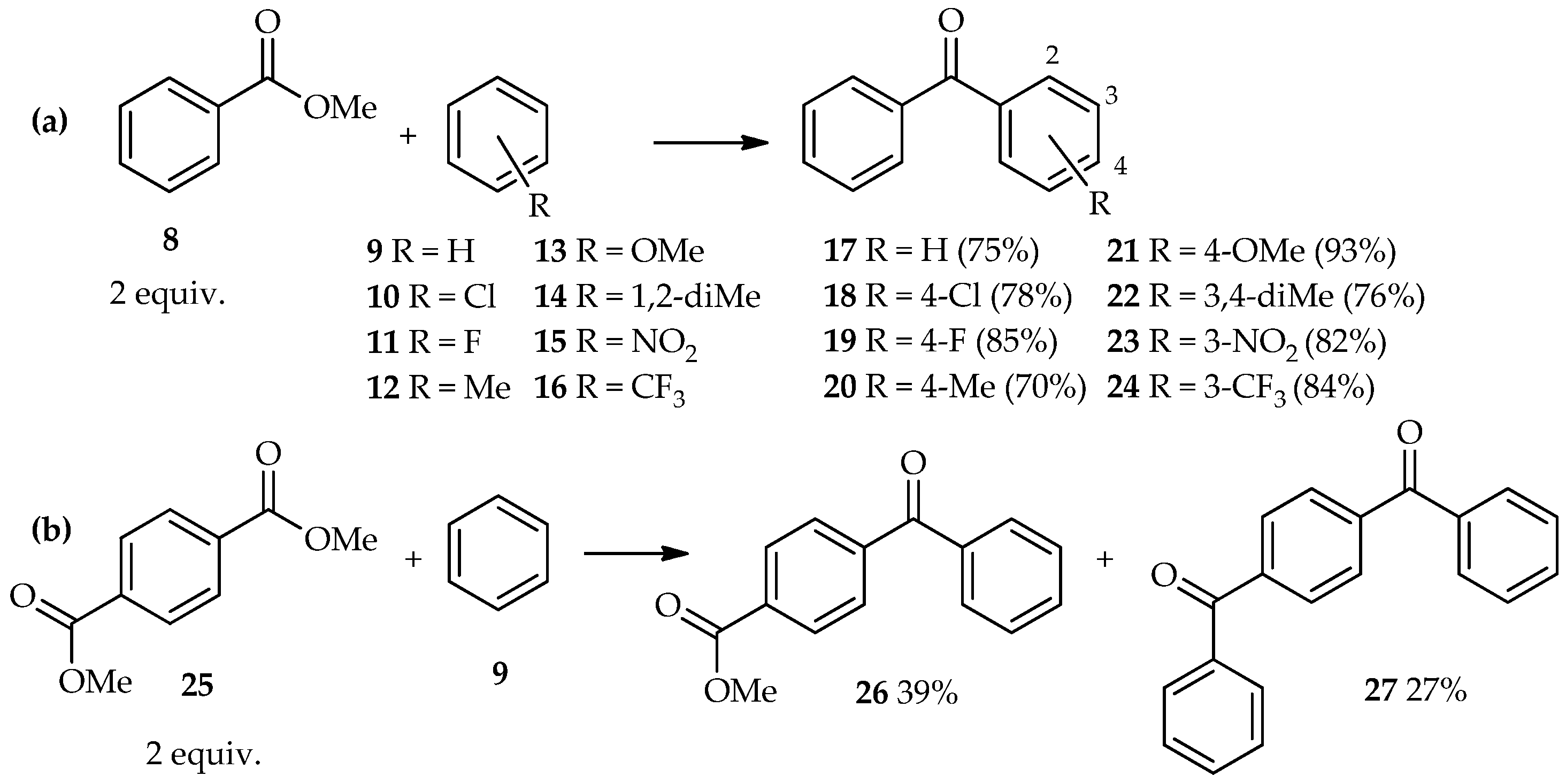
C-acylation, thus increasing the number of potential donors for this reaction. Direct
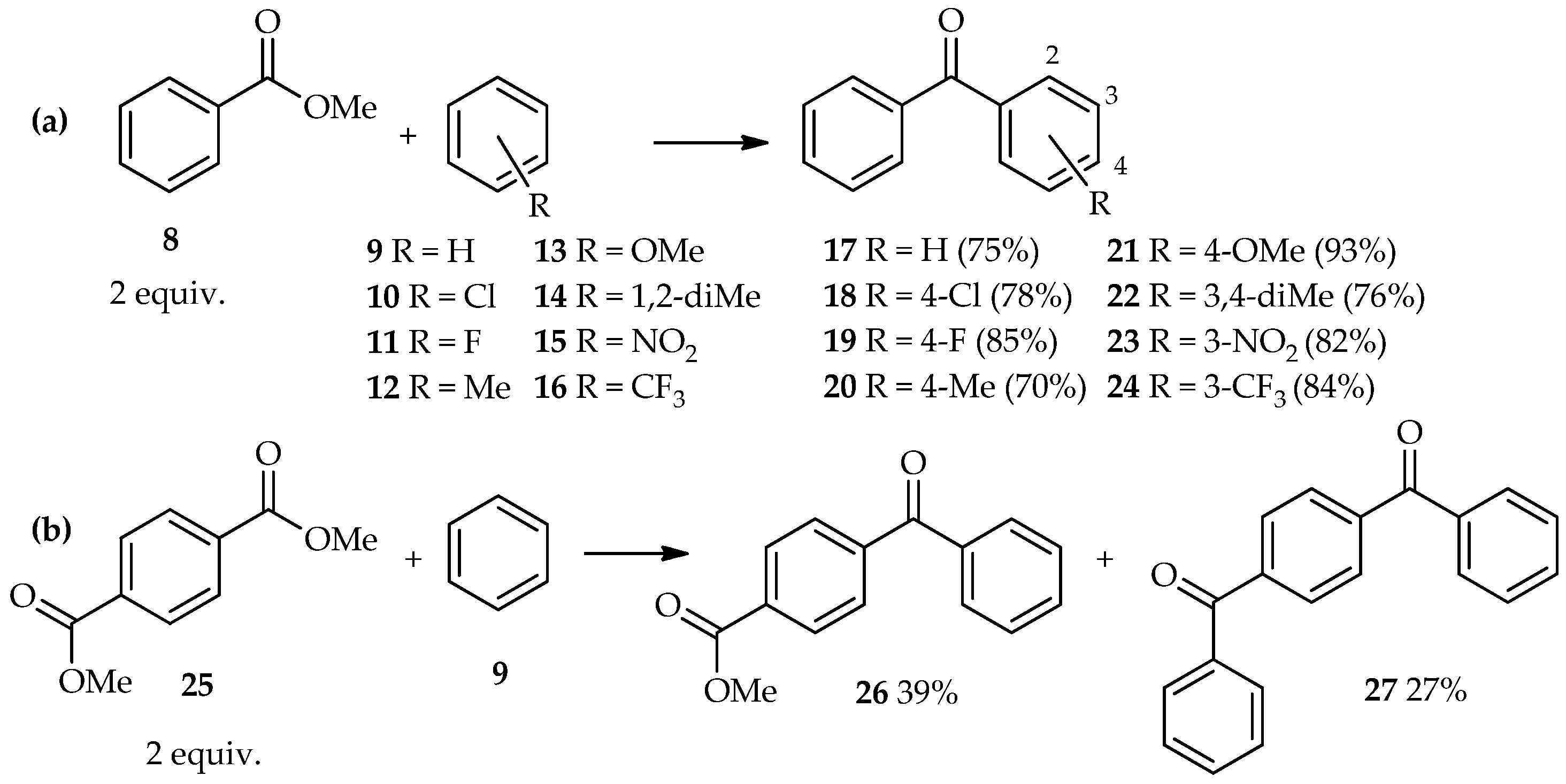
C-acylation of aromatic compounds using benzoic acid esters is extending this exploration series. At 85 °C, excess TfOH (5 equiv.) has been used for direct
C-acylation of methyl benzoate
8 (2 equiv.) and aromatic derivatives
9–
16, thus forming the benzophenone derivatives
17–
24 [
19]. In this reaction, no further electrophilic reaction of benzophenones with the aromatics was observed (
Scheme 1a). Even highly deactivated nitrobenzene
15 and benzotrifluoride
16 acted as acyl acceptors, affording the corresponding three-substituted benzophenones
23 and
24 in high yield. Furthermore, the TfOH system can also catalyze the acylation of benzene
9 with diesters such as 1,4-dimethyl terephthalate
25, which results in a mixture containing the major products 4-benzoyl methyl benzoate
26 and 1,4-dibenzoyl benzene
27, under similar reaction conditions (i.e., reaction for 8 h) (
Scheme 1b).
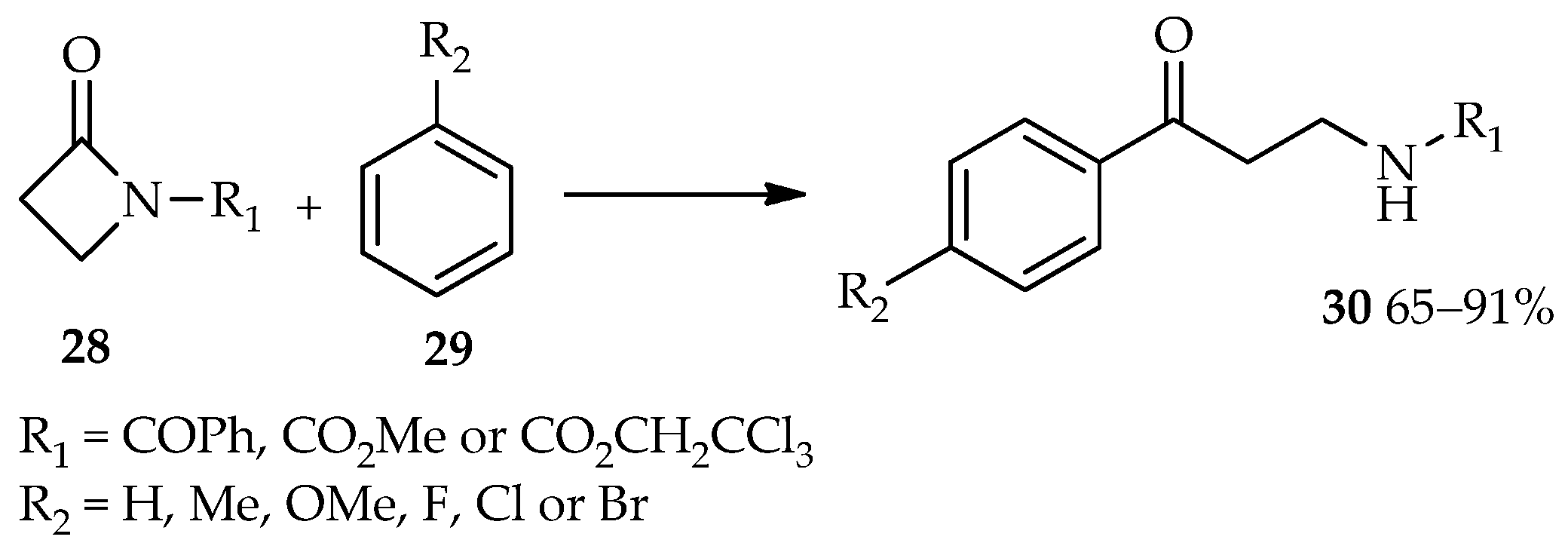
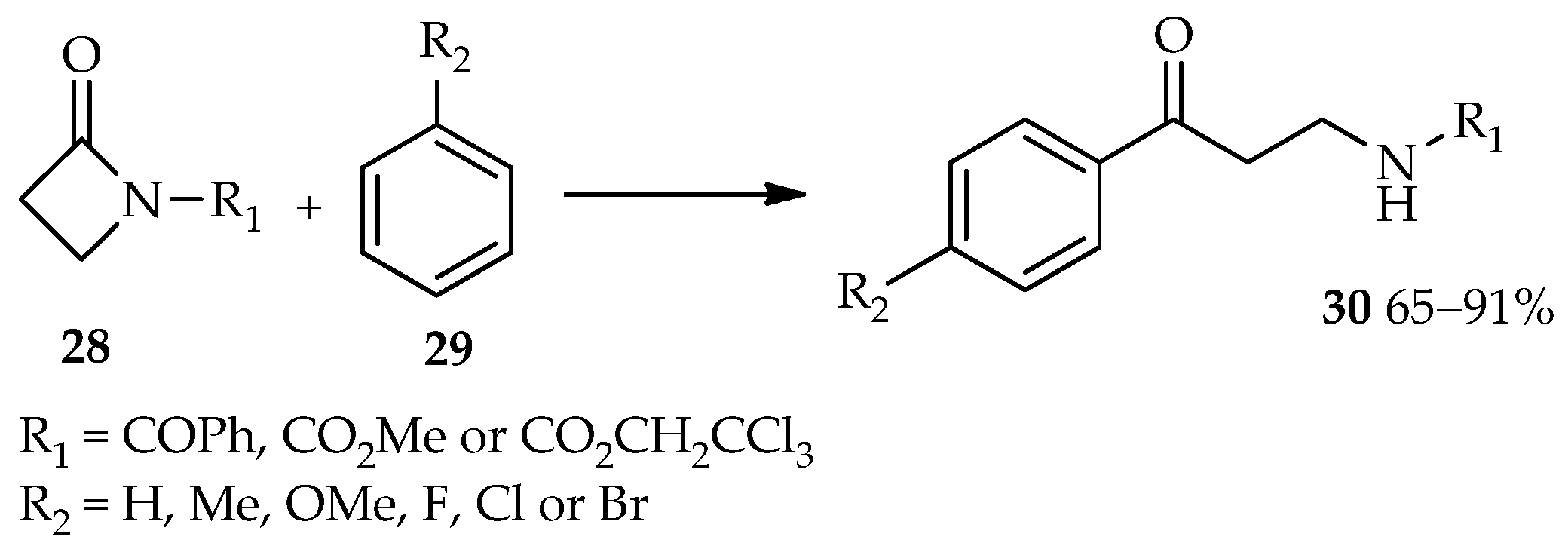
Efficient catalysis of reactions involving esters as acyl donors by using TfOH have led to the use of TfOH for the synthesis of β-amino aryl ketone derivatives from the acyl donors β-lactams (
28) [
20,
21]. The TfOH-catalyzed reaction between β-lactams
28 and arenes
29 produces the desired β-amino aryl ketone derivatives
30 in moderate to high yields (
Scheme 2). Meanwhile, no reaction was observed at room temperature using other common catalysts for the Friedel–Crafts acylation, such as AlCl
3, methanesulfonic acid (MSA), trifluoroacetic acid (TFA), BF
3·OEt
2, or SnCl
2. Reflux conditions did not produce the desired β-amino aryl ketone compounds; therefore, utilization of TfOH for this reaction at room temperature is preferred.
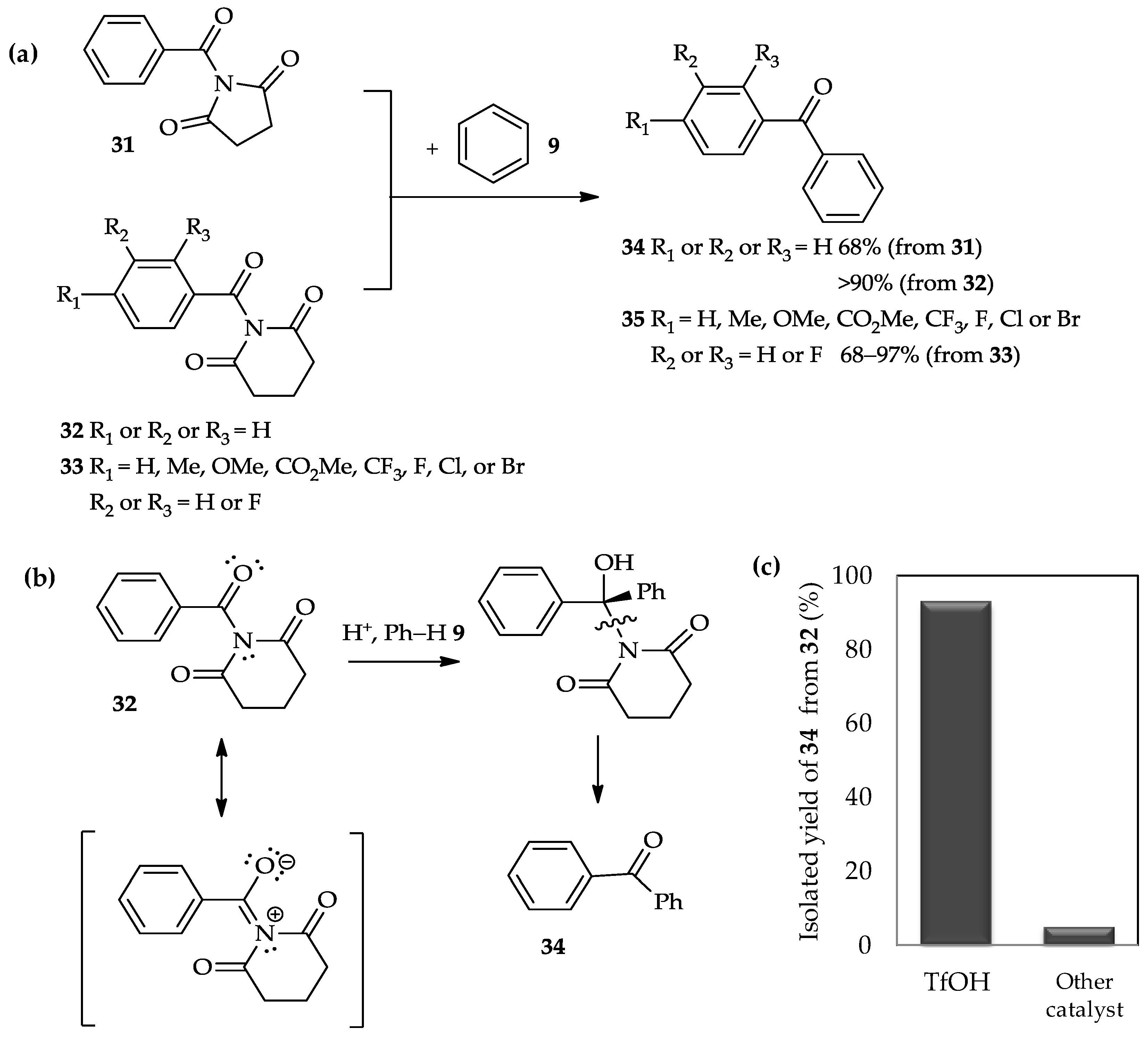
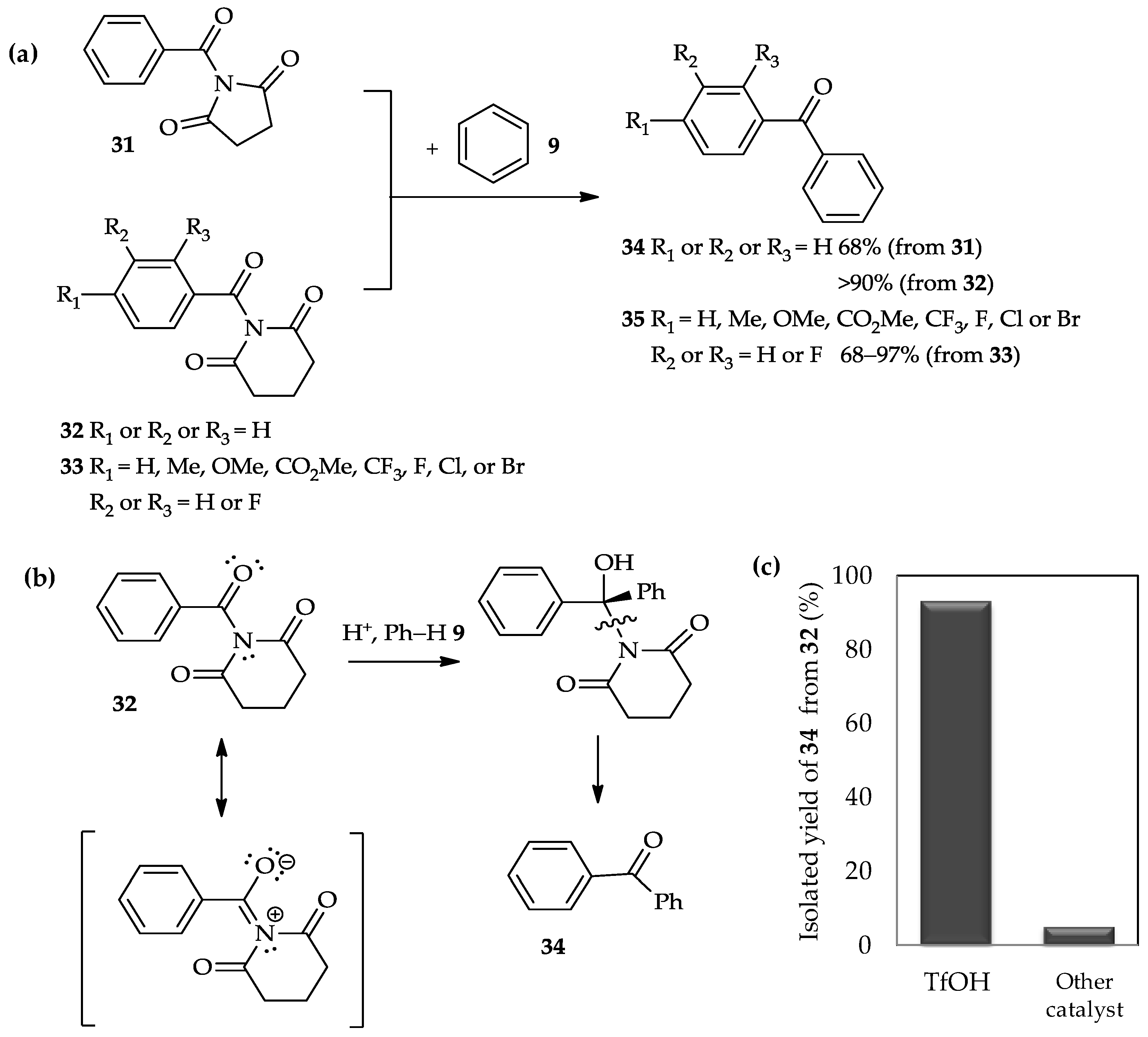
A recent study has reported the use of TfOH for the
C-acylation of arenes with twisted amides
31–
33 (
Figure 4a) [
22]. The acylation of benzene
9 with two different types of amide, i.e., benzoyl 2,5-pyrrolidinedione
31 and benzoyl 2,6-piperidinedione
32, had afforded ketone product
34 in good to excellent yields. Different to previous amides-type of acyl donor, β-lactams
28 (
Scheme 2), that involved the highly reactive acyl carbonium ion intermediate [
21], the acylation of arenes with twisted amides
31–
33 occurred exclusively at the endothermic N–CO bond. The high reactivity of the twisted amide bond might be tuned by N–C(O) bond rotation, and selective
N-protonation of the amide bond under acidic conditions may enhance the potential for nucleophilic addition (
Figure 4b). Therefore, the selection of the acid to promote this reaction is important. For example, acid-catalyzed acylation of benzene
9 with benzoyl 2,6-piperidinedione
32 is more efficient using TfOH, which results in up to 90% yield of ketone product
34 (
Figure 4c), than the other tested catalysts, such as HBF
4, HCl, TFA, BF
3·Et
2O, and TiCl
4. Moreover, the TfOH catalytic system is also suitable for the acylation of benzene with twisted amides
33, in which the aromatic rings carry various substituents (
Figure 4a). The mild condition using TfOH as catalyst is compatible with the wide array of functional groups introduced in twisted amides
33, indicated by up to a 70% yield of ketone product
35.
TfOH-catalyzed aromatic acylation using β-lactams as the acyl donors has broadened the application of TfOH as a catalyst for intramolecular
C-acylation. The typical Fries rearrangement of
N-arylazetidinones
36 can be carried out using TfOH to produce quinolones
37 at excellent yields (
Table 1, entry 1). Most protocols for quinoline-4-one synthesis require multistep procedures, a large amount of catalyst, and have low yields [
23,
24]; to address these limitations, TfOH has been utilized [
25]. The rearrangement of
trans-3-butadienyl-2-azetidinones
38 resulted in quinoline-4-ones
39 in moderate yields (55%–65%;
Table 1, entry 2). Initial protonation of the starting material
38 can lead to the generation of the carbenium ion intermediate, which can undergo the Fries rearrangement and subsequent oxidation to quinoline-4-ones
39. However, using TfOH can simplify the rearrangement process under mild conditions to a one-step procedure.
In
Table 1, the syntheses of 3-spirocycliquinon-4-ones involving intramolecular rearrangement of 3-spirocyclic β-lactam, which are used in the syntheses of important heterocyclic scaffolds, are described. Conditions for this synthesis uses quinolones
40 and
41 which have been optimized by using a combination of 20 mol % TfOH and 30 mol % FeCl
3 (
Table 1, entries 3 and 4) [
26]. Utilization of FeCl
3 without a Brønsted acid led to only 9% yield from quinolones
42 and
43, along with unreacted starting material. Due to the lower requisite amount of Brønsted acid and the lower reaction temperature required to effect high selectivity, TfOH is preferable to other acids such as TFA for these reactions.
In 1986, Roberts and Wells first used the TfOH catalytic system in the acylation of electron-rich metallocene and pyrenes using acetic anhydride
44 as an acyl donor (
Table 2, entries 1 and 2) [
27]. Treatment of excess acetic anhydride
44 (~6 equiv.) with 1 equiv. of TfOH at room temperature improved the acylation of ferrocenes
45 and
46 in comparison with the reaction using approximately 1 equiv. of the conventional catalyst AlCl
3 at room temperature [
28] or at ~50 °C [
29]. This system has been extended for the use of a limited number of possible acylation positions for metallocenes
47 and
48 [
30]. Monosubstitution of an acyl group at the two-position (
Table 2, entries 3 and 4) occurs with the TfOH catalytic system, in contrast to the disubstitution achieved with AlCl
3. Phenyl esters
49 and
50 have also been used as acylating agents for metallocene (
45,
46, and
51) and pyrene (
52) (
Table 2, entries 5–8). The substituted aromatic derivative of phenyl
p-methoxybenzoic acid (
49), which is more electron-rich than its corresponding phenyl benzoate (
50), had higher reactivity in the acylation of both ferrocene
51 and pyrene
52 (
Table 2, entries 5 and 8).
The application of functionalized or biologically important acids or their derivatives from the Friedel–Crafts reaction has been carried out by utilizing the so-called active esters. These esters (
53 and
54) are readily accessible, stable, and exhibit high electrophilic reactivity toward amino groups. The TfOH system has also been used to catalyze the direct acylation of the electron-rich aromatic rings ferrocene
51 and pyrene
52 using tetrafluorophenyl esters
53 and
N-hydroxysuccinimidyl esters
54 [
31]. Compounds
53 were found to be less reactive than the
N-hydroxysuccinimidyl esters
54 (
Table 2, entries 9–12). Despite the use of an excess of these esters, the acylation process was observed to be selective without formation of the diacylated compounds. Nevertheless, acylation using these esters as electron-rich donors seems to be identical with the process induced using TfOH as the catalyst. Another study found that acylation of an electron-rich acceptor using a carboxylic acid requires the combination TfOH and trifluoroacetic anhydride (TFAA) [
32]. This study also claimed that efficient acylation had been achieved due to the high reactivity of the electron-rich acceptors (e.g., ferrocene
51 and pyrene
52, which are, respectively, 3.3 × 10
6 and 220 times more reactive than benzene
9) [
33]. When a stochiometric amount of TFAA is reacted with carboxylic acid, the reaction mixture will form acyl trifluoroacetate intermediates. Next, these reactive electrophiles will react with the aromatic compound through a process that usually requires a strong acid (TFA formed in the reaction between carboxylic acid and TFAA may be not sufficiently strong to complete the reaction [
32]). Therefore, TfOH is needed in order to accelerate the acylation. Furthermore, the TfOH/TFAA catalytic system can also be applied to a vast number of acylation, such as the acylation of metallocenes with alkynoic acids which results in an acylated product that can undergo the impressive azide-alkyne “click” chemistry [
34].
2.2. Use of Neat Reagents and Mild Conditions
The TfOH catalytic system has been of interest because of its two properties that can enhance the reaction’s efficiency: (1) a solvent-free reaction that can be an alternative environment-friendly organic synthetic pathway; (2) a mild conditions, which are advantageous to reactions of valuable heat-sensitive substrates. For many years, the biotin–(strept)avidin system has been widely used for various applications. Biotin is a complex derivative of valeric acid that possesses a high binding affinity to avidin (a protein found in egg white) and streptavidin (a protein found in Streptomyces avidinni) [
35]. Previously, mixed anhydride generated in situ from biotin using the TfOH/TFAA catalytic system was reported to react with electron-rich acyl acceptors (ferrocene, ruthenocene, and pyrene) [
33]. The selected arenes are very reactive in the Friedel–Crafts acylation, thus providing an opportunity for using biotin in these reactions. At present, our laboratory can use neat TfOH for acylation reaction using acid chloride of biotin
55 as an acyl donor. It is estimated that the high reactivity to acylation, which is the key role in this reaction, is not only due to the reactive skeleton of the acyl acceptor, but also the proper selection of the acyl donor. As a result, the acid chloride of biotin
55 readily reacts with the less-electron-rich acyl acceptor
56, forming acylated product
57 in the presence of neat TfOH (
Scheme 3) [
36]. From the point of view of fundamental biological activity with the replacement of an avidin specific bound compound, it suggests that aromatic derivatives of acylated biotin have enough biological activity against the biotin skeleton. Furthermore, the resulting aromatic derivatives of acylated biotin can possibly be explored for functional bioanalysis-tag in the future.
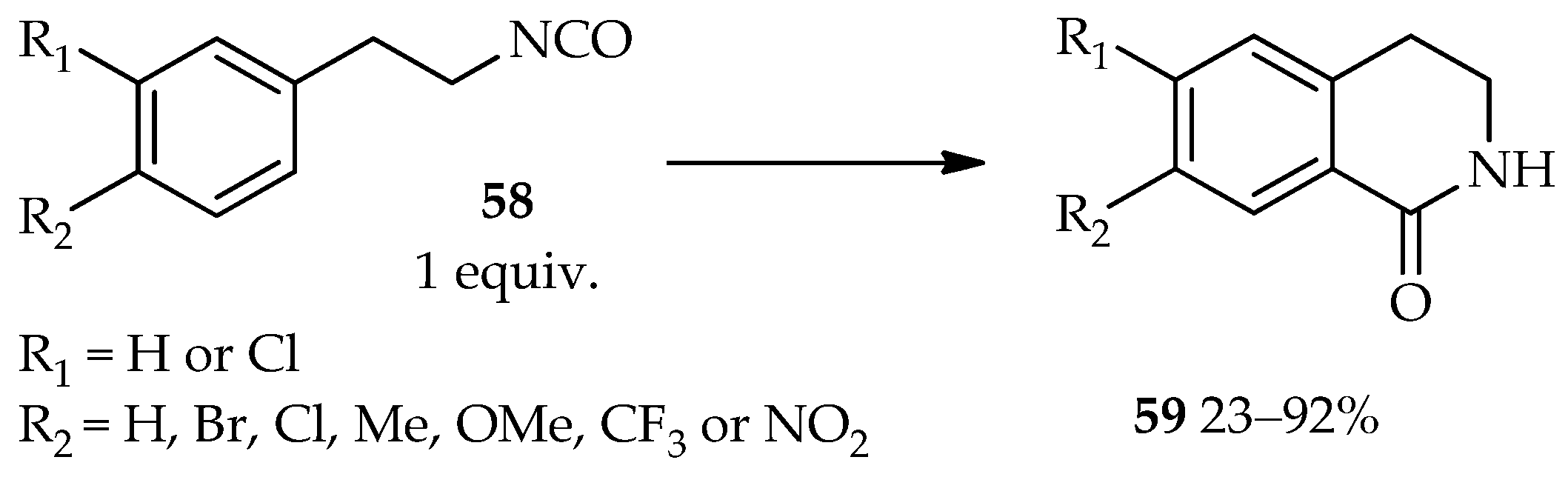
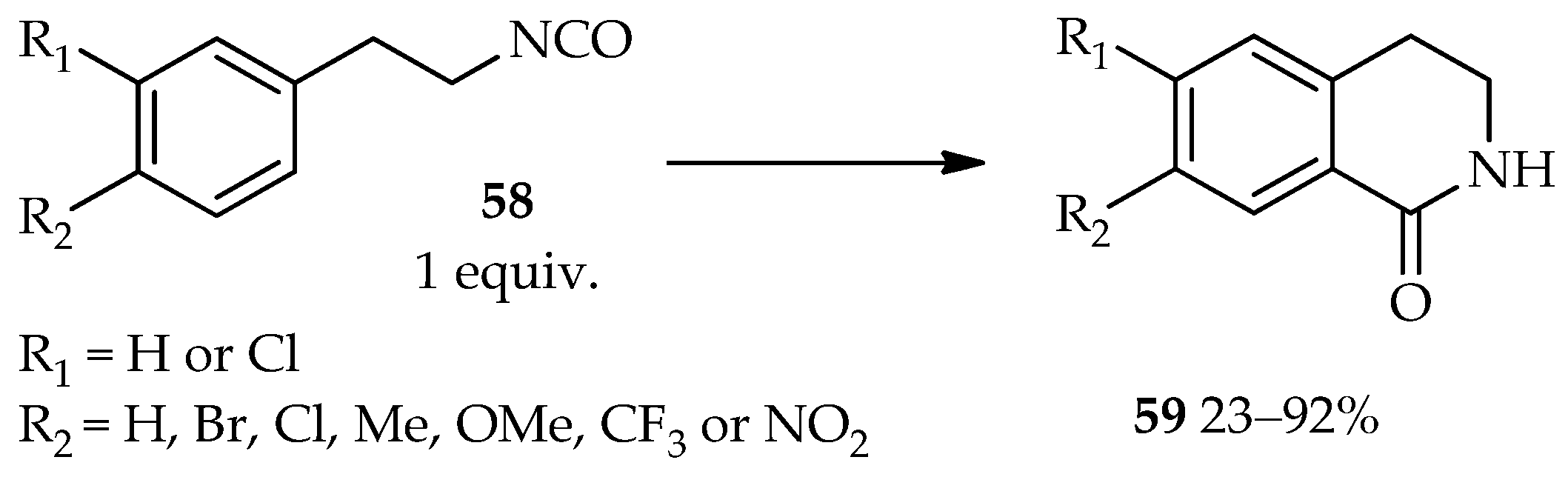
The development of an efficient intramolecular Friedel–Crafts reaction of 1-(2-isocyanatoethyl)benzene derivatives
58 using TfOH as catalyst (
Scheme 4) [
37] demonstrates the vast applications of the catalytic system based on neat TfOH. In contrast to neat TfOH utilization, other catalysts such as AlCl
3, ZnCl
2, TiCl
4, or sulfuric acid require the use of an organic solvent for this intramolecular reaction. The high temperatures and reflux conditions needed for these catalysts still result in the low yield of the desired product
59 and require a long reaction time. In fact, under certain conditions, the AlCl
3 and ZnCl
2 catalytic systems do not lead to the desired product
59.
Metal triflates can promote the acylation of benzene, chlorobenzene or fluorobenzene using benzoyl chloride [
38] and alcohol using benzoic anhydride [
39]. This reaction is known to be catalyzed mainly by TfOH (either added or released at the onset of the reactions); therefore, a metal-free reaction system utilizing neat TfOH is still of interest. The synthesis of aryl keto α-amino acids through straightforward and convenient methods has attracted more attention [
12] because these compounds are convenient intermediates that can be directly converted into the desired α-amino acids.
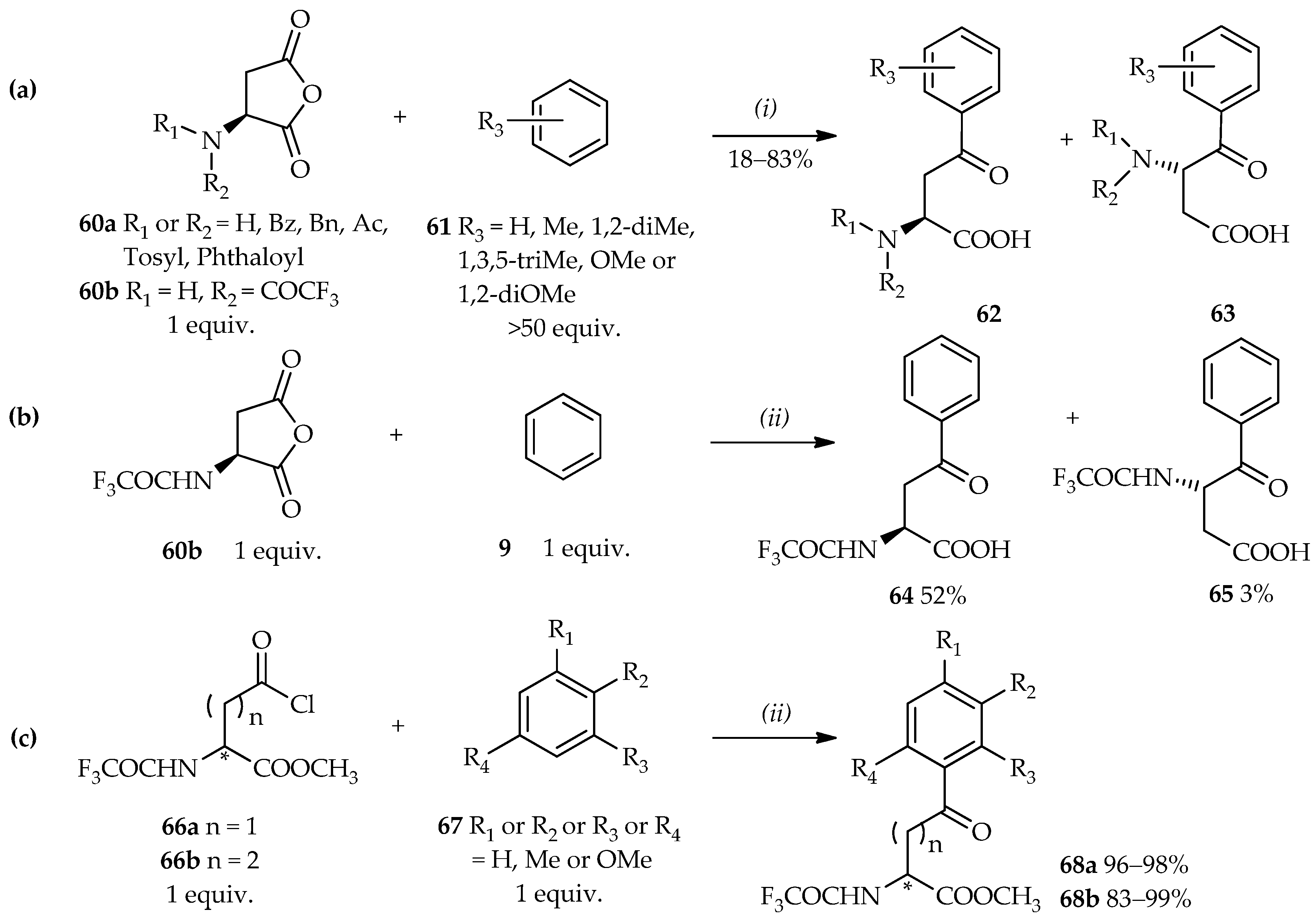
C-acylation involving the aromatic compounds and side chains of carboxylic derivatives is a potential pathway for the synthesis of aryl keto α-amino acids that obviates the requirement of special reagents or precursors. This reaction may also be used for the asymmetric synthesis of α-amino acids. Therefore, due to these purposes, aspartic acid derivatives have become popular acyl donors and have been well studied.
Homophenylalanine or bis-homophenylalanine, which differ in the addition of methylene or ethylene in the side chain of phenylalanine, is a functional biomolecule and the asymmetric synthesis of both compounds is important. One of the potential skeletons used in the synthesis of aryl keto α-amino acids is formed by using aspartic anhydride
60, which can undergo direct
C-acylation to arenes
61 under Friedel–Crafts conditions. When AlCl
3 was used as the catalyst for the acylation of aspartic anhydrides with arenes, common organic solvents such as CH
2Cl
2 [
40,
41] or MeNO
2 [
41,
42] were used under reflux for extended periods. When organic solvent was eliminated, arenes in excess amount were used because of their low solubility to solve some aspartic acid derivatives. For example, >50 equiv. of arenes
61 is needed to react with aspartic anhydride
60 (
Scheme 5a) in order to produce aryl ketone
62 or
63 (wherein the ratio depends on the type of aspartic anhydride derivative used in the reaction) [
43,
44]. Maintaining the ratio of aspartic anhydride
60b to
9 at a 1:1 ratio, as well as using AlCl
3 in CH
2Cl
2 for 12 h have also been shown to cause no reaction and the acyl donor remained precipitated. This result implies that the full dissolution of this system is essential and an excess amount of arenes is required for the reaction. Similarly, no reaction was observed when the conventional catalyst AlCl
3 was replaced with TiCl
4, sulfuric acid, or trifluoromethanesulfonic anhydride (Tf
2O) [
45].
In contrast to other catalysts for the Friedel–Crafts acylation, TfOH can behave both as a catalyst and solvent because of its high dissolving capacity [
12]. Aspartic anhydride derivatives
60b (1 equiv.) can dissolve well in TfOH, and the resulting homogenous system allows fast reaction with benzene
9 (1 equiv.), thus forming
64 (major product) or
65 within only an hour (
Scheme 5b). Moreover, both aspartic acid derivative
66a [
45] and glutamic acid derivative
66b [
46] are also potential acyl donors for direct
C-acylation for the synthesis of homophenylalanine or bis-homophenylalanine. Neat TfOH can catalyze acylation of the aromatic derivatives
67 by the acyl donors
66 (
Scheme 5c) with a good yield of acylated product
68 and a relatively short reaction time. Furthermore, this reaction is environmentally friendly since it does not use excess reagent. In the homogeneous reaction system, acyl donors
66 can undergo acylation smoothly without any hydrolysis and retain its enantiomer skeleton even after the reaction. TfOH acts as a catalyst and solvent, thus enhancing the effectiveness and efficiency of synthesis of the aryl keto α-amino acids by direct
C-acylation.
Photoaffinity labeling is a useful biochemical technique for investigating the structural and functional relationships between small biologically active compounds and biomolecules such as proteins (enzymes), RNA, and DNA [
47,
48]. In recent years, our laboratory has developed various photophores such as phenylazide, phenyldiazirine, and benzophenone for use in this type of analysis. One of these potential photophores is directly constructed from the benzophenone moiety on phenylalanine using a Friedel–Crafts reaction. Due to the low solubility of phenylalanine derivatives in organic solvents for direct
C-acylation [
49], neat TfOH was used [
50]. The neat TfOH catalytic system enabled the direct construction of a benzophenone moiety on optically pure phenylalanine via stereocontrolled Friedel–Crafts acylation (
Scheme 6a). When benzoyl chloride
6 and phenylalanine derivatives
69a were used for the reaction system, a complex reaction mixture was observed with a low yield of the desired product
70a. When
N-protected phenylalanine
69b was used to suppress any competing reactions, it was found to increase the product yield without any optical loss (up to 40% yield of
70b was achieved with 98% ee). Here, benzoic anhydride [
51] can also be used for the production of benzophenone derivatives via direct
C-acylation. Reaction of this compound with
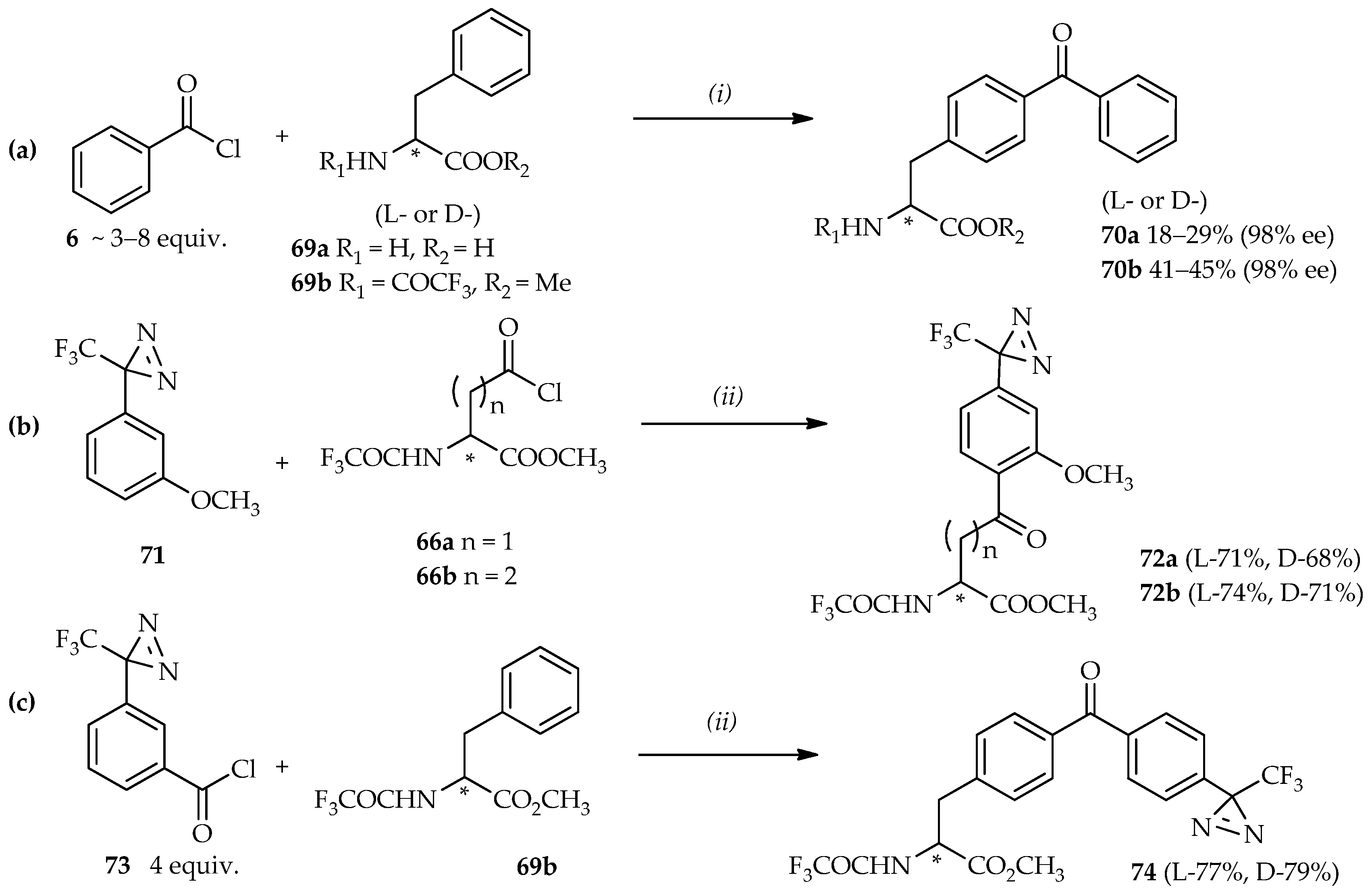
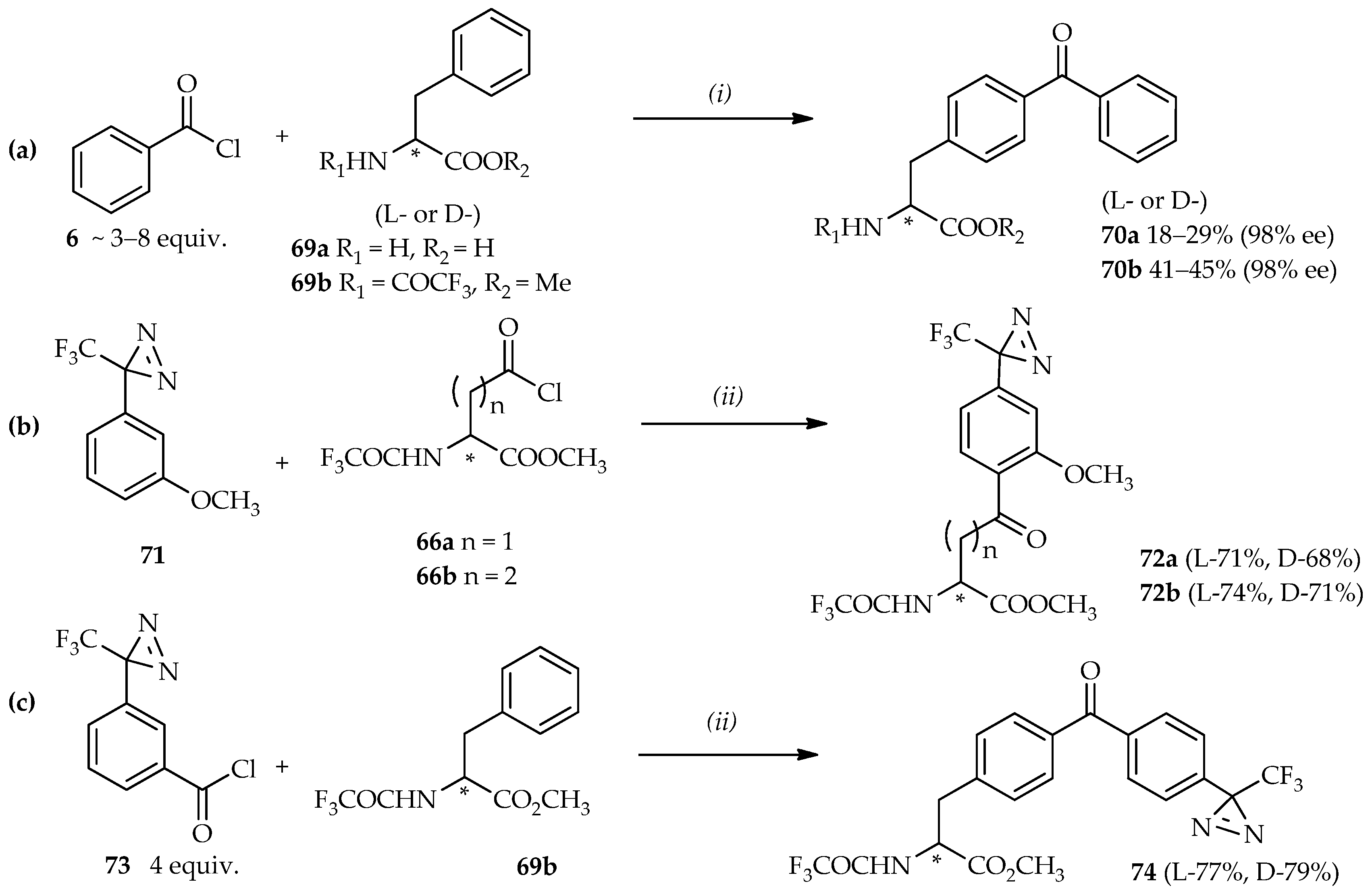
N-protected phenethylamine using neat TfOH resulted in a good yield (86%) at room temperature.
The use of the (3-trifluoromethyl)phenyldiazirinyl (TPD) group as a photoreactive group in photoaffinity labeling confers selectivity for probe activation without damaging the peptides and proteins under radiation of 350 nm wavelength [
48,
52,
53]. The strategy for the synthesis of homophenylalanine or bis-homophenylalanine containing TPD moiety starts with the introduction of a TPD moiety in anisole
71 into an aspartic acid analogue
66a [
48] or a glutamic acid analogue
66b [
46] (
Scheme 6b). In this reaction, high temperatures can result in the decomposition of the starting material
71 due to the thermolability of TPD under strongly acidic conditions [
50,
54]. A temperature of 0 °C is optimal for synthesis using these valuable heat-sensitive aromatic compounds. Furthermore, the reaction between
71 and
66a did not proceed when neat TiCl
4 was used, whereas utilization of TfOH afforded the acylated product
72a up to a 68% yield. The acylated products
72 are intermediates that can be reduced and deprotected to produce the photoaffinity labeling probes based on homophenylalanine or bis-homophenylalanine derivatives. The mild conditions offered by TfOH catalysis can also be applied to construct not only one, but also two kinds of photophores to introduce in one probe. The strategy for this synthesis is the direct acylation of benzoyl chloride containing TPD
73 and
N-protected phenylalanine
69b at room temperature (
Scheme 6c). Enhancement of this reaction was evidenced by no decomposition of the diazirine moiety (product
74); and no loss of optical purity was observed after the deprotection of product
74 by NaOH (99% ee).
2.3. Forced Conditions
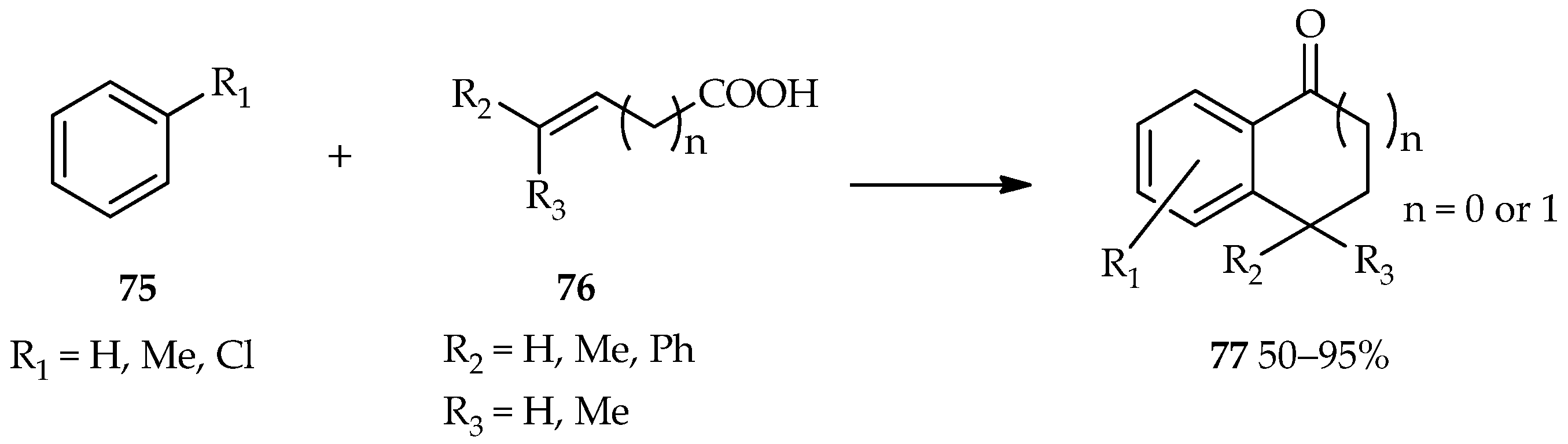
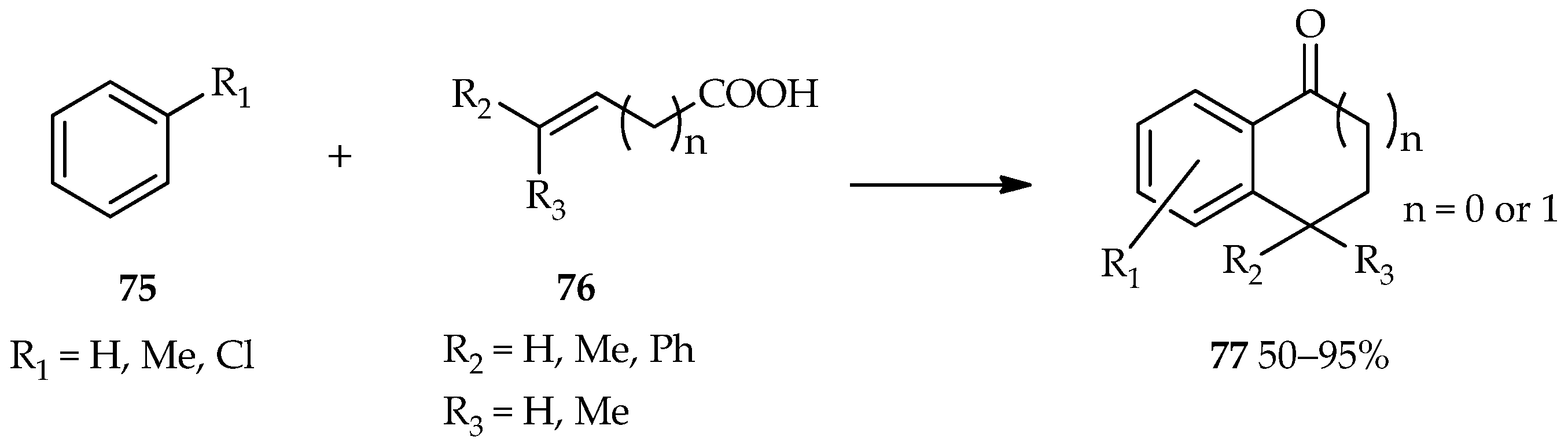
A bifunctional substrate for both the acylation and alkylation of aromatic compounds draws a lot of attention for the proposes of useful cyclic aromatic ketones formation [
55]. The process, known as cyclic acylalkylation, can be formed by the TfOH catalytic system under forced conditions. A high temperature and excess catalyst suppresses the competitive intermolecular reaction to make the cyclization possible. The reaction between aromatics
75 with unsaturated carboxylic acids
76 catalyzed by TfOH has been found to produce 1-indanone (
n = 0) or 1-tetralone (
n = 1) forms of
77 (
Scheme 7) [
55]. The high temperature (at 80 °C) contributes to the induction of the cyclic acylalkylation in this reaction. In comparison, a low temperature (at 20 °C) favors the multiple terminals acylation of the alkyne moiety and carboxylic moiety of unsaturated carboxylic acid
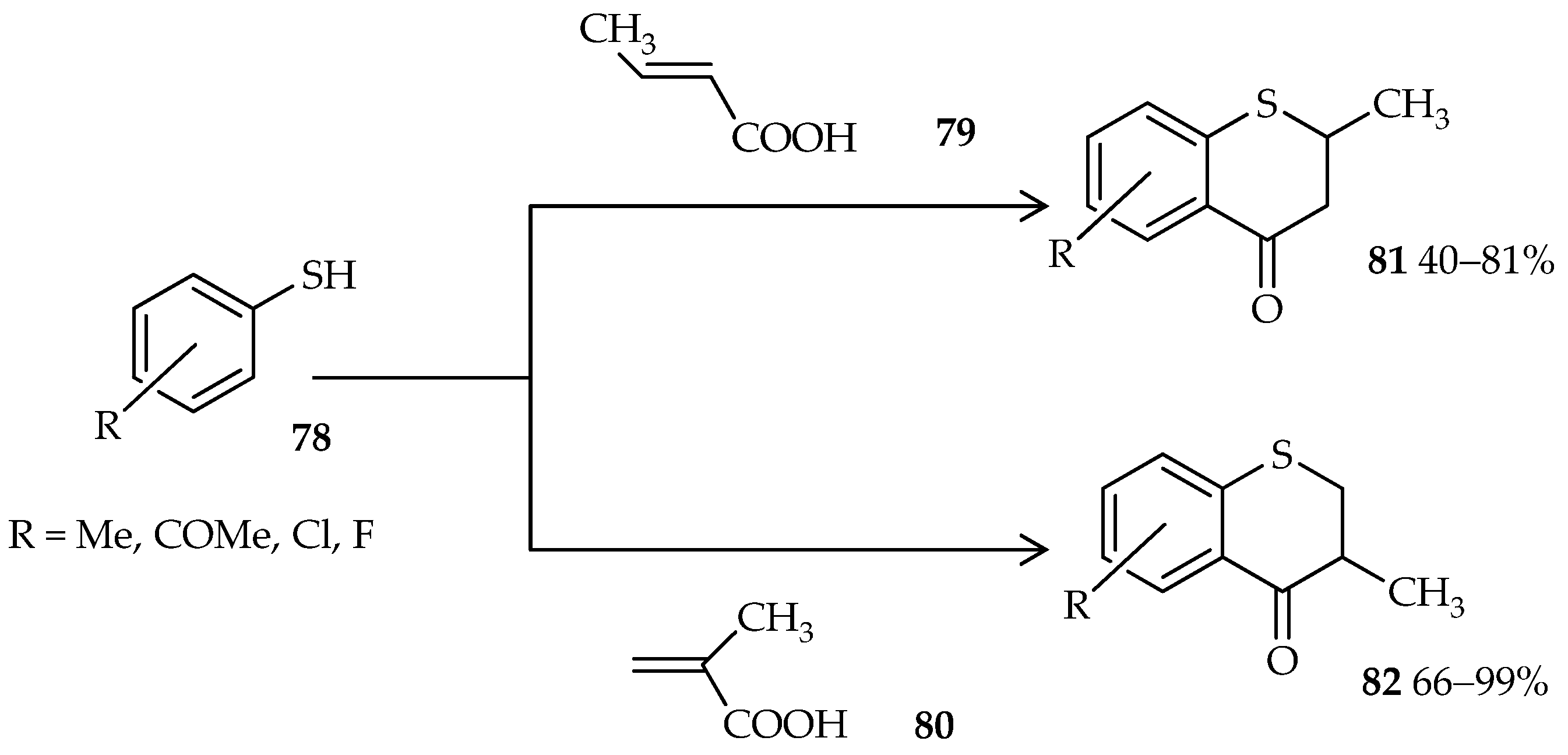
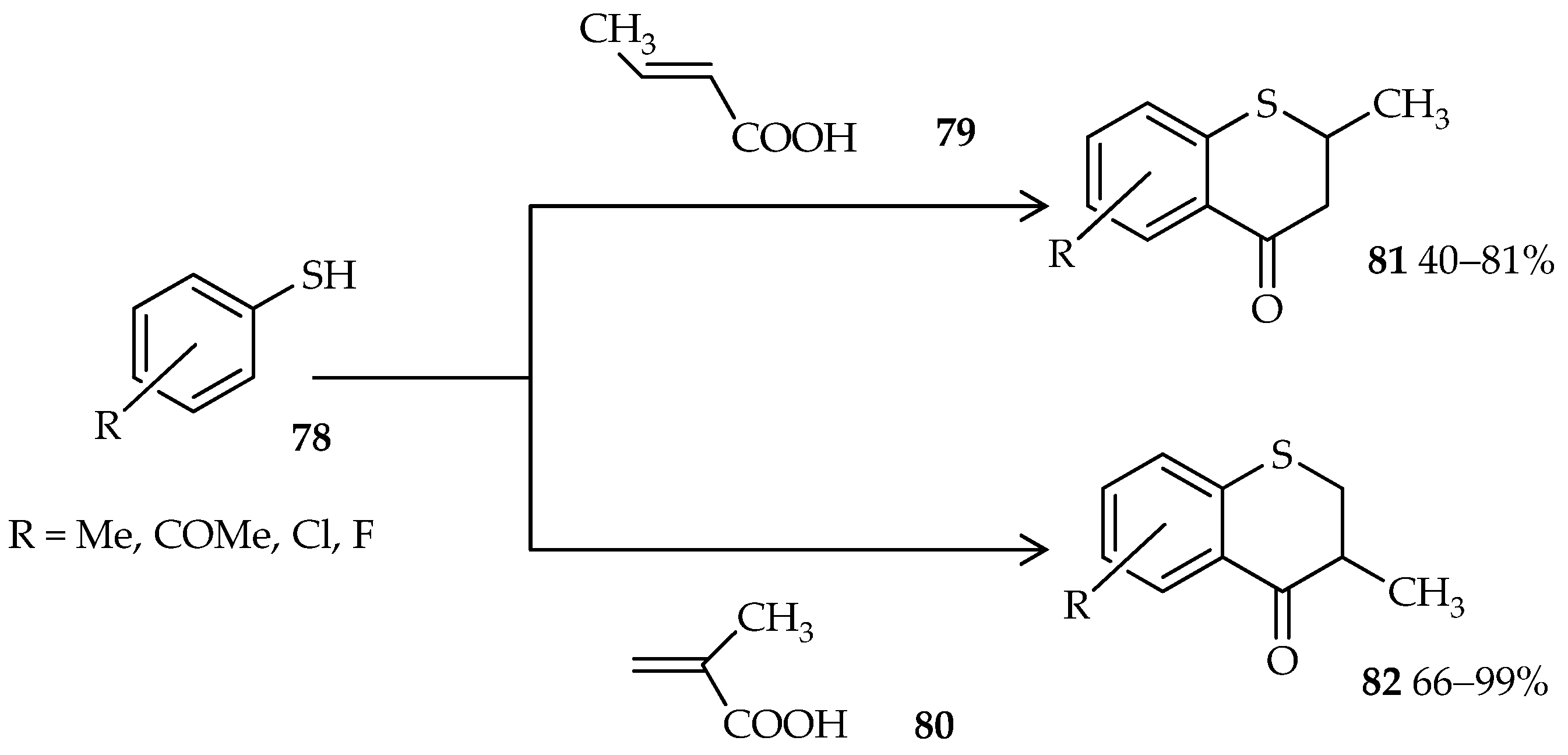
76. Moreover, thiochroman-4-ones, which are valuable synthons and important precursors in organic syntheses, are generally synthesized via a multistep reaction; a one-pot approach was uncovered through the use of TfOH [
56]. One-pot cyclic acylalkylation of thiophenols
78 and crotonic or methacrylic acid (
79 or
80) under microwave irradiation and in the presence of excess TfOH gives good yields of thiochroman-4-ones
81 and
82 (
Scheme 8).
3. General Features of the TfOH Catalytic Systems for O-Acylation
Esterification, ester condensation, or so-called
O-acylation feature is clearly identified as the general utilization of carboxylic acids and alcohols that are usually activated by Brønsted acids. Thus, esterifications do not require superacid catalysis [
9]. Indeed, typical superacids have been studied and observed as convenient catalyst which can improve the method for direct esterification, without any changes of catalytic activity even for prolonged reaction times [
9,
57]. This characteristic has thus led to an interest in the use of TfOH as a catalyst for
O-acylation.
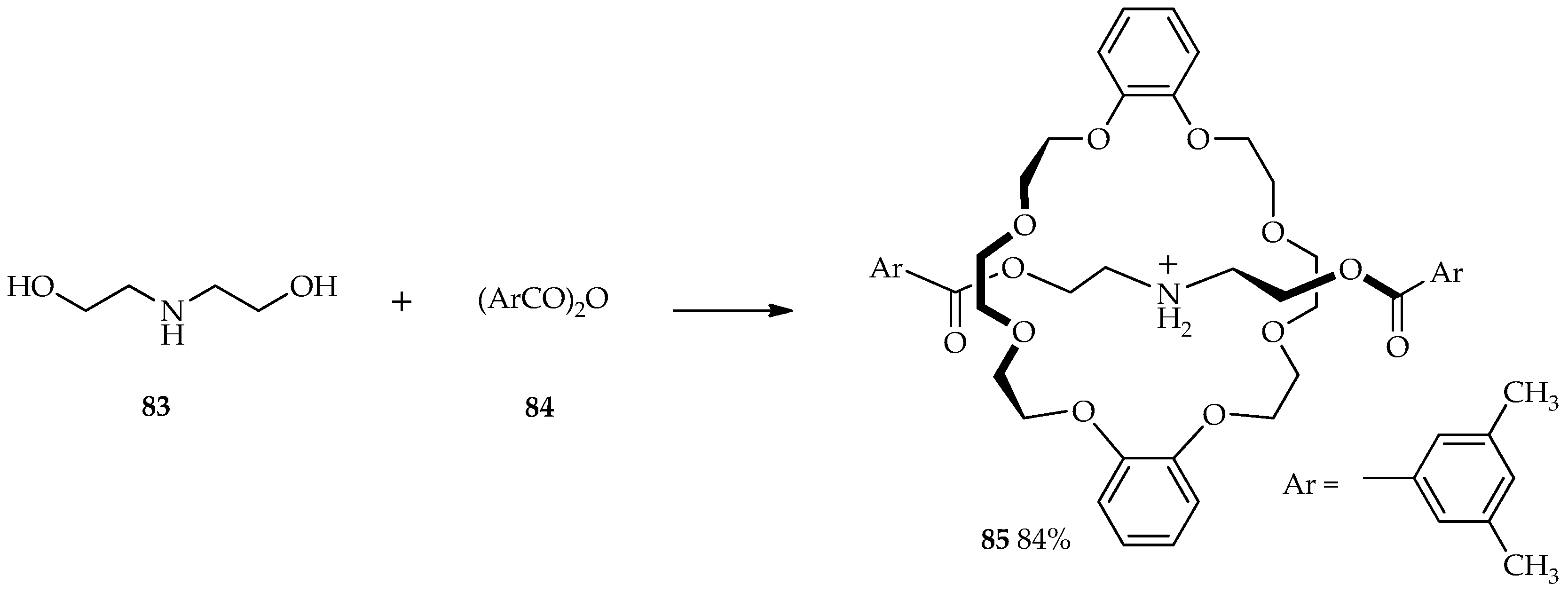
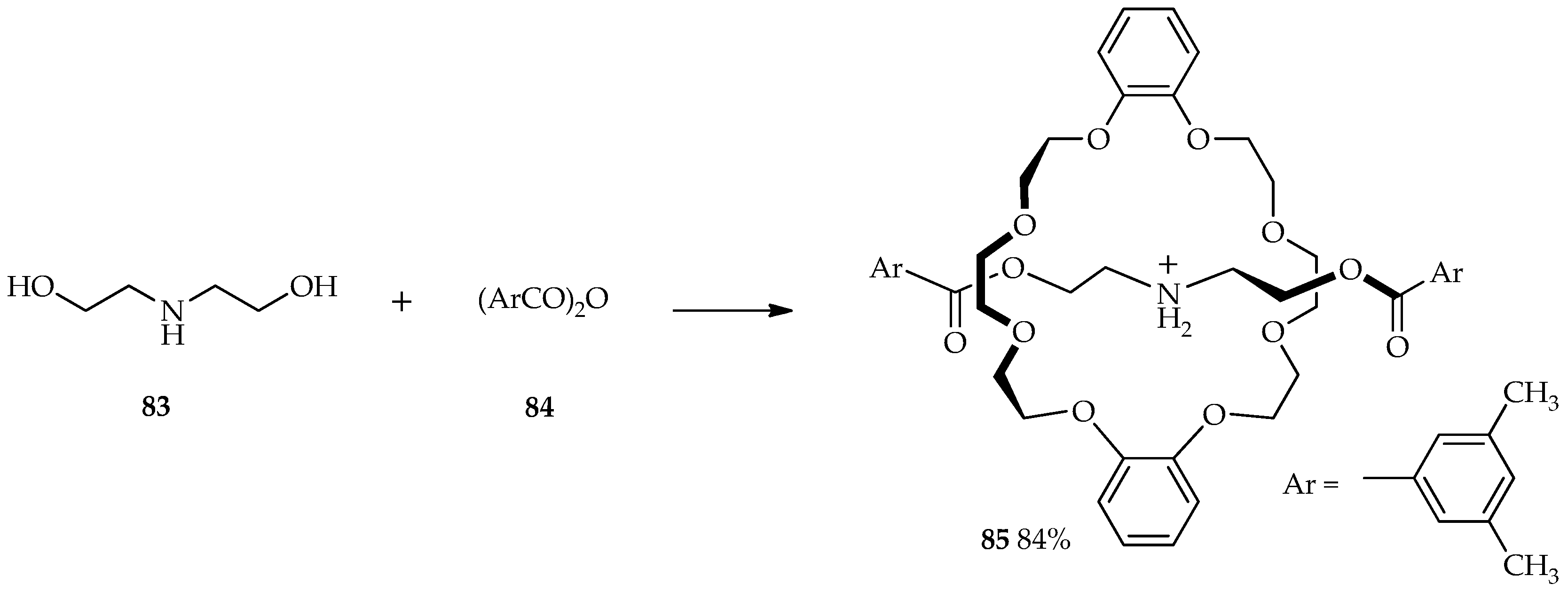
Rotaxanes are molecules composed of a linear dumbbell-shaped substituent flanked by one or more macrocycles. For many years, chemists have been intrigued by the challenge of synthesizing these mechanically interlocked molecules; traditional synthetic methods can only produce poor yields of rotaxanes [
58]. The selective
O-acylation of diethanolamine
83 by aromatic acid anhydride
84 was carried out in the presence of 1.5 equiv. of TfOH and dibenzo-24-crown-8 to afford [2]rotaxane
85 at 0 °C (
Scheme 9). The use of TFA and MSA did not allow the formation of
85 and only formed the esterification product from starting material
83. Relative to the acidity of other catalysts, TfOH is sufficient for the protection of the amino group via protonation and esterification. Thus, the TfOH catalytic system enables high production yields of
85 at low temperature [
59].
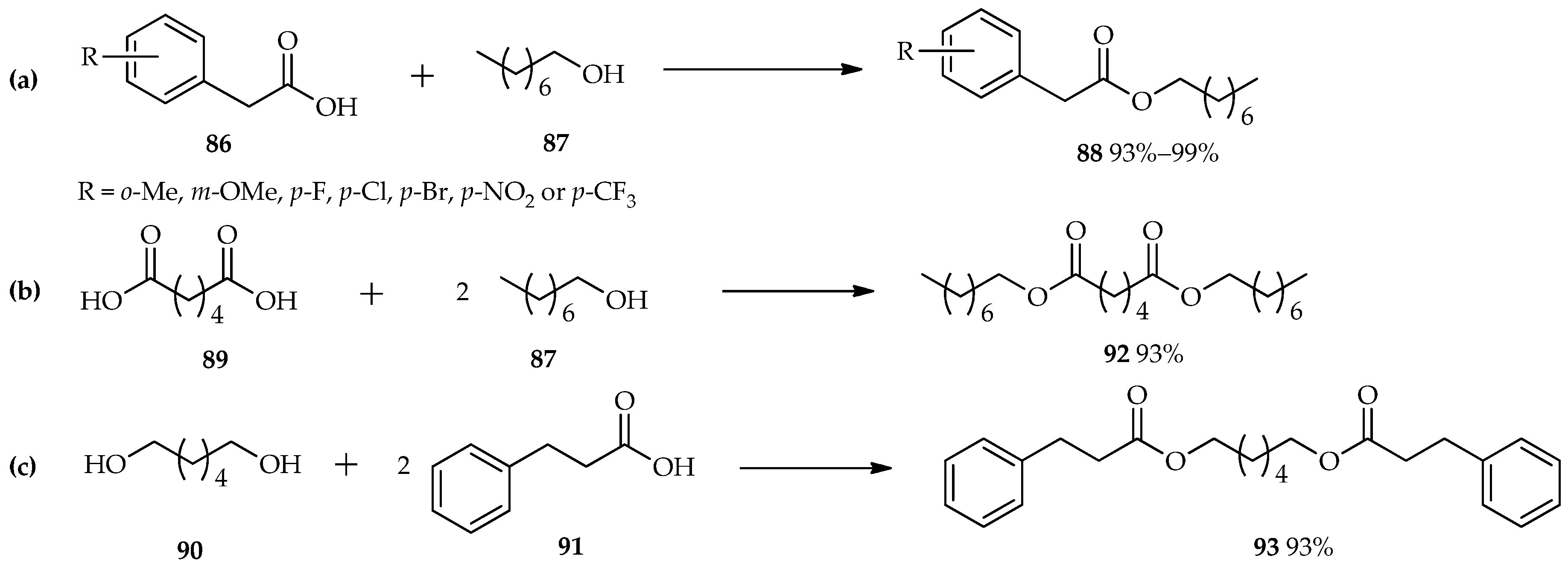
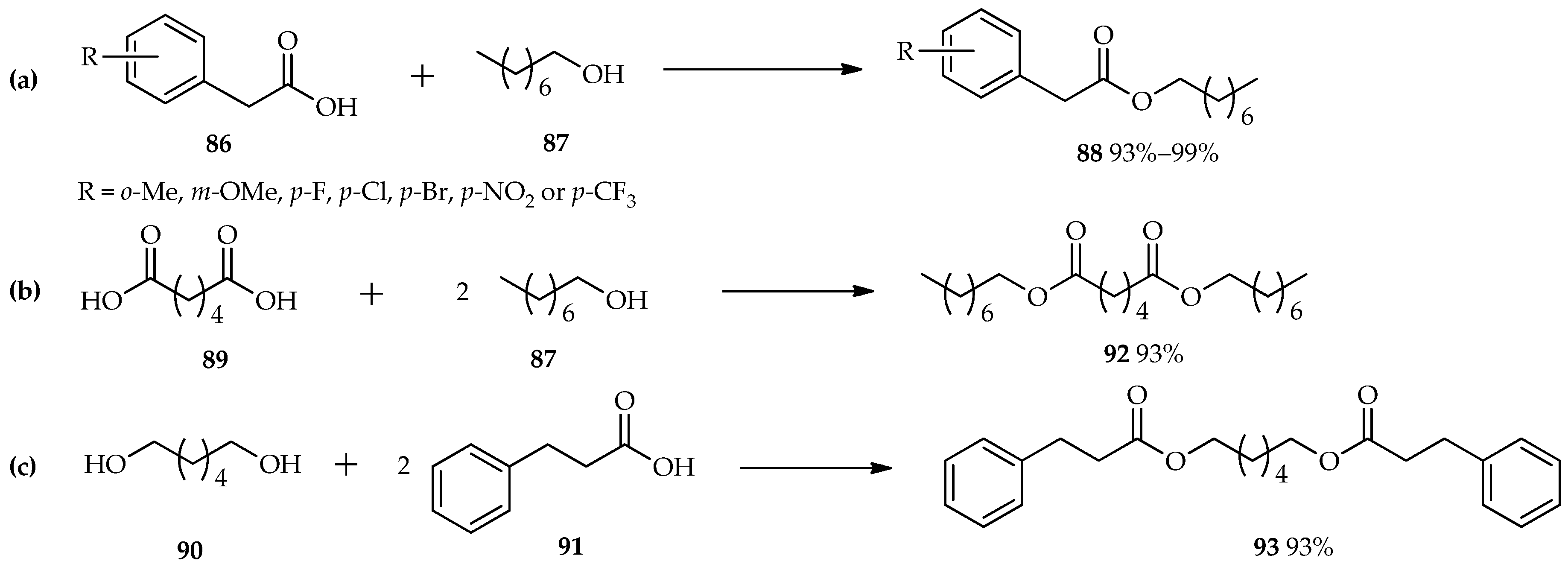
TfOH as an efficient catalyst in the esterification reaction was also studied by Shibata et al. [
60]. Reactions between various benzoic acid derivatives (
86) and octan-1-ol (
87) were carried out using a catalytic amount of TfOH in 1,1,1,3,3-pentafluorobutane as a solvent. The unique properties of TfOH make it a more active catalyst than sulfonic acid; thus, 0.2 mol % of TfOH is sufficient for the excellent conversion of the reactants to
88 (
Scheme 10a). This condition is also suitable for the
O-acylation of either the diacid
89 with an alcohol
87 (
Scheme 10b), or the diol
90 with carboxylic acid
91 (
Scheme 10c). Both of these reactants were converted to their diesters,
92 or
93, respectively, with excellent yields without the need for extended reaction times.
4. Special Features of TfOH in Selective C- and/or O-Acylations
C- and/or
O-acylations using various substrates naturally depend on the acyl donor and acceptor used. Both reactions can be controlled by changing the reaction temperature, which is inherently related to the reaction time. The catalyst, and sometimes the solvent, also plays an important role in these reactions. One of the unique characteristics of TfOH as a catalyst is that conditions for TfOH-catalyzed acylation may be determined and adjusted to increase the reaction’s selectivity. Denoting into this term, it can be mentioned as a “special” catalytic feature of TfOH. For example, TfOH can be used in the transacylation and deacylation between several types of hindered acetophenones and anisole
1 at 70 °C in the presence of imidazolium-based ionic liquids [
61]. The high yield conversion (>98% for one moiety of acyl group in hindered acetophenones), high selectivity, minimal side reactions, and the obviated use of excess TfOH illustrate its superiority over other acids utilized in these reactions.
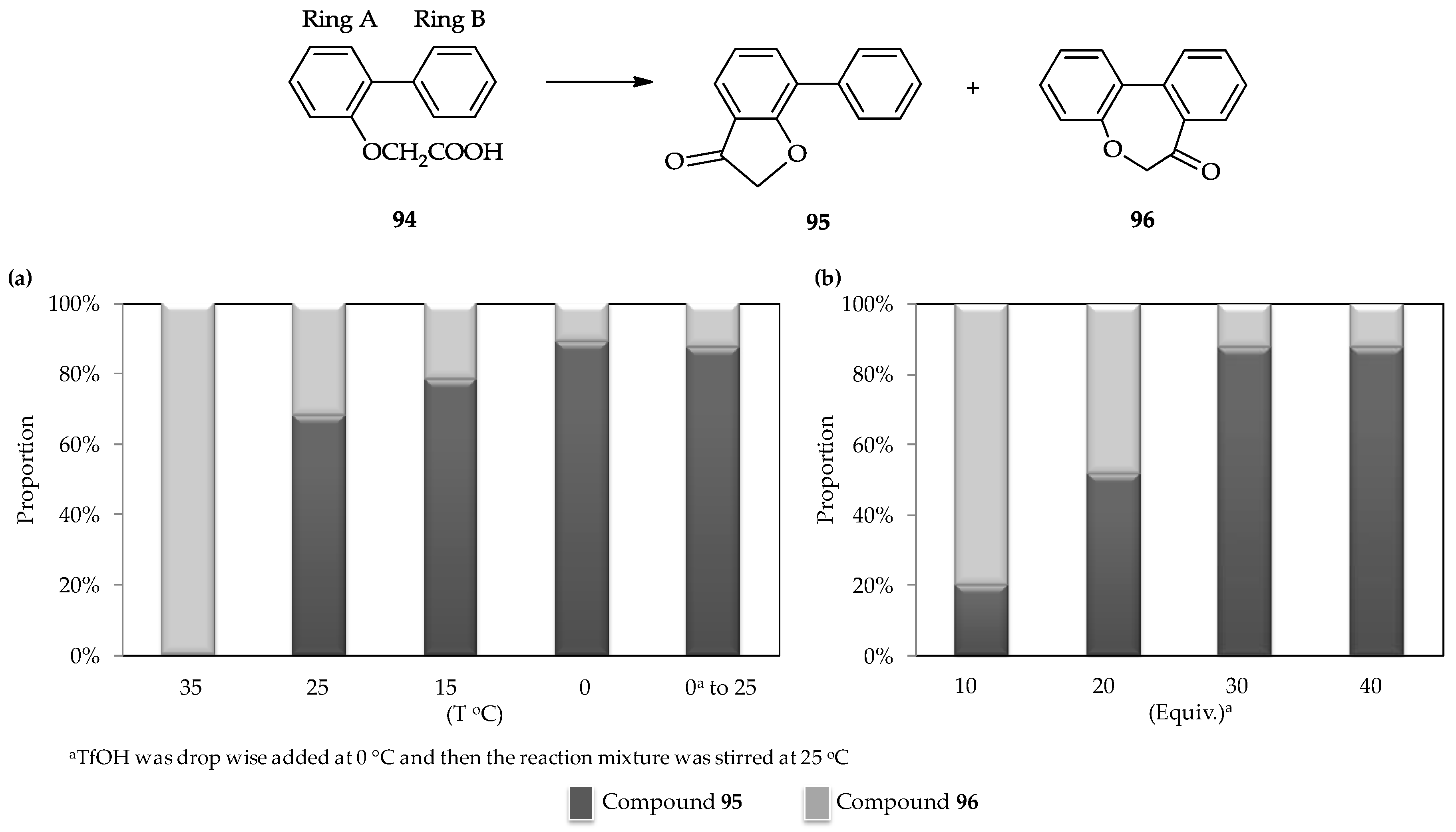
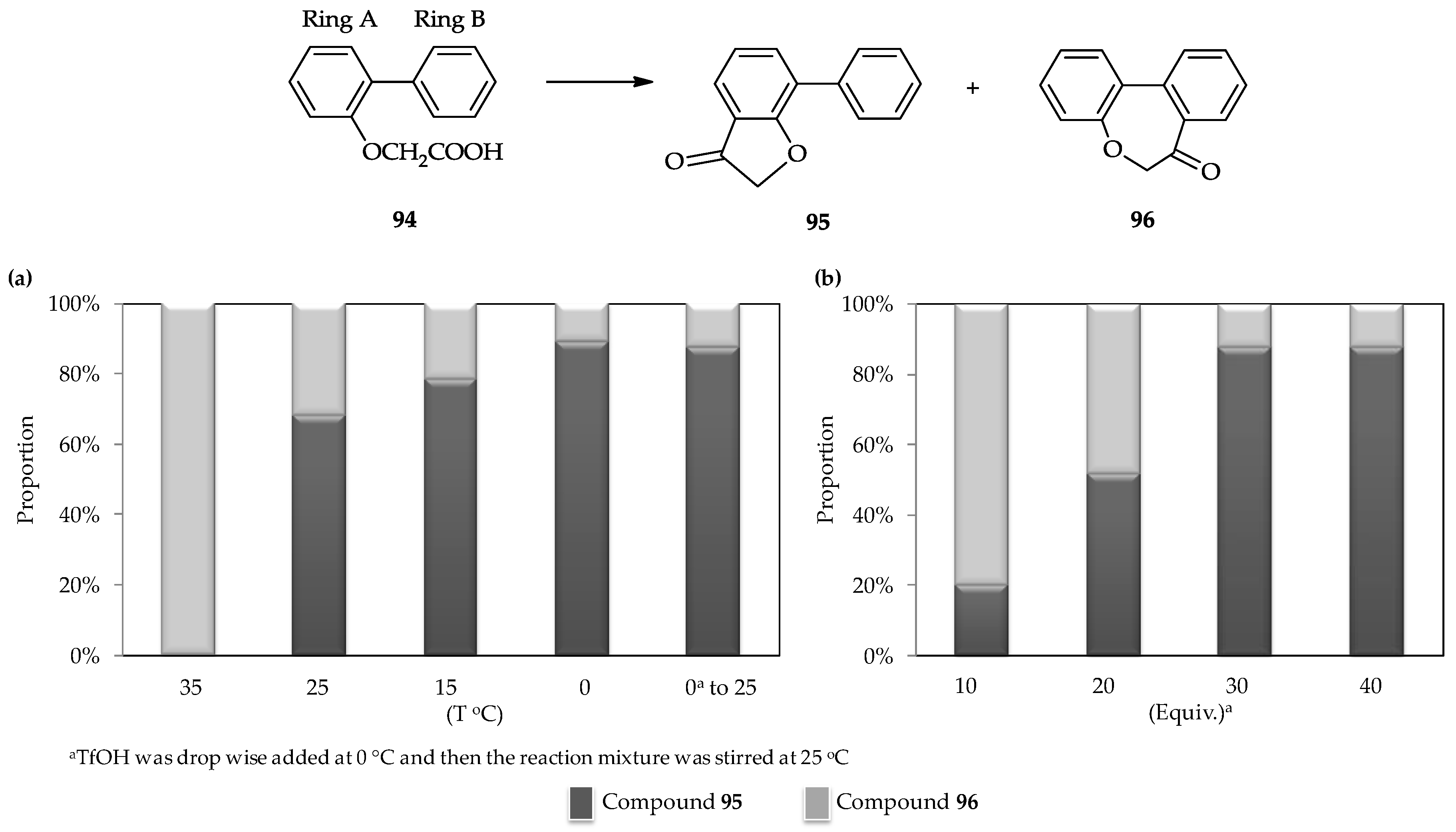
The selective intramolecular cyclic acylation of the 2-arylphenoxyacetic acid
94, which contains two phenyl ring moieties (which are denoted as rings A and B,
Figure 5) [
62], is another unique characteristic of the TfOH catalytic system. Cyclic acylation of the carboxylic moiety of
94 into ring A to form arylcoumaranone
95 or ring B to form dibenzoxepine
96 can be controlled (
Figure 5). Under the action of the strong acid TfOH, the intermediate of mixed anhydride with
94 can form and then further transformed into an ionic intermediate by removing the possible leaving group as –OTf. This ionic intermediate exhibits the less steric hindrance; therefore, the formation of
95 or
96 is achievable, depending upon the conditions. A low temperature is preferred for the selective formation of
95 (
Figure 5a) using a similar amount of TfOH (30 equiv.) added dropwise. Meanwhile, under the same temperature, utilization of a low amount of TfOH (10 equiv.) can result in the formation of
96 (
Figure 5b).
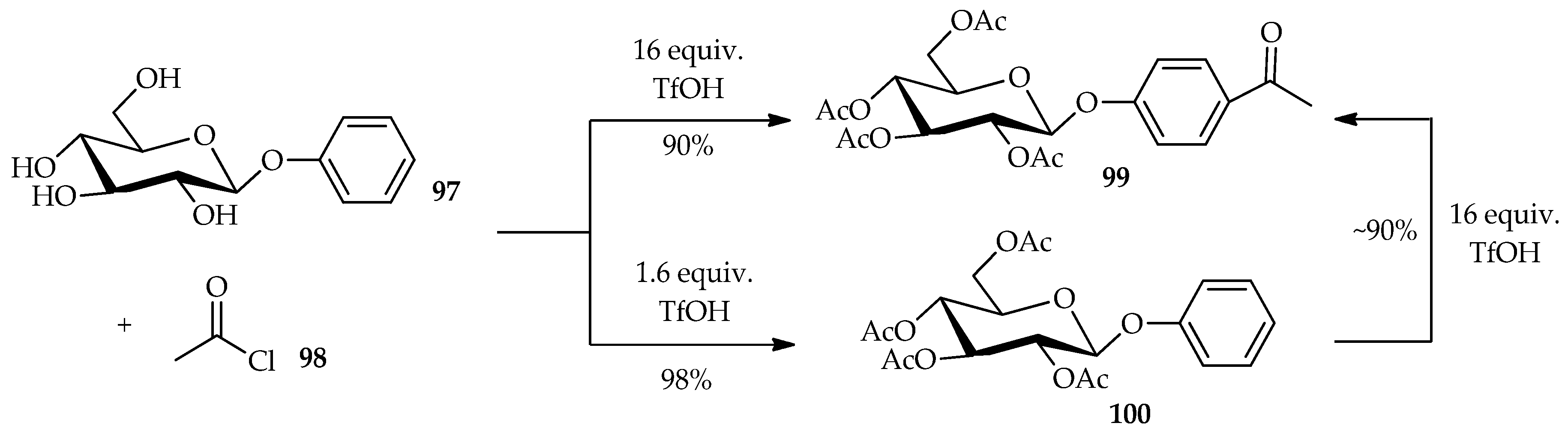
The derivatization of
O- and
C-arylglycoside is hampered by the low solubility of the unprotected aromatic glycosides in the reaction medium and the competitive deglycosidation under acidic conditions for
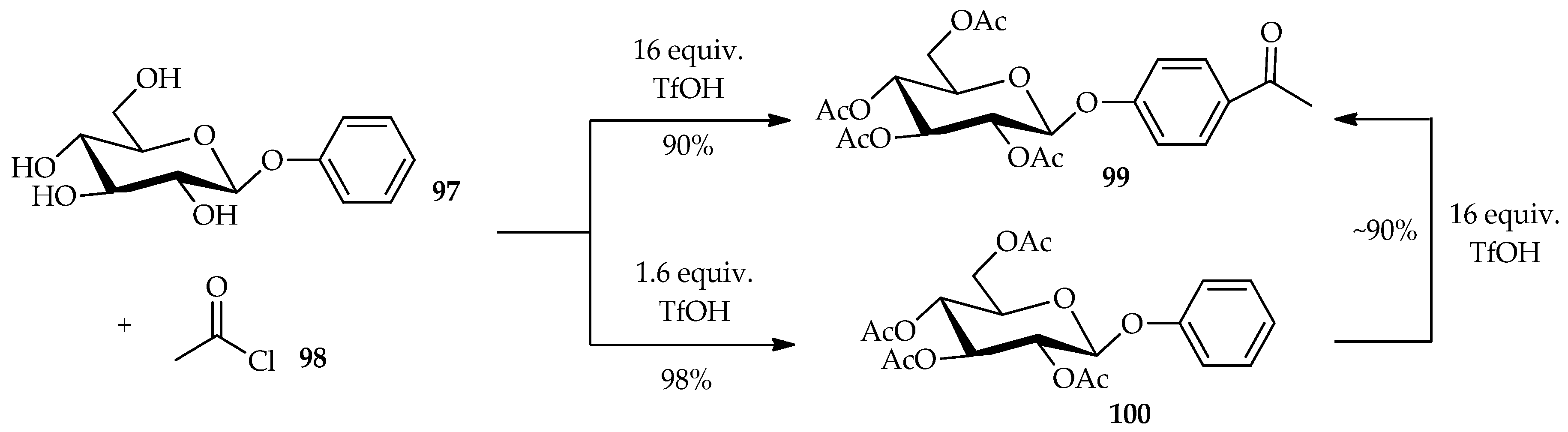
O-glycosides. TfOH was found to easily dissolve the carbohydrates without causing their decomposition. The low solubility of unprotected aromatic glycosides can be overcome by using TfOH as a Friedel–Crafts acylation promoter. This technique can be extended to other reactions such as direct acylation of
O- and
C-arylglycosides. The Friedel–Crafts acylation of the aromatic moiety and
O-acylation of the glucose moiety of aryl glycoside is dependent upon the proportion of TfOH used [
63]. The reaction of phenyl-β-
d-glucoside
97 with acetyl chloride
98 in the presence of excess TfOH (16 equiv.) can result in the production of
98 with good yield (
Scheme 11). In this case, the excess TfOH promotes not only the Friedel–Crafts acylation of the aromatic moiety, but also the
O-acylation of the glucose moiety. Treatment of
97 with 1.6 equiv. of TfOH, produces only the per-
O-acetylated compound
100, indicating that the
O-acylation is faster than the Friedel–Crafts acylation. This result is supported by the re-treatment of
100 with a large amount of TfOH, which afforded an ~90% yield of the Friedel–Crafts product
99.The Friedel–Crafts
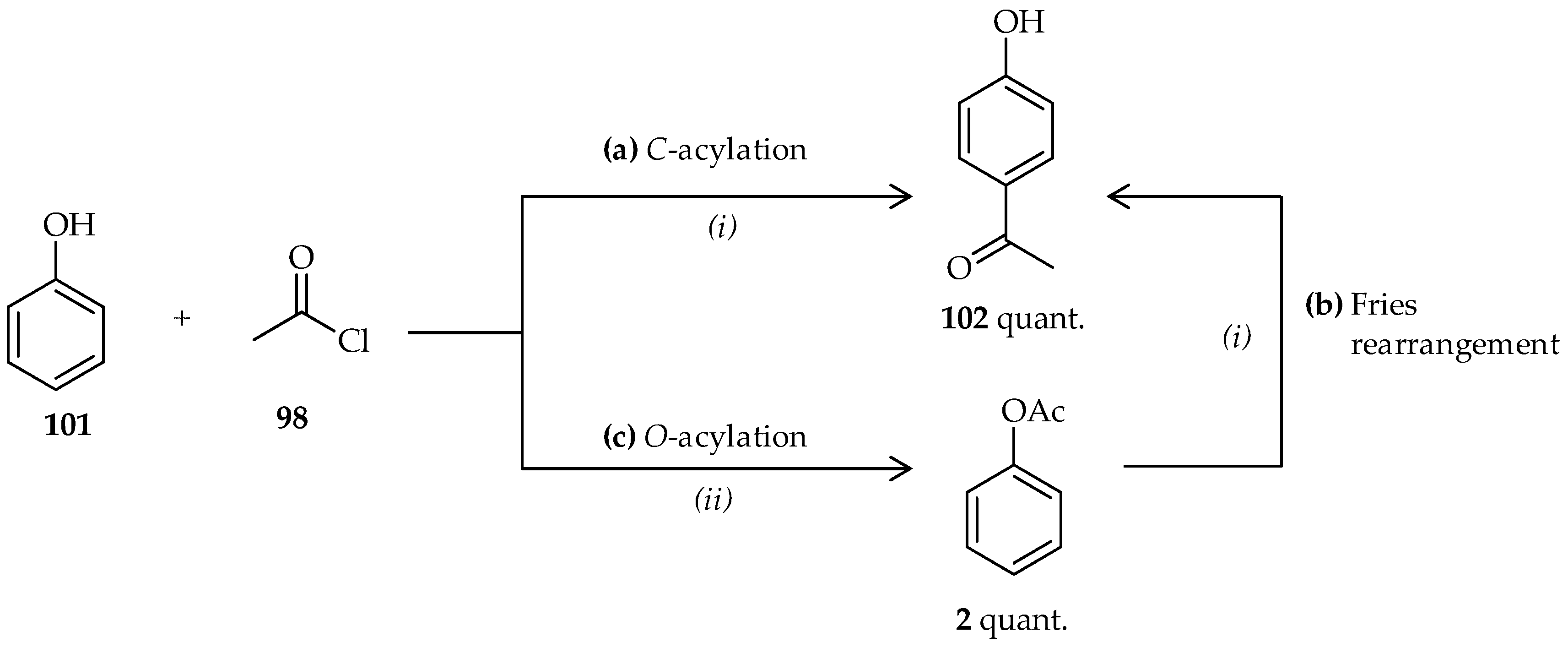
C-acylation of phenol derivatives and acetyl agents and the Fries rearrangement of phenyl ester derivatives via an appropriate catalyst are useful approaches to the synthesis of
o- or
p-hydroxyaryl ketones [
1,
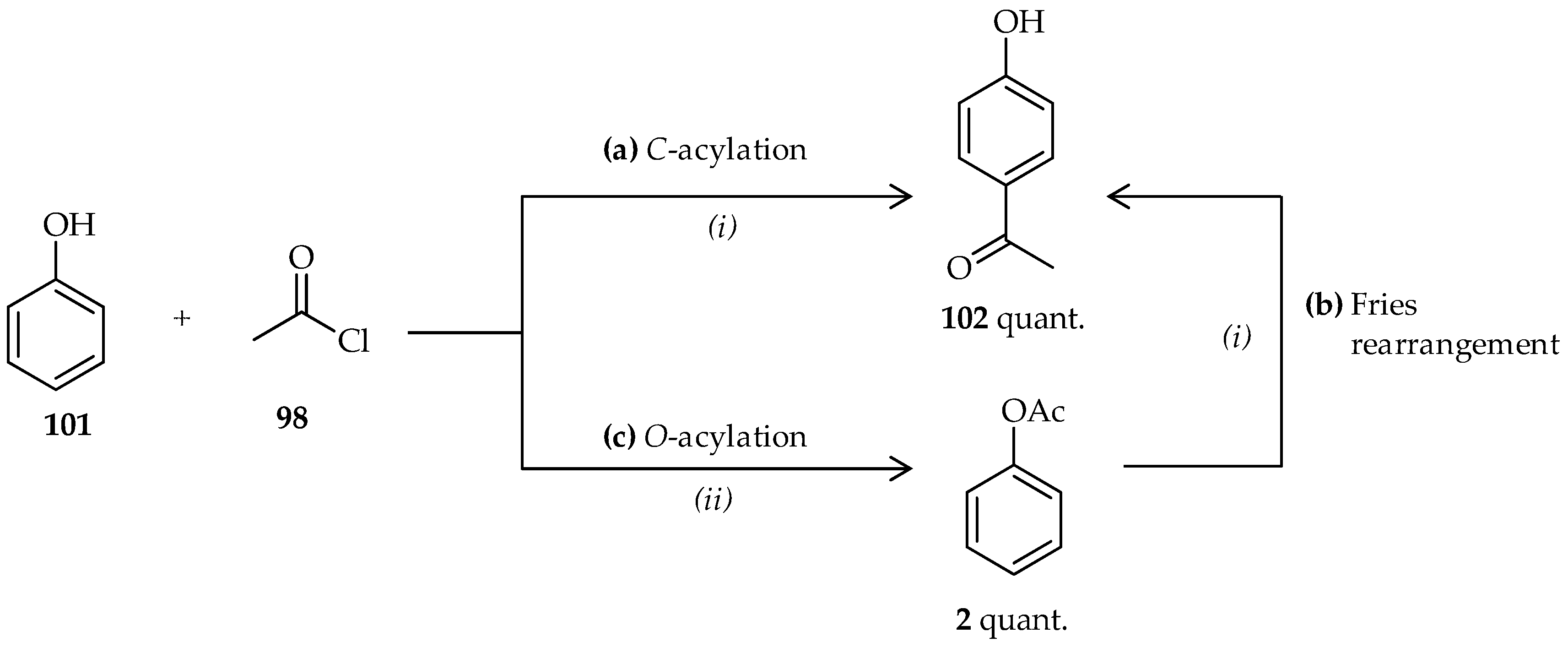
2]. In these reactions, TfOH may act as both catalyst and solvent, resulting in high efficiencies of these reactions. When phenol
101 was reacted with 1 equiv. of acetyl chloride
98 using an excess of neat TfOH (>30 mmol),
p-hydroxyphenyl ketone
102 was formed (
Scheme 12a) [
64]. Despite the reaction being conducted at room temperature, the reaction was relatively fast and had excellent yield. Under the same conditions, neat TfOH can also be used in the Fries rearrangement of phenyl acetate
2 to produce
p-hydroxyphenyl ketone
102 (
Scheme 12b). In contrast to the conventional Fries rearrangement, which usually involves heating the reaction mixture to 80–180 °C [
2], the Fries rearrangement of phenyl acetate
2 occurs at room temperature under the mild conditions offered by TfOH. The appropriate conditions for catalysis using neat TfOH can also be applied to direct
C-acylation of acyl chlorides having various side chains to phenol derivatives. Their application to the Fries rearrangement of various phenyl carboxylate derivatives also results in high yield.
The mixed carboxylic trifluoromethanesulfonic anhydride is formed from the reaction of TfOH with acyl chloride [
65] or carboxylic acid [
66,
67]. It is known as an extremely powerful acylating agent [
65,
67]; its reaction with toluene is highly selective when a bulky side chain is used [
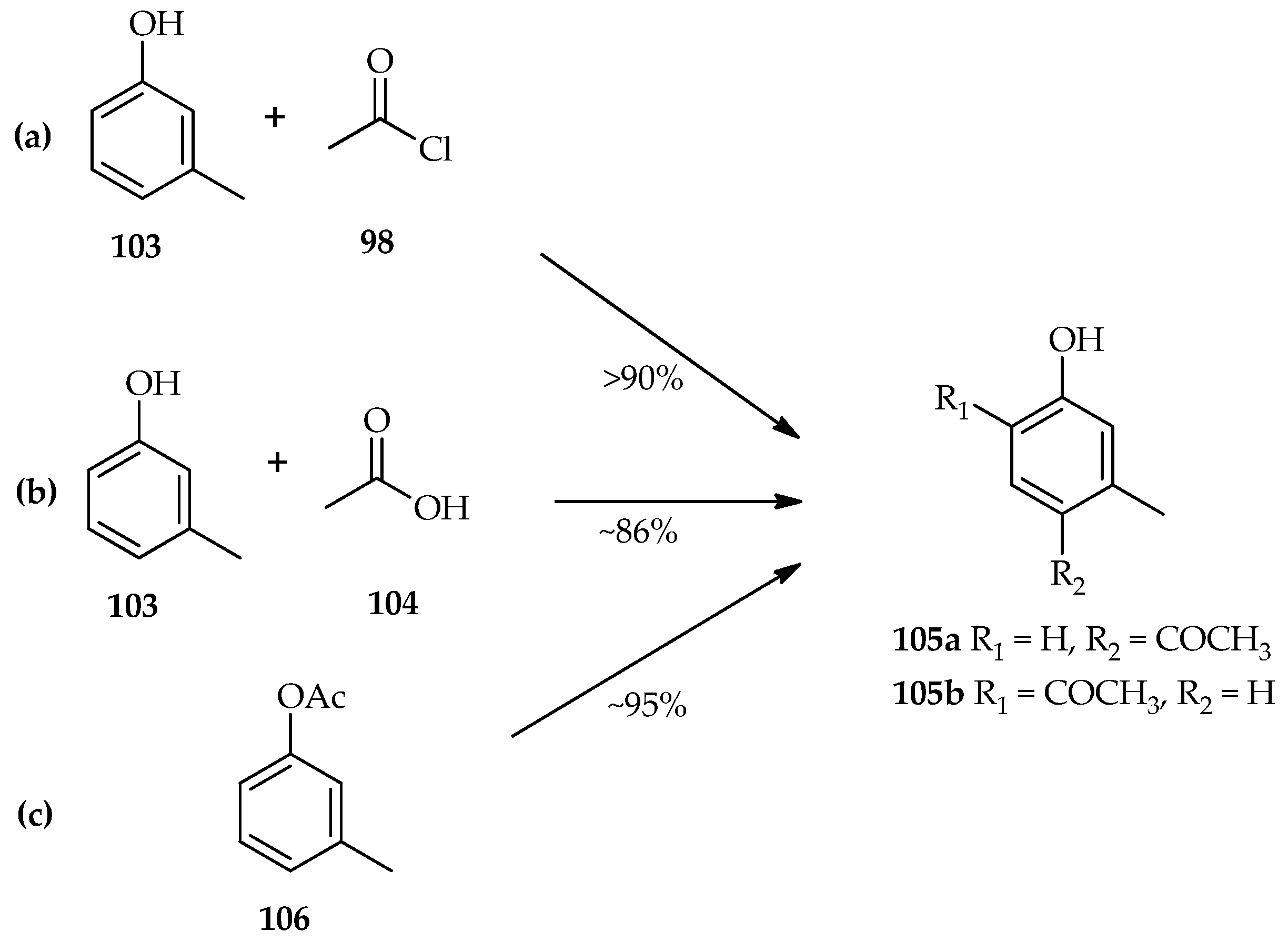
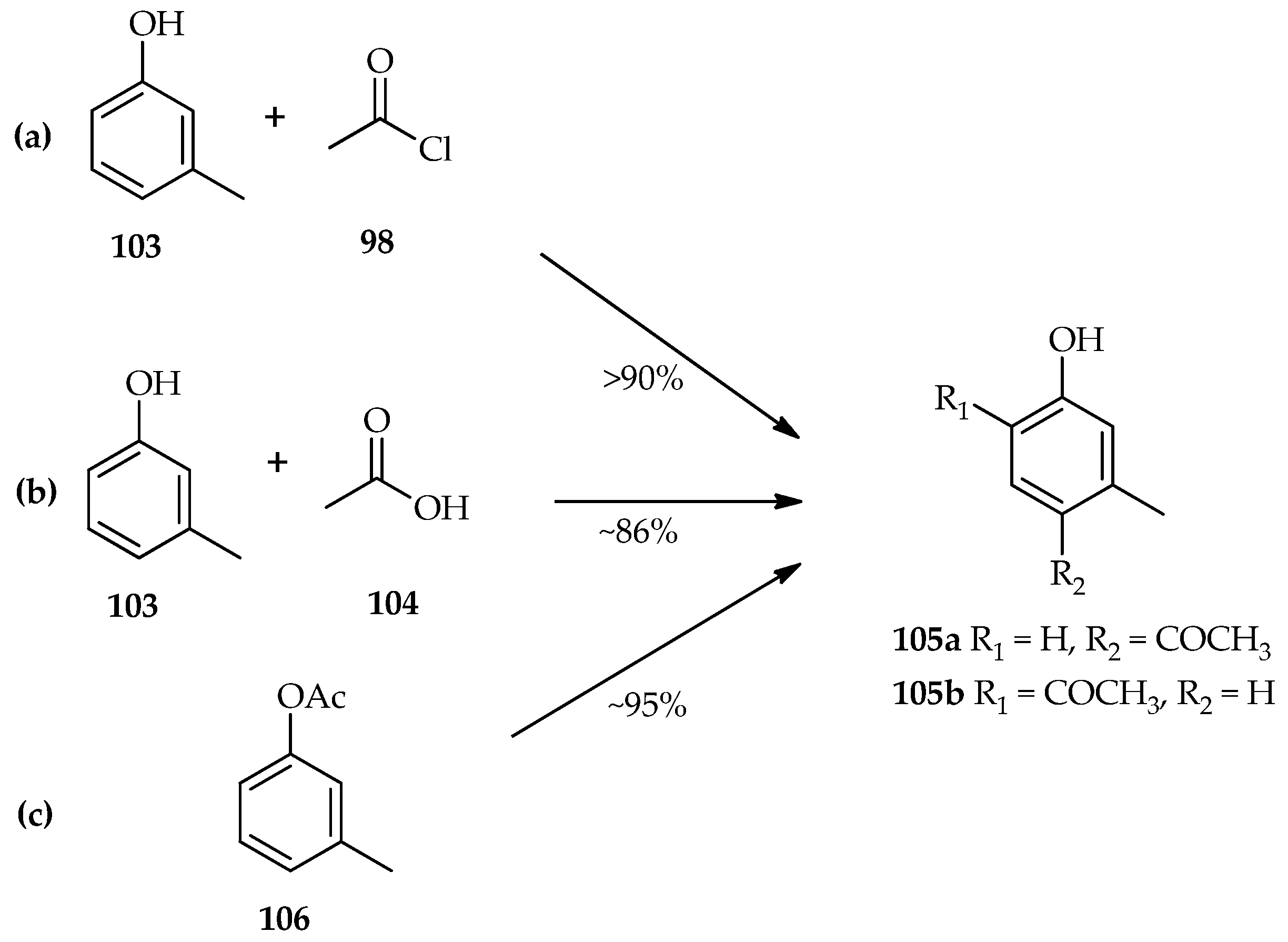
67]. The TfOH catalytic system for acylation of
m-cresol
103 using either acetyl chloride
98 (
Scheme 13a) or acetic acid
104 (
Scheme 13b) has been found to have similar yields of the isomeric products (
105a/
105b ratio of 1.4–2:1) [
64]. This result indicates that the acylation of phenolic compounds with either acetyl chloride
98 or acetic acid
104 have the same active species that plays an important role in exceeding the reaction. When
m-tolyl acetate
106 was reacted with TfOH (
Scheme 13c), both the Fries rearrangement and hydrolysis were observed. Even though the Fries rearrangement of
106 was reported to be faster than the reaction of
m-cresol
103 and acetic acid 98 in an hour, after 16 h reaction at room temperature the yield of the products was almost the same. These results indicate competition between the Fries rearrangement and hydrolysis of
m-tolyl acetate
106. However, the hydrolyzed acyl donor again underwent a Friedel–Crafts type reaction with
m-cresol
103 over an extended period [
64].
When phenol derivatives were reacted with a selected acyl donor using a catalyst, direct
C-acylation was achieved and the
O-acylation for the formation of phenyl ester derivatives was also observed (
Scheme 12)
. A catalytic amount of TfOH in CH
3CN (1%) used in the room-temperature reaction between phenol
101 and acetyl chloride
98 afforded phenyl acetate
2 in excellent yields (
Scheme 12c) [
64]. Increasing the proportion of acetyl chloride
98 (3 equiv.) resulted in a quantitative yield of phenyl acetate
2. A high proportion of the acylating agent was utilized to prevent other reactions when the available TfOH in the reaction system was very low. The interaction between CH
3CN, which also functions as a solvent, and TfOH has already been studied and a wide variety of complicated structures that can be formed according to the ratio of the two compounds are known [
68]. A previous study suggested that an excess amount of neat TfOH, acting as both catalyst and solvent, can catalyze the direct
C-acylation of phenol
101 with acetyl chloride
98. This study showed that the unique properties of TfOH, which depends on its proportion, affects the phenol
101 acylation process and can be controlled for
C- or
O-acylated product synthesis [
64]. However,
O-acylated product of phenyl acetate
2 is also can be followed by the Fries rearrangement to
p-hydroxyaryl ketones, which are known as useful reactants and/or intermediates for the manufacture of agrochemicals and pharmaceuticals.
Direct
C-acylation of naphthol
107 with acetyl chloride
98 and acetic acid
104, as well as the Fries rearrangement of naphtyl acetate
108 can be conducted by using the TfOH catalytic system (
Table 3). Kobayashi et al. [
69] reported that the direct acylation of naphthol
107 required the use of 20% TfOH in a toluene–nitromethane (6.7:1) solution and a temperature of 100 °C (
Table 3, entry 1). As mentioned above, catalysis by 1% TfOH in CH
3CN at room temperature produces product
108 (
Table 3, entry 2). In a study of thermodynamic analysis of phenol
101 acylation with acetic acid
104 by Sobrinho et al. [
70], they found that temperature played an important role in determining the acylation product; phenyl acetate
2 is favored over hydroxyphenyl ketone derivatives at high temperature (>800 K). The product distribution was also very clear in which ~100% formation of hydroxyphenyl ketone derivatives were selective when the temperature was up to 800 K. Therefore, when the utilization of a limited proportion of TfOH was applied to naphthol
107 at a high temperature, the two parallel pathways that resulted in the production of hydroxyaryl ketones [
71,
72] might be pursued (the solvent effect, however, is not discussed here). First is the direct
C-acylation of phenolic compound. Second is the
O-acylation of phenolic compounds, which form the ester as an intermediate, and directly transform to hydroxyaryl ketones via the Fries rearrangement. Neat TfOH can also smoothly catalyze the direct
C-acylation of naphthol
107 and the Fries rearrangement of
108, which affords good yields of the two isomers
109 and
110 (
Table 3, entries 3 and 4). In summary, the controllability of TfOH catalysis in direct
C- and/or
O-acylations can be exploited to obtain the desired aromatic products from thermolabile substrates, an approach that would be of interest for further study.
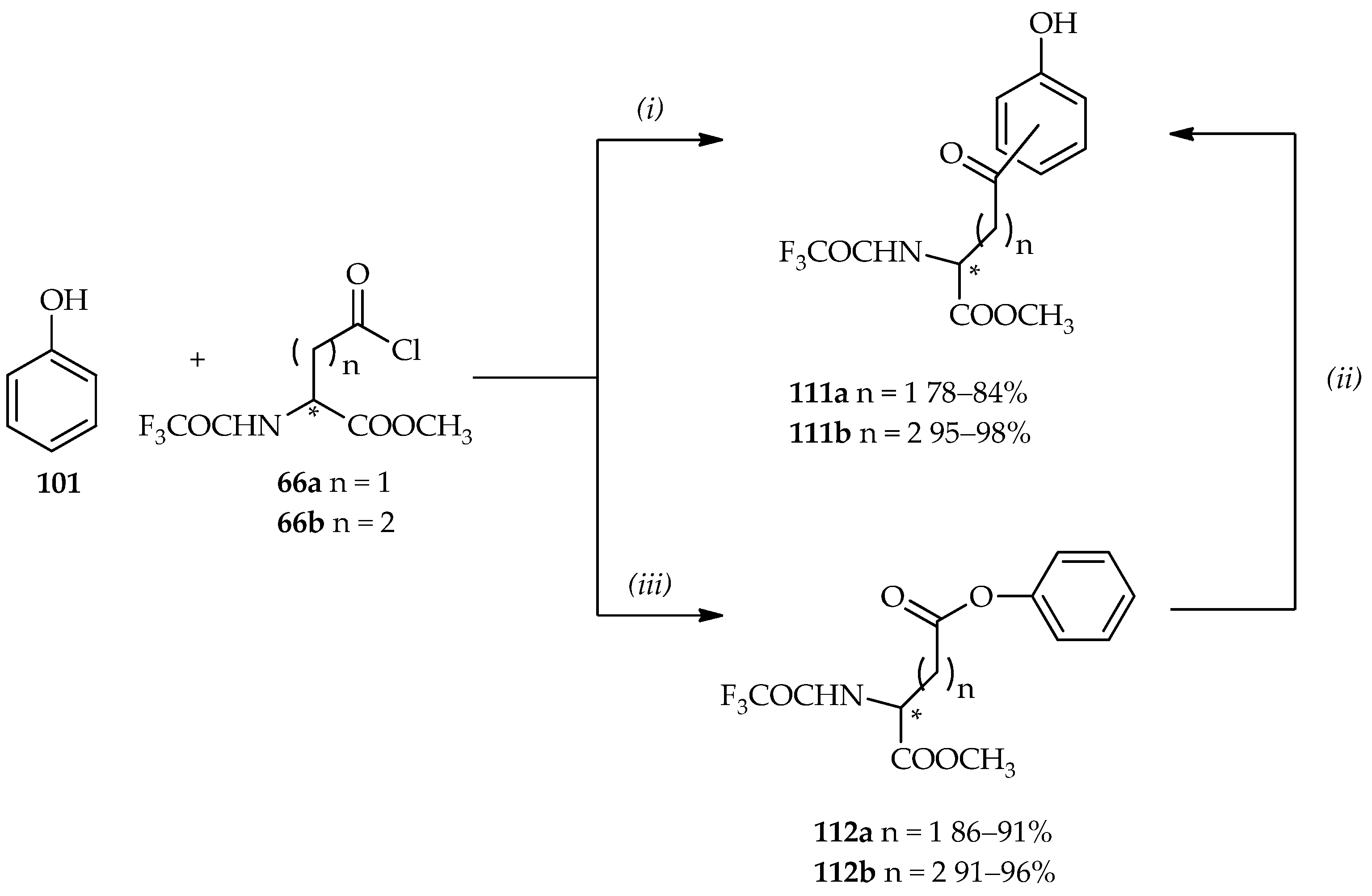
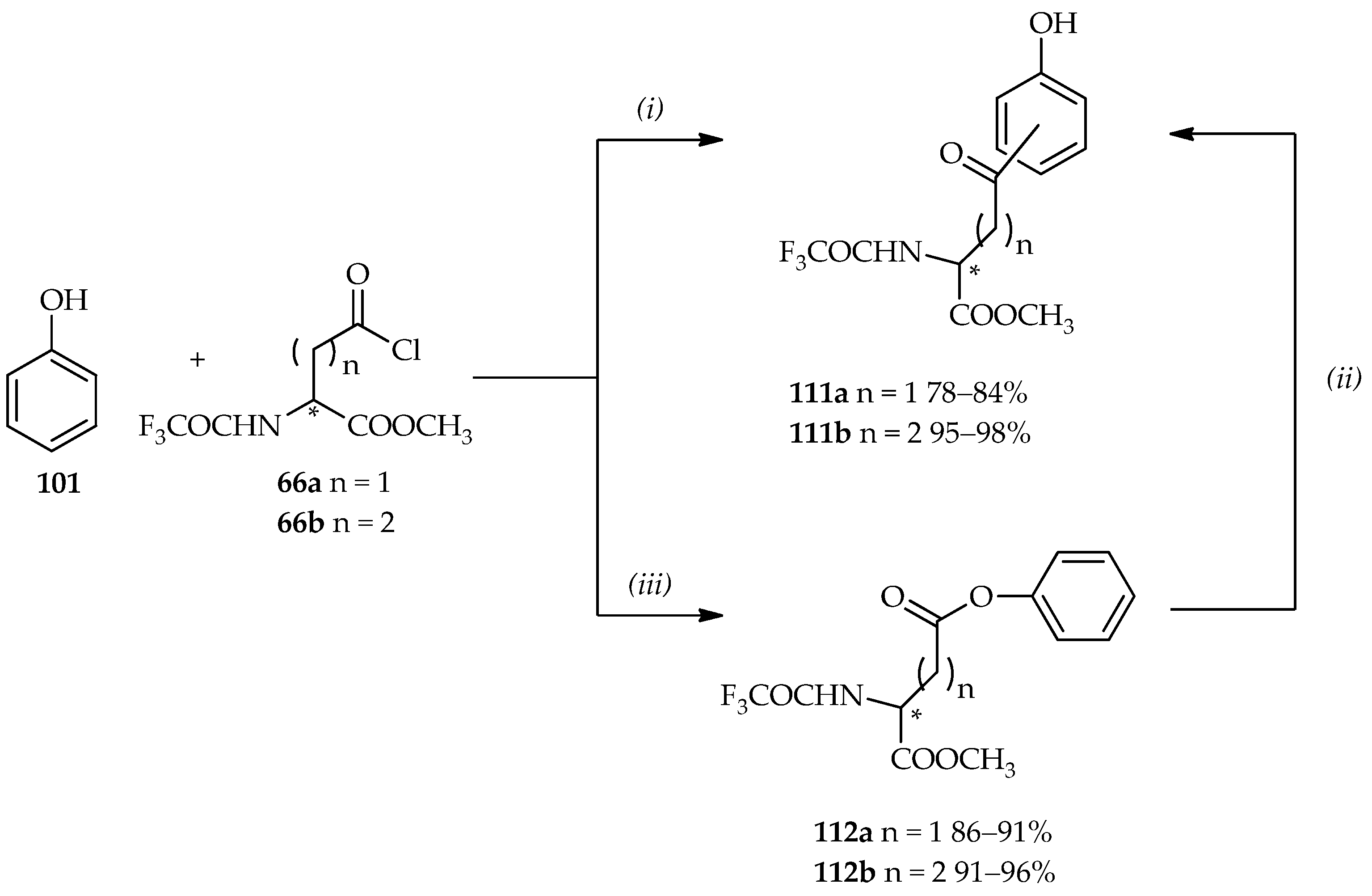
The applications of controlled TfOH-catalyzed
C- and/or
O-acylations of phenol
101 are showed in
Scheme 14 [
64,
73]. In a previous section, the use of neat TfOH under mild conditions in the acylation of aromatic derivatives using acylating agent
66 (
Scheme 5c) is discussed. When 1 equiv. of acylating agent
66 is reacted with phenol
101 under neat TfOH at room temperature, the
C-acylation product, benzyl carbonyl derivative
111, is observed in good yield. The Fries rearrangement step can also be conducted from the phenyl ester
112 by the use of neat TfOH to produce two
C-acylated isomers
111 (
o- or
p-position with ratio of 1.4:1 for compound
111a and 9:1 for compound
111b).
O-acylation can be proceeded in the reaction between 3 equiv. of acylating agent
66 and phenol
101 by using diluted TfOH in CH
3CN (3%–5%) at room temperature, producing the phenyl ester
112 in good yield. The
O-acylation product can also undergo the Fries arrangement to the regioisomers
111, with isolation yields and proportions identical to those of the direct Friedel–Crafts
C-acylation products. Compound
111 can then undergo reduction and deprotonation to optically pure multifunctional products, homophenylalanine or bis-homophenylalanine derivatives (>99% ee), which can be used for further analytical studies. Hence, the TfOH catalytic system has a number of functions, acting not only as a catalyst but also as a solvent. Furthermore, it can be controlled for the desired
C- and/or
O-acylations products synthesis.
5. Multifunctional Features of Deuterated TfOH (TfOD)
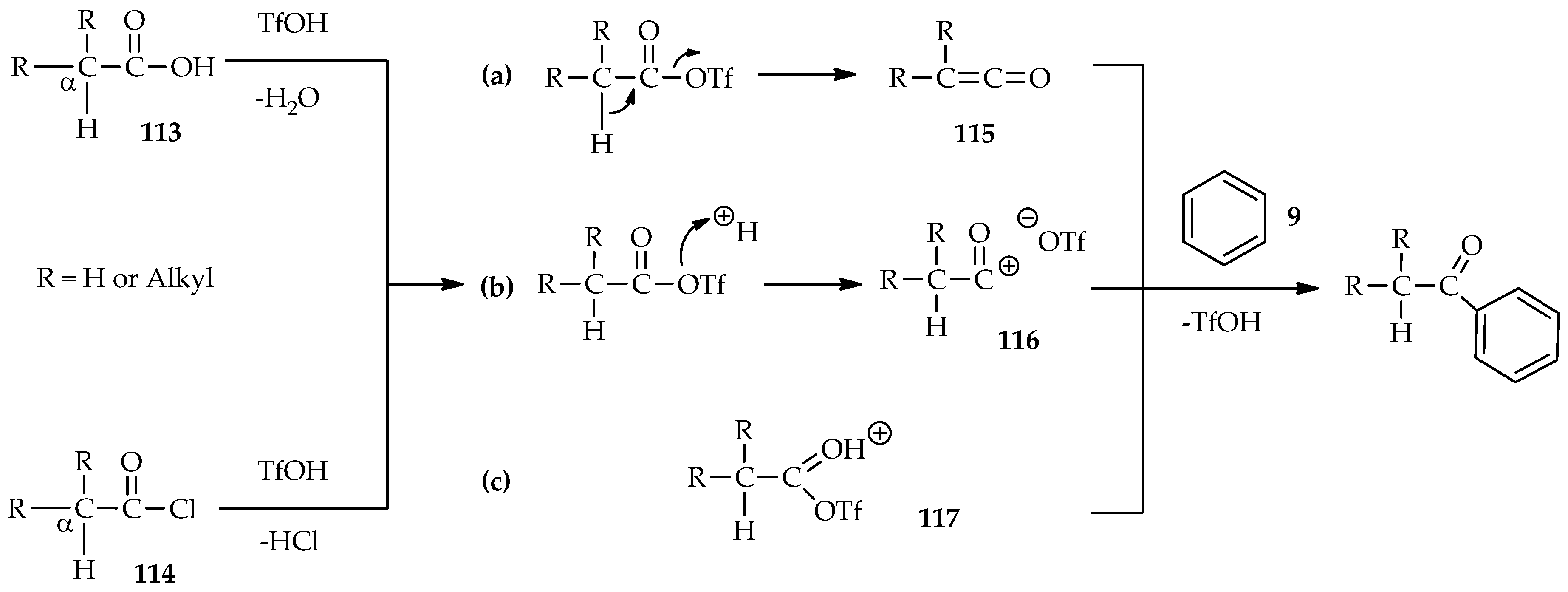
Even though catalytic amounts of TfOH are known to enable selective
O- and/or
C-acylation in organic solvents, the solubility of some substrates such as amino acids, which was previously mentioned above, should be taken into consideration. Selection of suitable conditions for these reactions is necessary, especially the reactions for optically active amino acids. Several plausible routes using various active species [
65,
66,
67] for aromatic acylation exist (
Scheme 15). The mixed carboxylic trifluoromethanesulfonic anhydride may be obtained by treatment of TfOH with acyl chloride [
65] or carboxylic acid [
66,
67]. The acylation pathway for an acyl donor in the form of alkyl carboxylic acid
113 or acyl chloride
114 can produce several species. When mixed, carboxylic trifluoromethanesulfonic anhydride is deprotonated and ketene
115 may be formed (
Scheme 15a). Next, if no proton is eliminated during the process, the dissociated acyl carbonium ion
116 seems more reliably formed. (
Scheme 15b). Another possible species is the protonated carbonyl of the mixed anhydride
117 (
Scheme 15c), in which the detailed spectroscopy has been studied. Ascertaining which intermediate has formed is a crucial step in preventing the formation of a racemic adduct during acylation. One of the advantages of TfOH potential features are the
2H-labelling experiments by using deuterated TfOH (TfOD). The use of TfOD as a catalyst, solvent, and source of deuterium in hydrogen/deuterium (H/D) exchange in the direct
C-acylation of aromatic substrates provides insight into the mechanisms of these reactions. Deuterium incorporation at the α-proton of carbonyl during TfOD-catalyzed reaction between acyl donor and substrate indicates the formation of the intermediate ketene
115. Otherwise, non-deuterium incorporation at the α-proton of carbonyl indicates formation of either the acyl cabonium ion
116 or protonated carbonyl of mixed anhydride
117.
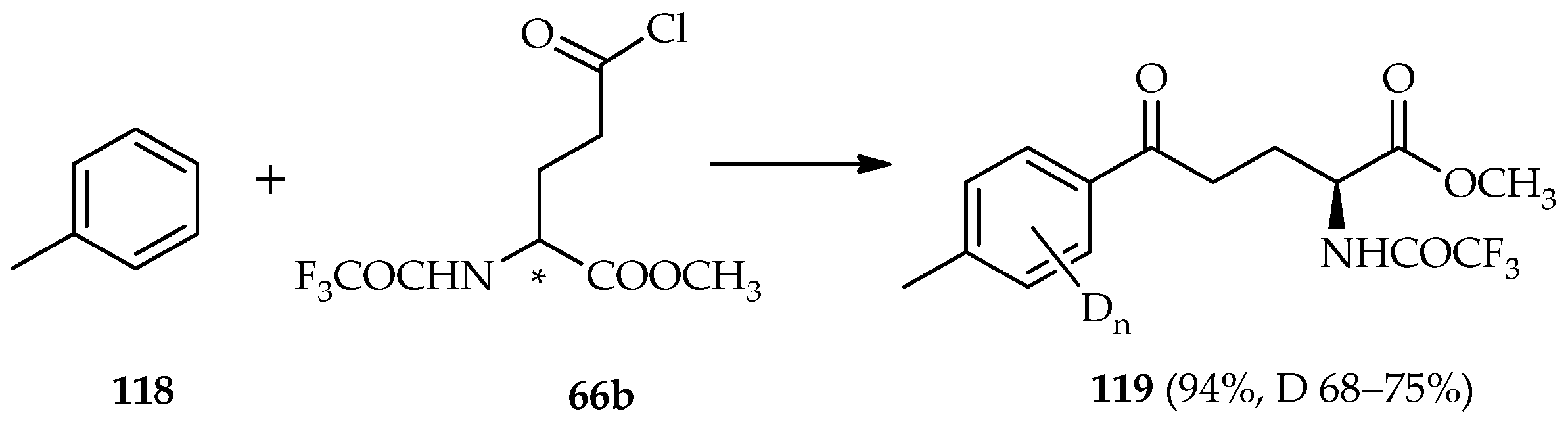
TfOD catalysis of the direct
C-acylation of toluene
118 and glutamic acid derivatives
66b has been previously established (
Scheme 16) [
74]. The deuterium incorporation was observed only on the aromatic protons (68%–75% deuterium incorporation) at a fine yield (94%) of the acylated product
119. The active species in the typical Friedel–Crafts acylation/electrophilic aromatic substitution is very important, especially the
N-alkylated α-amino acids that behave as acyl donors. If the deprotonation of the α-proton of the carbonyl favors ketene species
115 (
Scheme 15a), racemization can occur because of the loss of optical purity of the α-amino acid. Experiments for elucidating reaction mechanisms using catalysis by TfOD have revealed intramolecular cyclization of methyl-cyano-3-arylpropionate and non-deuterium incorporation of the α-proton of the recovered starting material was observed [
75]. These findings suggest that acid-catalyzed deprotonation of the α-proton of methyl-cyano-3-arylpropionate does not reach equilibrium even under strongly acidic conditions. In conclusion, in the reaction process, the exclusion of the loss α-proton is feasible. A similar tendency has also been observed in the intramolecular cyclization of 3-nitro-3-phenylpropionate, which found no contribution of proton elimination during the reaction [
76]. In the TfOD-catalyzed acylation of toluene
118 with glutamic acid derivatives
66b, the deuterium incorporation occurred only at aromatic protons, indicating the absence of α-proton deprotonation and that formation of ketene
115 formations was not possible.
H/D exchange methods can generally be divided into those utilizing D
2O as the deuterium source, those involving the addition of an acid or base, and those using metal-promoted H/D exchange. Commonly, heterogeneous catalytic systems are frequently used under harsh conditions and followed by dehalogenation, hydrogenation, hydrolysis, as well as epimerization and racemization [
77,
78]. Since a homogenous system is preferable in H/D exchange, especially in the case of optically active amino acids, the organic solvent must consider no observation of amino acid racemization during the reaction. A study that used a D
2O/D
2SO
4 system found that conditions will lead to either partial or complete racemization of phenylalanine H/D exchange, which is indicated by the exchange of α-protons [
79]. Deuterated TFA (TFA-
d) is appropriate for slow H/D exchange [
80], but its application to aromatic amides and amines [
81] is less effective for more electron-poor substrates such as acetophenone and its derivatives [
82]. In
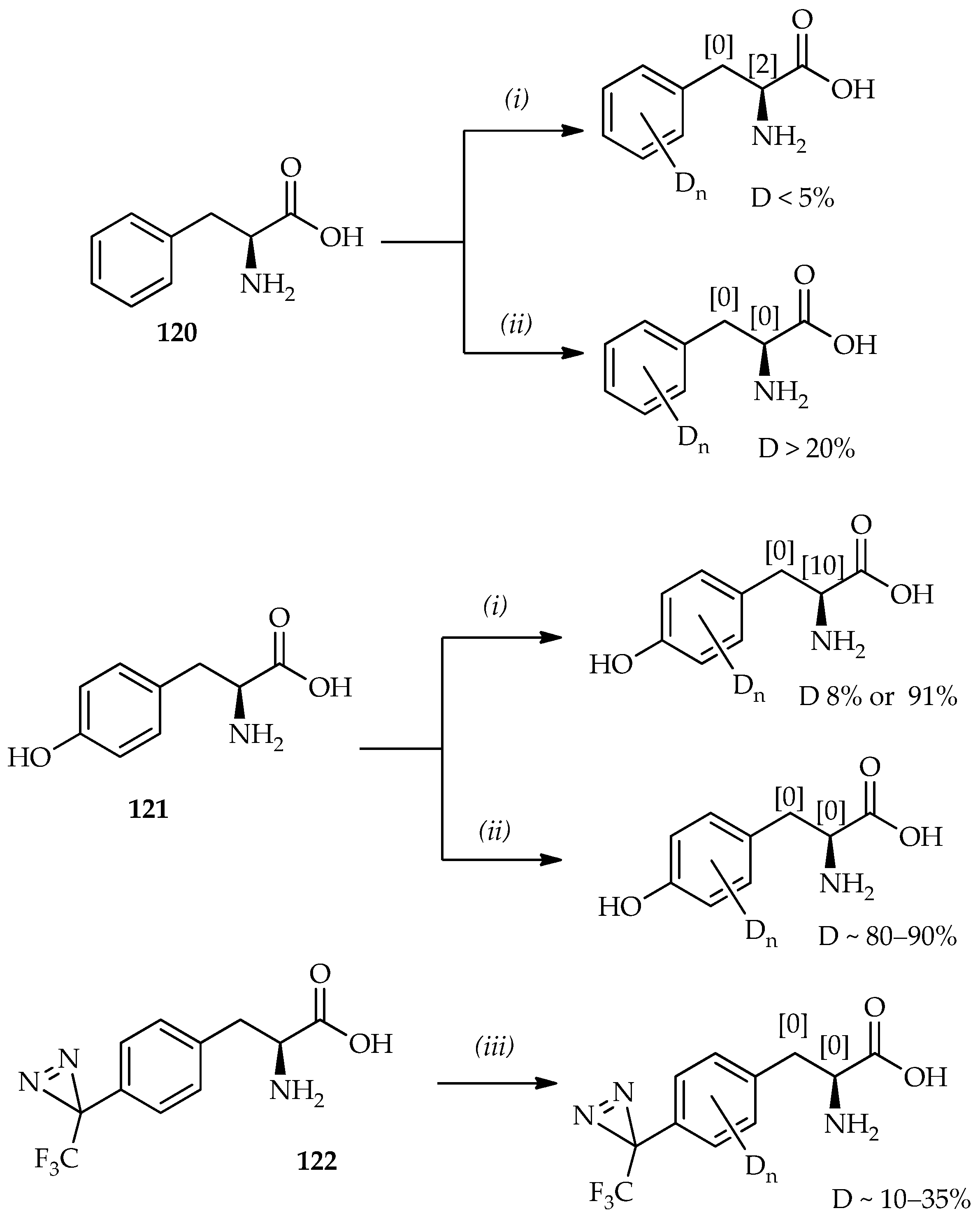
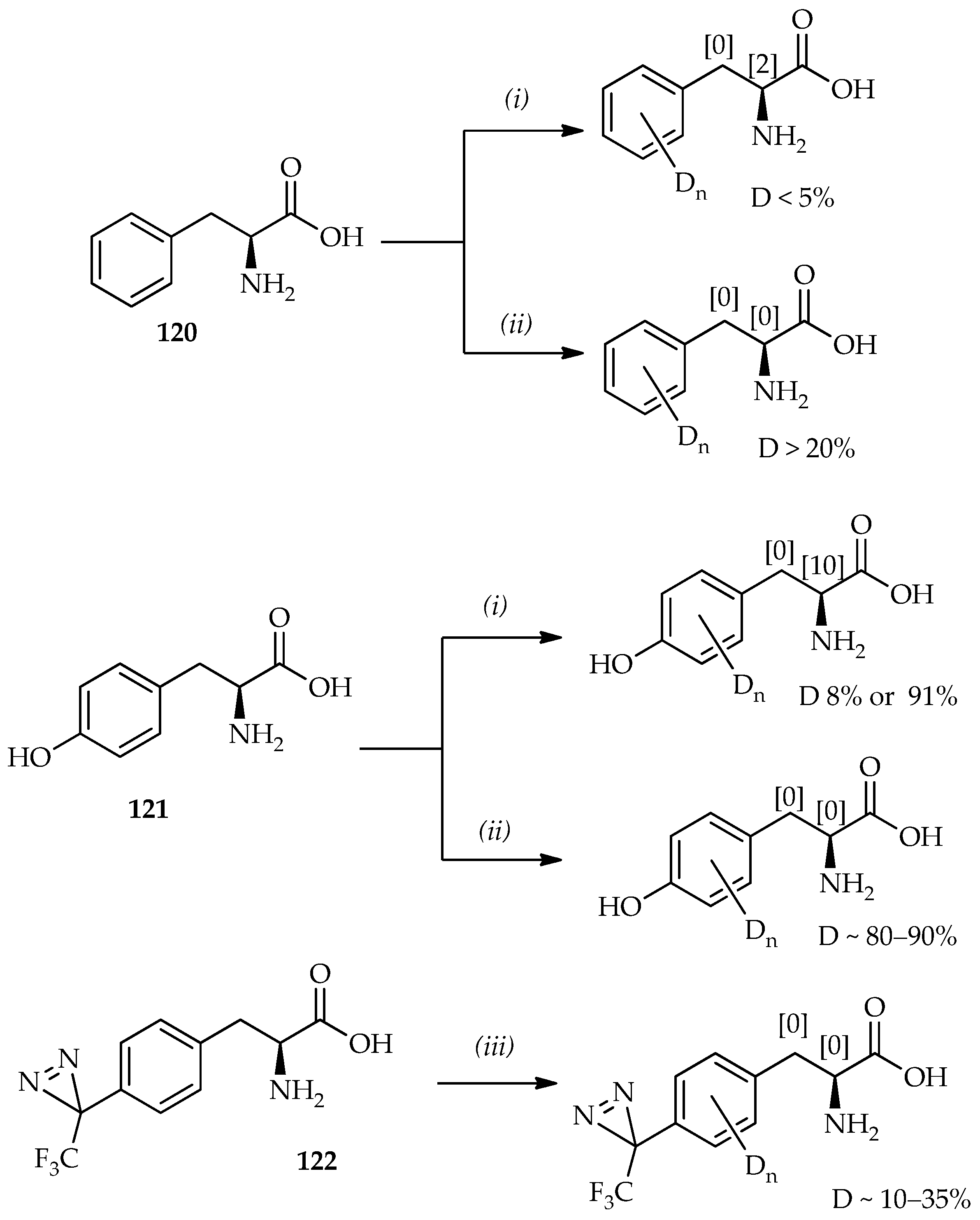
Scheme 17, a comparison can be made between phenylalanine
120 and tyrosine
121 treated with TFA-
d [
82] and TfOD [
74,
83]. It shows that the aromatic proton deuterium incorporation is generally larger when TfOD is used. Thus, the absence of α-proton exchange with TfOD indicates no loss of optical purity of amino acids. On the basis of the time dependence of H/D exchange using TfOD as monitored by mass spectroscopy, the favored sites for H/D exchange can be summarized as the
o-position with respect to the hydroxyl group on tyrosine > the hydrogen atoms of phenylalanine > the
m-position with respect to the hydroxyl group on tyrosine.
As mentioned previously, TfOH can be used in mild reaction conditions for
C-acylation synthesis of probes containing valuable heat-sensitive photophores for photoaffinity labeling (
Scheme 6). TfOD was thus also used in H/D exchange for aromatic amino acids or peptides containing photophores. The special feature utility of TfOD under mild conditions for H/D exchange of phenylalanine derivatives containing photophores has been established [
83]. In this system, the phenylalanine derivative containing TPD (
122;
Scheme 17) can dissolve well in TfOD, and the low temperature allows H/D exchange of these valuable heat-sensitive photophores. The selective aromatic-protons deuterium incorporation by TfOD in the synthesis of other important photophores based on benzophenone derivatives via direct
C-acylation of an aromatic substrate and benzoyl chloride has also been studied [
84]. Fast H/D exchanges that occur only on the aromatic protons of the substrate is proposed from the exclusion of benzyl carbonium ion formation from benzoyl chloride. Intramolecular H/D exchanges between benzene and TfOD via 1,2-hydride shifts, that have been previously studied, are known to occur rapidly at room temperature [
85]. However, aromatic-protons H/D exchange on the substrates containing photophores can contribute to comprehensive photoaffinity labeling studies in the future.
H/D exchange can be used not only in investigations of reaction mechanisms, but also for selective protonation/deuteration, which can greatly simplify the spectra of even the largest protein moieties [
86]. Several simple exchange techniques for the selective deuteration of aromatic amino acids have been developed [
79,
86,
87], yet α-amino acids are known to interfere in these methods because of their solubility in many organic solvents [
83,
88,
89]. The substrate solubility in the homogenous catalytic system for H/D exchanges influences the rate of hydride transfer between the substrate and its deuterated derivative, which can be analyzed from isotopolog distributions obtained by mass spectroscopy. For example, the low solubility of isobutene in D
2SO
4 can limit hydride transfer to the gas–liquid interface. Thus, the cation solvated in the acid has a long residence time, which undergoes repeated deprotonation and reprotonation, in turn leading to fast H/D exchange before hydride transfer. In contrast, the solubility of isobutane in TfOD is much higher; as a result, hydride transfer may occur in the liquid phase [
90] and result in fast H/D exchange. This example is in agreement with the high dissolving capacity of TfOH [
12]. Therefore, TfOD can be used to dissolve most α-amino acid derivatives and H/D exchange study can be broadened into peptides utilization.
According to Wishart et al. [
86], there are several criteria that must be fulfilled for improving the selectivity of deuteration of aromatic amino acids. First, the solubility of very hydrophobic amino acids must be enhanced in order to increase their labeling efficiency. Second, the labeling efficiency must not be lost and amino acid decomposition must not occur. Third, the methodological improvements should make large-scale protonation and deuteration of aromatic amino acids accessible and affordable to any well-equipped biochemistry laboratory, and finally, the method must provide significant savings in time and contribute to greater laboratory safety. The simple, safe, and high product yield of this process, as well as its ability to produce non-racemic deuterated amino acids, can be achieved because of the unique properties of TfOD as discussed above. Rapid room-temperature H/D exchange with peptides without any stereochemical changes has been achieved by controlling the amount of TfOD [
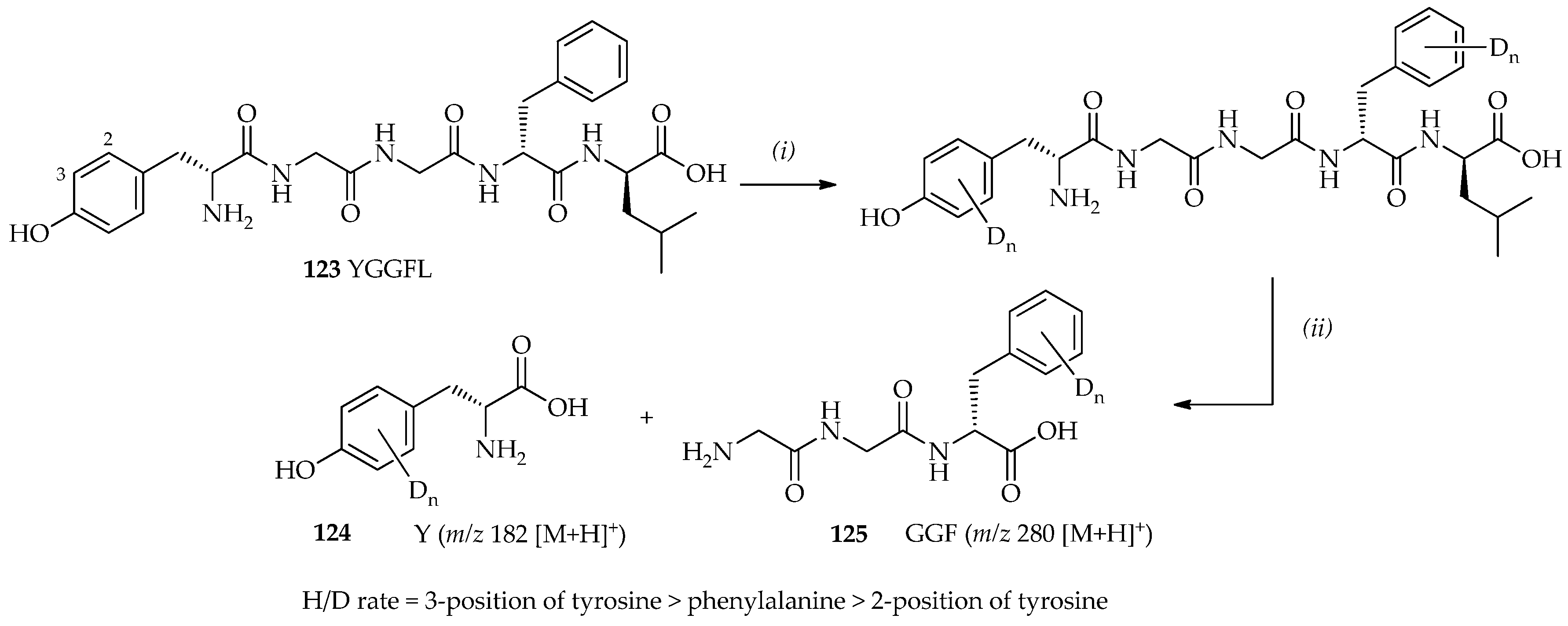
74]. Leucine enkephalin
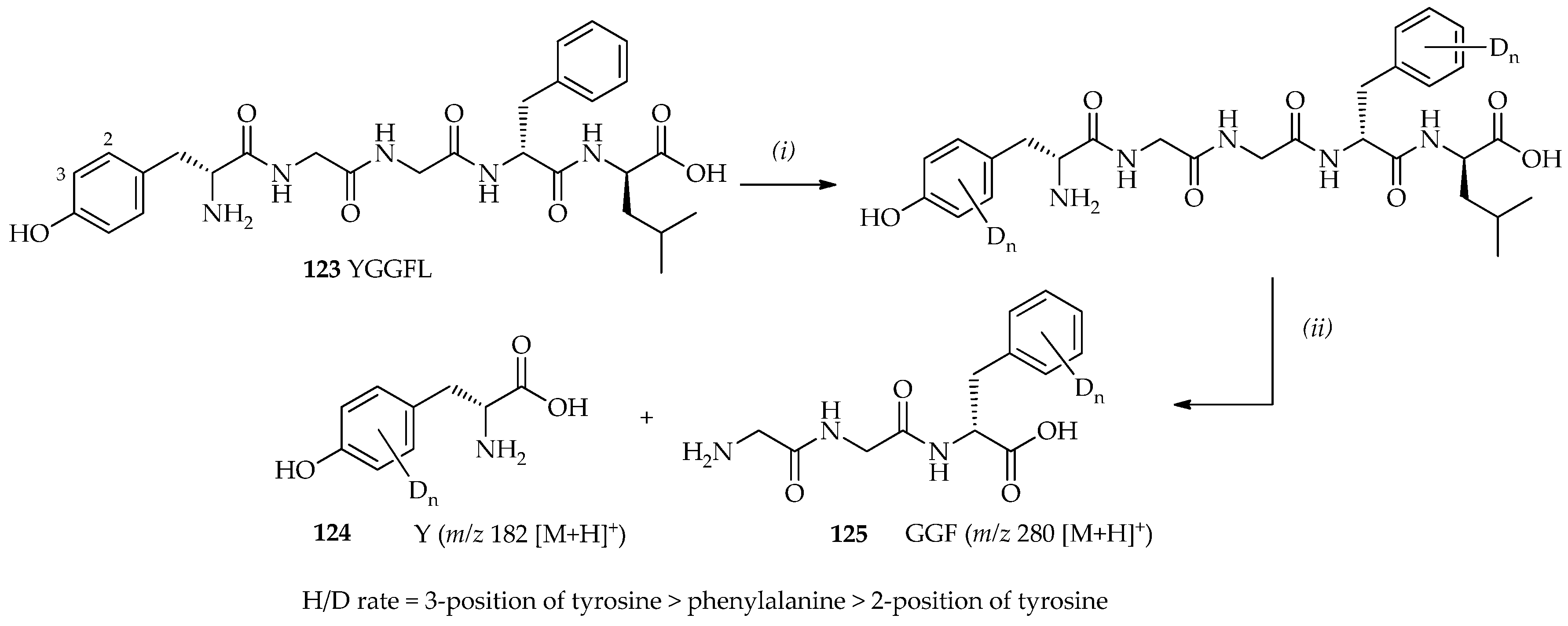
123 (Tyr-Gly-Gly-Phe-Leu/YGGFL) is a pentapeptide with two aromatic rings (tyrosine and phenylalanine;
Scheme 18) that has been studied by mass spectrometry [
91,
92]. Leucine enkephalin
123 has been previously shown to undergo H/D exchange with ND
3 [
93] in the gas phase [
94]. Additionally, a homogenous system may be obtained using TfOD (as shown by mass spectrometry and nuclear magnetic resonance (NMR) spectroscopy). In this system, leucine enkephalin
123 can dissolve well in TfOD even at low temperatures. Within 9 h at room temperature, four and five exchanges sites were observed. Characterizations of deuterated leucine-enkephalin
123 via digestion with chymotrypsin have shown that the favored H/D exchange sites in the order of most to least favorable are as follows: the 3-position of tyrosine > phenylalanine > 2-position of tyrosine (
Scheme 18). TfOD catalytic systems may also be utilized in the development of H/D exchange methods, which may extend the application of H/D exchange to a vast range of substrates, including compounds that are valuable, heat-sensitive, and sparingly soluble in many organic solvents.



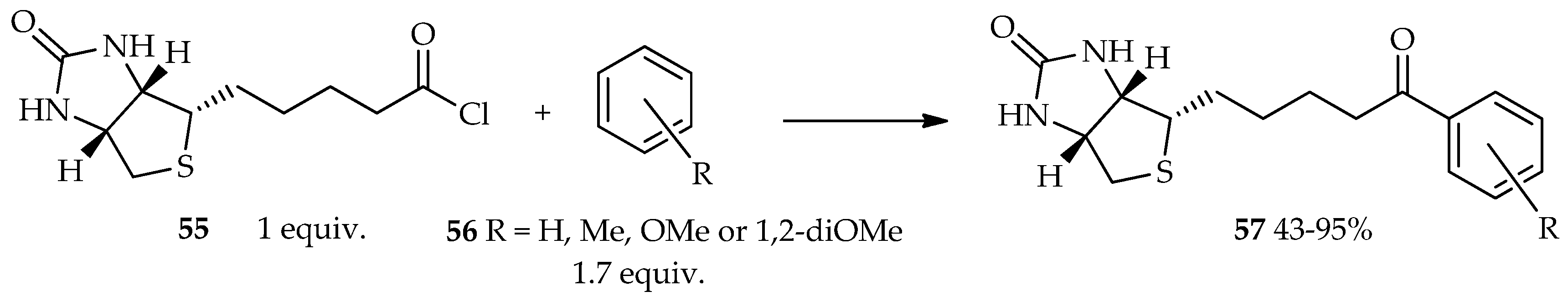



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
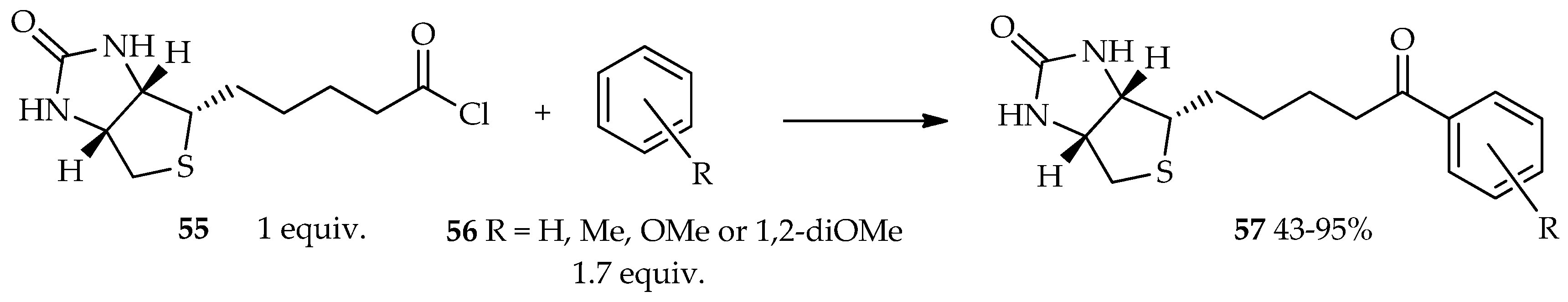{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
























