
Transition Metal-Modified Zirconium Phosphate Electrocatalysts for the Oxygen Evolution Reaction

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
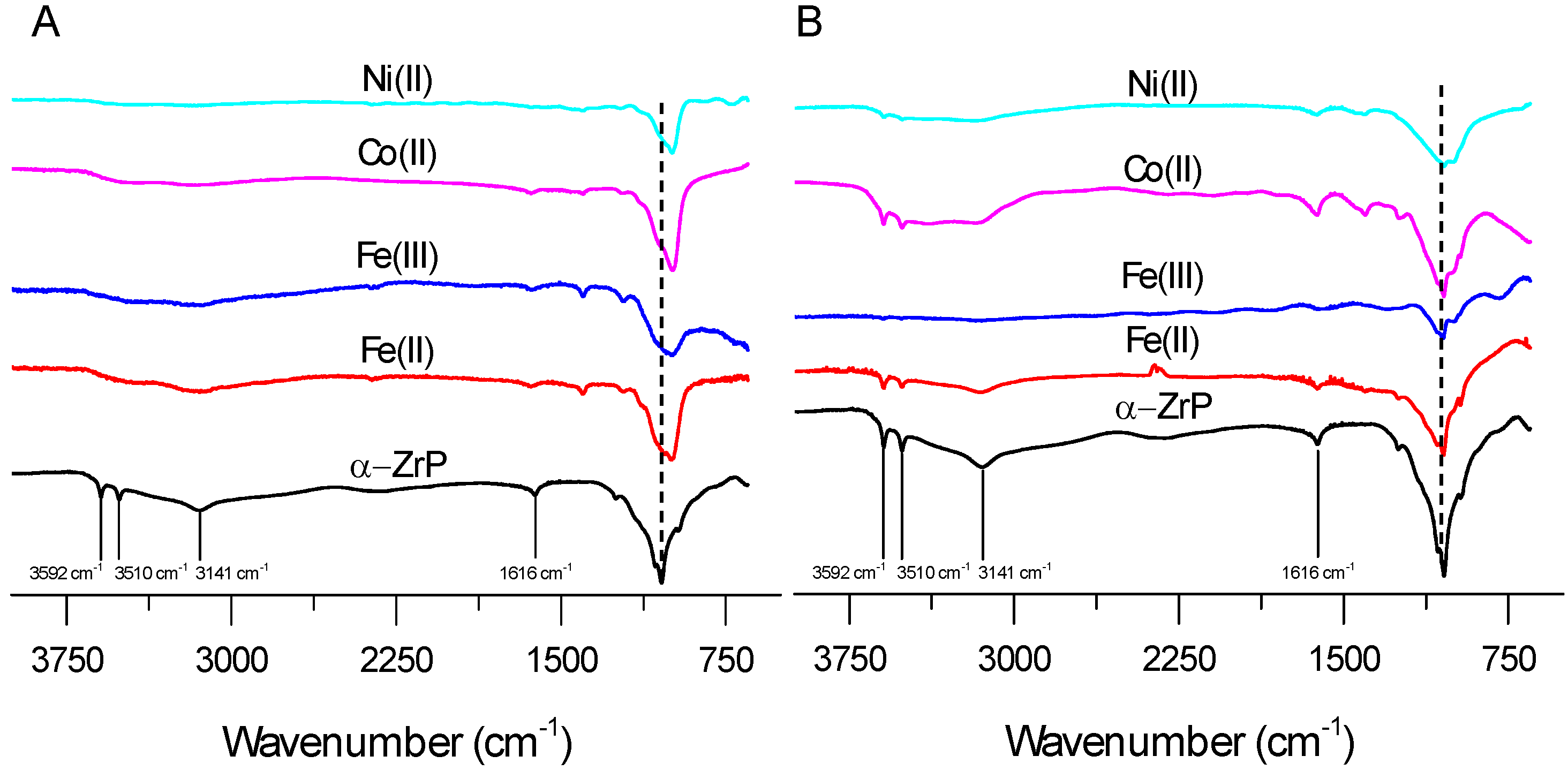
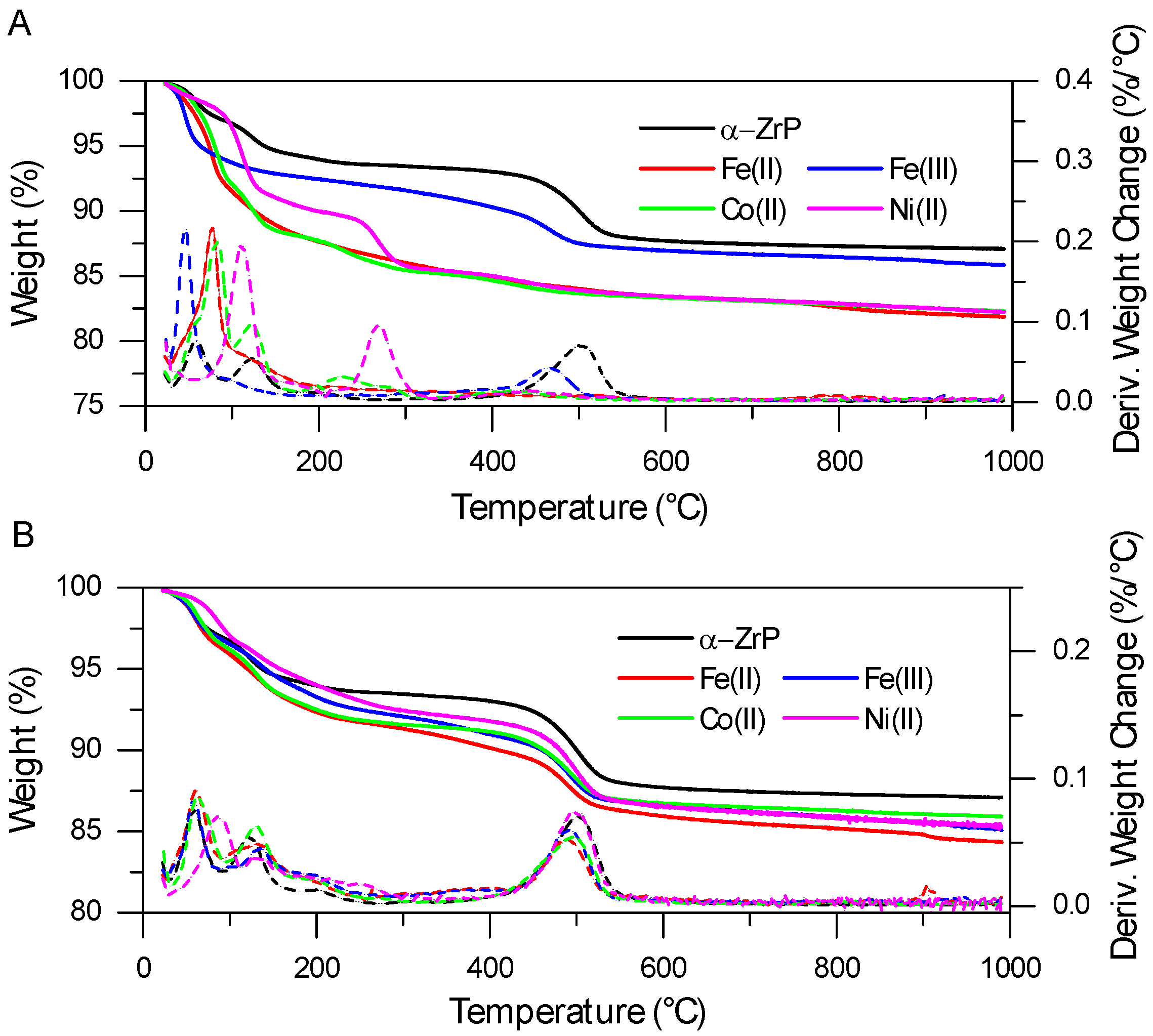
2.1. FTIR and TGA Analysis of Interlayer and Surface Modification of ZrP
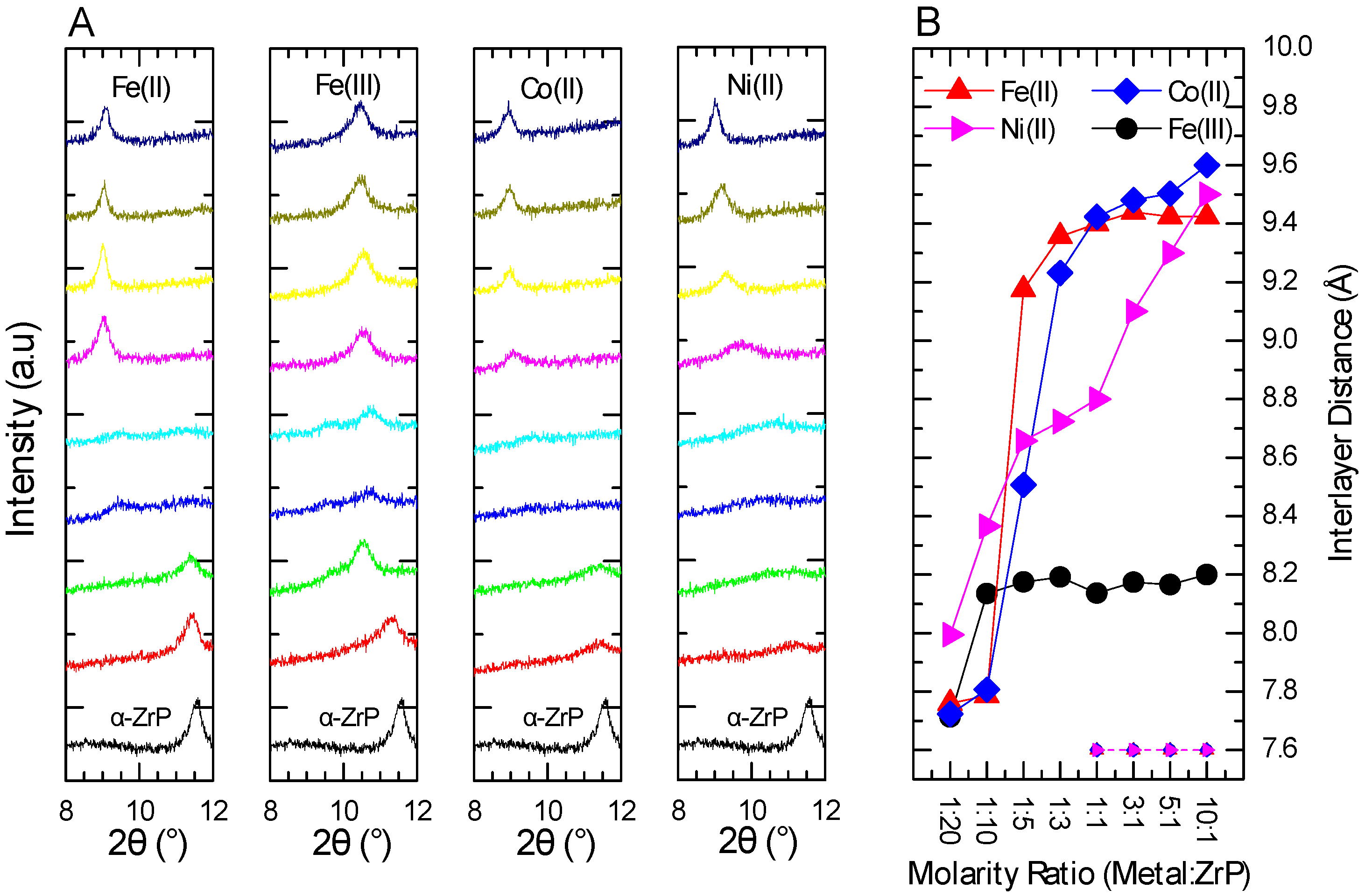
2.2. XRPD Interlayer Expansion Analysis of Metal-Modified ZrP
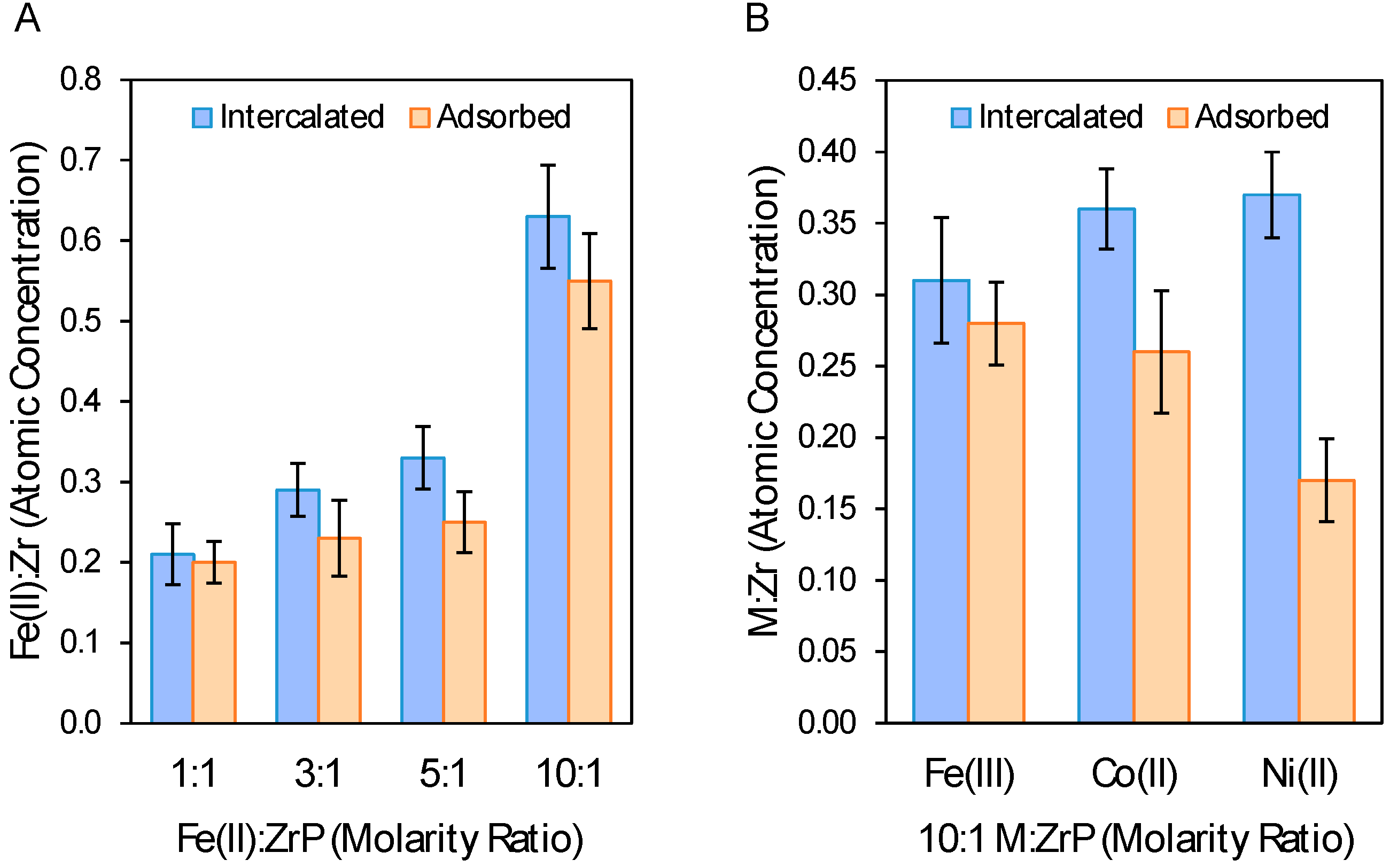
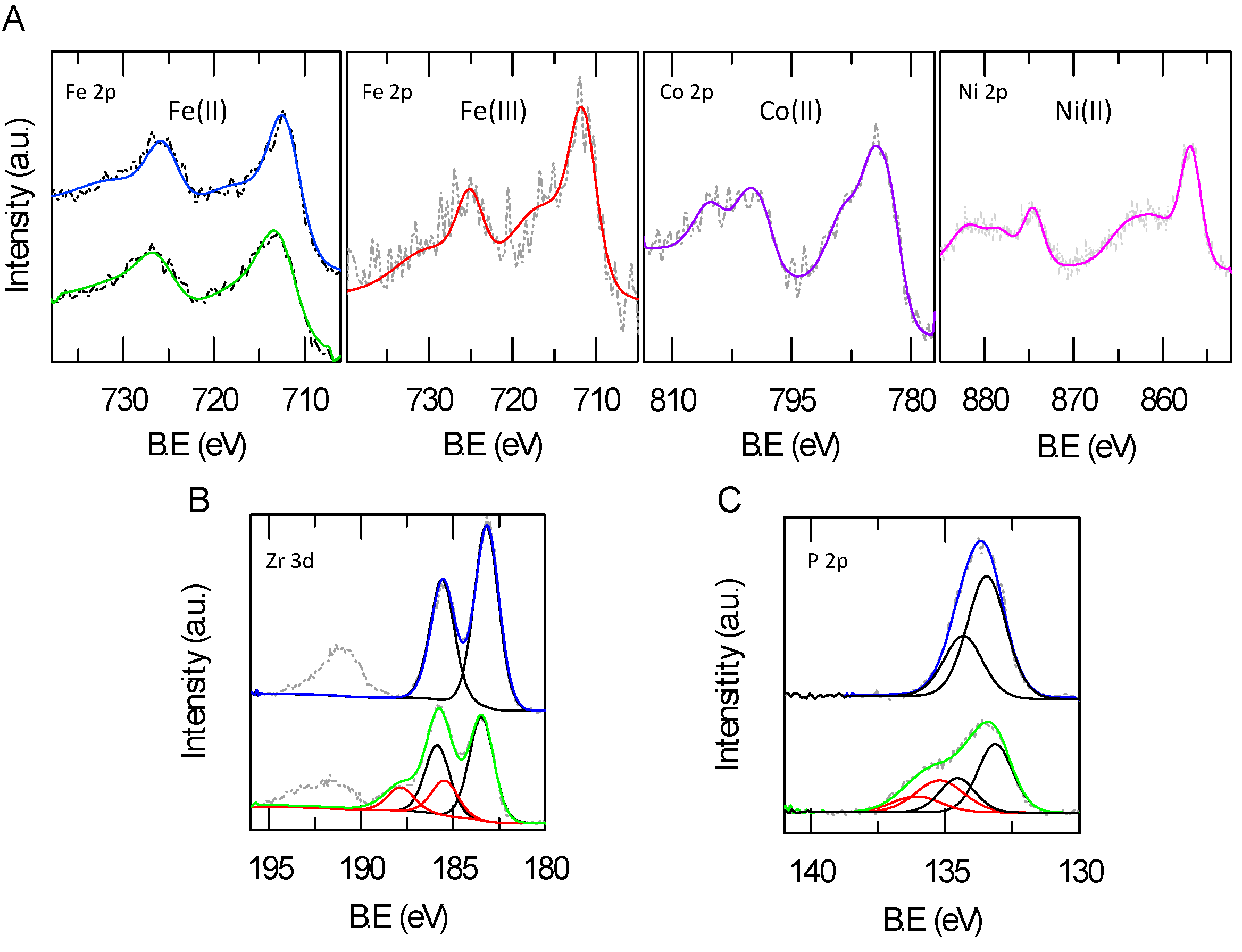
2.3. XPS Analysis of Metal Loading and Chemical Composition of Metal-Modified ZrP
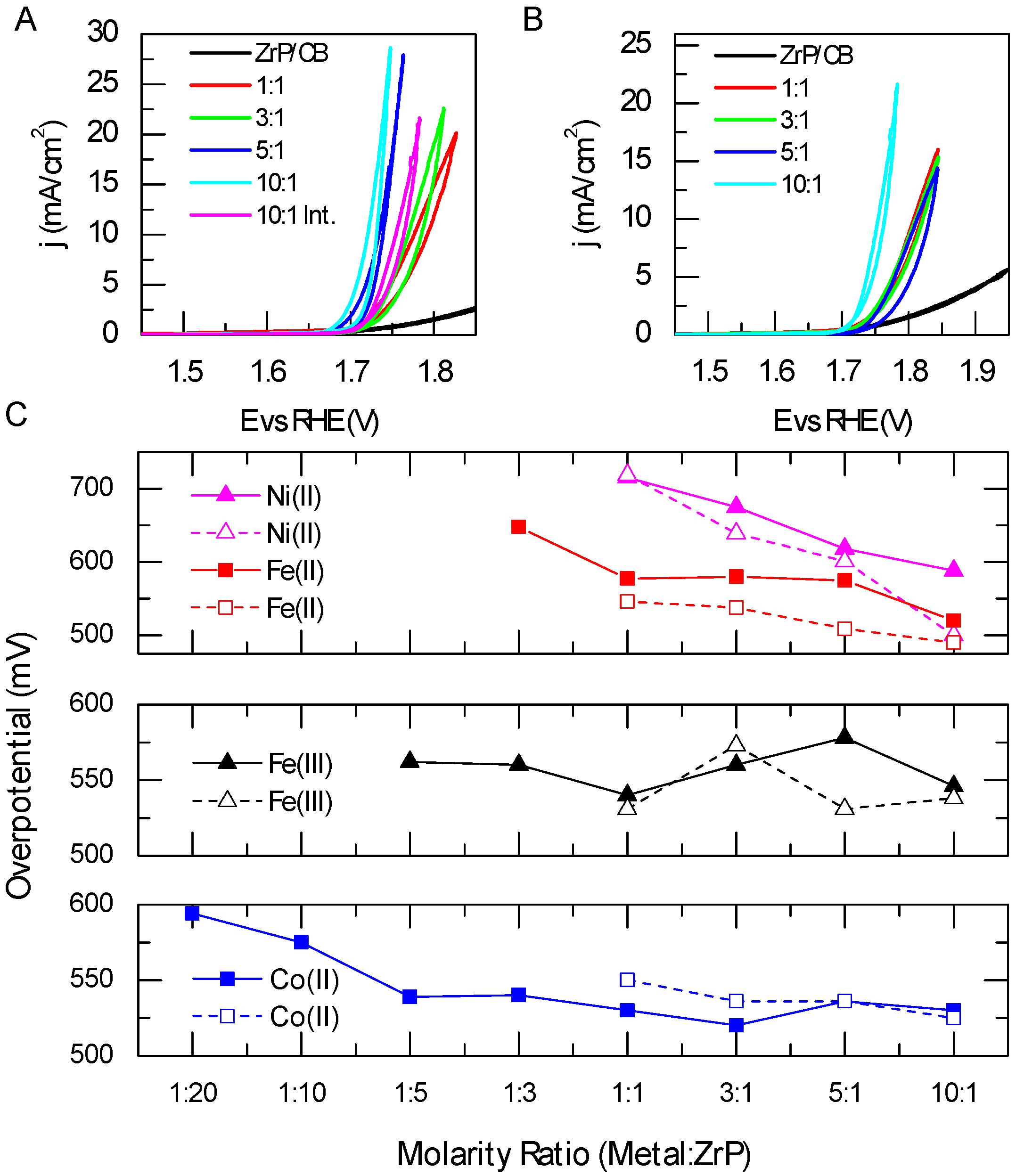
2.4. Electrochemical Studies of Metal-ModifiedZrP
3. Conclusion
4. Materials and Methods
4.1. Materials
4.2. θ-, α-, and Metal-Modified ZrP Synthesis
4.3. Physical and Chemical Characterization (TEM, FTIR, TGA, XRPD, XPS)
4.4. Ink Preparation and Electrochemical Measurements
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ursua, A.; Gandia, L.M.; Sanchis, P. Hydrogen production from water electrolysis: Current status and future trends. Proc. IEEE 2012, 100, 410–426. [Google Scholar] [CrossRef]
- Gahleitner, G. Hydrogen from renewable electricity: An international review of power-to-gas pilot plants for stationary applications. Int. J. Hydrogen Energy 2013, 38, 2039–2061. [Google Scholar] [CrossRef]
- Ehteshami, S.M.M.; Chan, S.H. The role of hydrogen and fuel cells to store renewable energy in the future energy network—Potentials and challenges. Energy Policy 2014, 73, 103–109. [Google Scholar] [CrossRef]
- Lewis, N.S.; Nocera, D.G. Powering the planet: Chemical challenges in solar energy utilization. Proc. Natl. Acad. Sci. USA 2006, 103, 15729–15735. [Google Scholar] [CrossRef] [PubMed]
- Gray, H.B. Powering the planet with solar fuel. Nat. Chem. 2009, 1, 7. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, J.M.; Huang, D.L.; Crabtree, R.H.; Brudvig, G.W. Iridium-based complexes for water oxidation. Dalton Trans. 2015, 44, 12452–12472. [Google Scholar] [CrossRef] [PubMed]
- Browne, M.P.; Nolan, H.; Duesberg, G.S.; Colavita, P.E.; Lyons, M.E. G. Low-overpotential high-activity mixed manganese and ruthenium oxide electrocatalysts for oxygen evolution reaction in alkaline media. ACS Catal. 2016, 6, 2408–2415. [Google Scholar] [CrossRef]
- Enman, L.J.; Burke, M.S.; Batchellor, A.S.; Boettcher, S.W. Effects of intentionally incorporated metal cations on the oxygen evolution electrocatalytic activity of nickel (oxy)hydroxide in alkaline media. ACS Catal. 2016, 6, 2416–2423. [Google Scholar] [CrossRef]
- Burke, M.S.; Enman, L.J.; Batchellor, A.S.; Zou, S.; Boettcher, S.W. Oxygen evolution reaction electrocatalysis on transition metal oxides and (oxy)hydroxides: Activity trends and design principles. Chem. Mater. 2015, 27, 7549–7558. [Google Scholar] [CrossRef]
- Burke, M.S.; Kast, M.G.; Trotochaud, L.; Smith, A.M.; Boettcher, S.W. Cobalt–iron (oxy)hydroxide oxygen evolution electrocatalysts: The role of structure and composition on activity, stability, and mechanism. J. Am. Chem. Soc. 2015, 137, 3638–3648. [Google Scholar] [CrossRef] [PubMed]
- Galán-Mascarós, J.R. Water oxidation at electrodes modified with earth-abundant transition-metal catalysts. ChemElectroChem 2015, 2, 37–50. [Google Scholar] [CrossRef]
- Antolini, E. Iridium as catalyst and cocatalyst for oxygen evolution/reduction in acidic polymer electrolyte membrane electrolyzers and fuel cells. ACS Catal. 2014, 4, 1426–1440. [Google Scholar] [CrossRef]
- Fabbri, E.; Habereder, A.; Waltar, K.; Kötz, R.; Schmidt, T.J. Developments and perspectives of oxide-based catalysts for the oxygen evolution reaction. Catal. Sci. Technol. 2014, 4, 3800–3821. [Google Scholar] [CrossRef]
- Suntivich, J.; May, K.J.; Gasteiger, H.A.; Goodenough, J.B.; Shao-Horn, Y. A Perovskite oxide optimized for oxygen evolution catalysis from molecular orbital principles. Science 2011, 334, 1383–1385. [Google Scholar] [CrossRef] [PubMed]
- McCrory, C.C.L.; Jung, S.; Ferrer, I.M.; Chatman, S.M.; Peters, J.C.; Jaramillo, T.F. Benchmarking hydrogen evolving reaction and oxygen evolving reaction electrocatalysts for solar water splitting devices. J. Am. Chem. Soc. 2015, 137, 4347–4357. [Google Scholar] [CrossRef] [PubMed]
- Man, I.C.; Su, H.-Y.; Calle-Vallejo, F.; Hansen, H.A.; Martínez, J.I.; Inoglu, N.G.; Kitchin, J.; Jaramillo, T.F.; Nørskov, J.K.; Rossmeisl, J. Universality in oxygen evolution electrocatalysis on oxide surfaces. ChemCatChem 2011, 3, 1159–1165. [Google Scholar] [CrossRef]
- Weng, B.; Xu, F.; Wang, C.; Meng, W.; Grice, C.R.; Yan, Y. A layered Na1−xNiyFe1−yO2 double oxide oxygen evolution reaction electrocatalyst for highly efficient water-splitting. Energy Environ. Sci. 2017, 10, 121–128. [Google Scholar] [CrossRef]
- Lu, Z.; Wang, H.; Kong, D.; Yan, K.; Hsu, P.-C.; Zheng, G.; Yao, H.; Liang, Z.; Sun, X.; Cui, Y. Electrochemical tuning of layered lithium transition metal oxides for improvement of oxygen evolution reaction. Nat. Commun. 2014, 5, 4345. [Google Scholar] [CrossRef] [PubMed]
- Doyle, A.D.; Montoya, J.H.; Vojvodic, A. Improving oxygen electrochemistry through nanoscopic confinement. ChemCatChem 2015, 7, 738–742. [Google Scholar] [CrossRef]
- Oh, H.-S.; Nong, H.N.; Reier, T.; Bergmann, A.; Gliech, M.; Ferreira de Araújo, J.; Willinger, E.; Schlögl, R.; Teschner, D.; Strasser, P. Electrochemical catalyst–support effects and their stabilizing role for IrOx nanoparticle catalysts during the oxygen evolution reaction. J. Am. Chem. Soc. 2016, 138, 12552–12563. [Google Scholar] [CrossRef] [PubMed]
- Wei Seh, Z.; Kibsgaard, J.; Dickens, C.F.; Chorkendorff, I.; Nørskov, J.K.; Jaramillo, T.F. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 2017, 355. [Google Scholar] [CrossRef]
- Wu, Z.-S.; Yang, S.; Sun, Y.; Parvez, K.; Feng, X.; Müllen, K. 3D Nitrogen-doped graphene aerogel-supported Fe3O4 nanoparticles as efficient electrocatalysts for the oxygen reduction reaction. J. Am. Chem. Soc. 2012, 134, 9082–9085. [Google Scholar] [CrossRef] [PubMed]
- Kou, R.; Shao, Y.; Wang, D.; Engelhard, M.H.; Kwak, J.H.; Wang, J.; Viswanathan, V.V.; Wang, C.; Lin, Y.; Wang, Y.; et al. Enhanced activity and stability of Pt catalysts on functionalized graphene sheets for electrocatalytic oxygen reduction. Electrochem. Commun. 2009, 11, 954–957. [Google Scholar] [CrossRef]
- Ma, T.Y.; Dai, S.; Jaroniec, M.; Qiao, S.Z. Graphitic carbon nitride nanosheet-carbon nanotube three-dimensional porous composites as high-performance oxygen evolution electrocatalysts. Angew. Chem. Int. Ed. 2014, 53, 7281–7285. [Google Scholar] [CrossRef] [PubMed]
- Troup, J.M.; Clearfield, A. On mechanism of ion exchange in zirconium phosphates. 20. Refinement of the crystal structure of α-zirconium phosphate. Inorg. Chem. 1977, 16, 3311–3314. [Google Scholar] [CrossRef]
- Alberti, G.; Costantino, U.; Gupta, J.P. Crystalline insoluble acid salts of tetravalent metals—XIX: Na+-catalyzed H+-Mg2+ and H+-Cs2+ ion exchanges on α-zirconium phosphate. J. Inorg. Nucl. Chem. 1974, 36, 2109–2114. [Google Scholar] [CrossRef]
- Alberti, G.; Bertrami, R.; Costantino, U. Crystalline insoluble acid salts of tetravalent metals—XXII: Effect of small amounts of Na+ on the ion exchange of alkaline earth metal ions on crystalline Zr(HPO4)2·H2O. J. Inorg. Nucl. Chem. 1976, 38, 1729–1732. [Google Scholar] [CrossRef]
- Alberti, G.; Costantino, U. Recent progress in the intercalation chemistry of layered α-zirconium phosphate and its derivatives, and future perspectives for their use in catalysis. J. Mol. Catal. 1984, 27, 235–250. [Google Scholar] [CrossRef]
- Marti, A.; Colón, J.L. Direct ion exchange of tris(2,2′-bipyridine)ruthenium(II) into an α-zirconium phosphate framework. Inorg. Chem. 2003, 42, 2830–2832. [Google Scholar] [CrossRef] [PubMed]
- Alberti, G.; Costantino, U.; Gill, J.S. Crystalline insoluble acid salts of tetravalent metals—XXIII: Preparation and main ion exchange properties of highly hydrated zirconium bis monohydrogen orthophosphates. J. Inorg. Nucl. Chem. 1976, 38, 1733–1738. [Google Scholar] [CrossRef]
- Atienzar, P.; Victoria-Rodríguez, M.; Juanes, O.; Rodríguez-Ubis, J.C.; Brunet, E.; García, H. Layered γ-zirconium phosphate as novel semiconductor for dye-sensitized solar cells: Improvement of photovoltaic efficiency by intercalation of a ruthenium complex-viologen dyad. Energy Environ. Sci. 2011, 4, 4718–4726. [Google Scholar] [CrossRef]
- Santiago, M.B.; Declet-Flores, C.; Díaz, A.; Vélez, M.M.; Zoé-Bosques, M.; Sanakis, Y.; Colón, J.L. Layered inorganic materials as redox agents: Ferrocenium-intercalated zirconium phosphate. Langmuir 2007, 23, 7810–7817. [Google Scholar] [CrossRef] [PubMed]
- Santiago, M.B.; Daniel, G.A.; David, A.; Casañas, B.; Hernández, G.; Guadalupe, A.R.; Colón, J.L. Effect of enzyme and cofactor immobilization on the response of ethanol oxidation in zirconium phosphate modified biosensors. Electroanalysis 2010, 22, 1097–1105. [Google Scholar] [CrossRef]
- Rivera, E.J.; Barbosa, C.; Torres, R.; Grove, L.; Taylor, S.; Connick, W.B.; Clearfield, A.; Colón, J.L. Vapochromic and vapoluminescent response of materials based on platinum(ii) complexes intercalated into layered zirconium phosphate. J. Mater. Chem. 2011, 21, 15899–15902. [Google Scholar] [CrossRef]
- Díaz, A.; Saxena, V.; González, J.; David, A.; Casañas, B.; Carpenter, C.; Batteas, J.D.; Colón, J.L.; Clearfield, A.; Hussain, M.D. Zirconium phosphate nanoplatelets: A novel platform for drug delivery in cancer therapy. Chem. Commun. 2012, 48, 1754–1756. [Google Scholar] [CrossRef] [PubMed]
- Horsley, S.E.; Nowell, D.V.; Stewart, D.T. The infrared and Raman spectra of α-zirconium phosphate. Spectrochim. Acta Part A Mol. Spectrosc. 1974, 30, 535–541. [Google Scholar] [CrossRef]
- Mosby, B.M.; Díaz, A.; Bakhmutov, V.; Clearfield, A. Surface functionalization of zirconium phosphate nanoplatelets for the design of polymer fillers. ACS Appl. Mater. Interfaces 2014, 6, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Szirtes, L.; Szeleczky, A.M.; Rajeh, A.O.; Kuzmann, E. Thermal behaviour of transition metal-containing zirconium phosphate. J. Therm. Anal. Calorim. 1999, 56, 447–451. [Google Scholar] [CrossRef]
- Alberti, G.; Casciola, M.; Costantino, U.; Vivani, R. Layered and pillared metal(IV) phosphates and phosphonates. Adv. Mater. 1996, 8, 291–303. [Google Scholar] [CrossRef]
- Alagna, L.; Prosperi, T.; Tomlinson, A.A.G.; Ferragina, C.; La Ginestra, A. Extended X-ray absorption fine structure investigation of local symmetry changes in Co2+- and Ni2+-exchanged zirconium phosphates. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Ph. 1983, 79, 1039–1047. [Google Scholar] [CrossRef]
- Alagna, L.; Tomlinson, A.A.G.; Ferragina, C.; La Ginestra, A. Zirconium phosphates partially exchanged with transition-metal ions: Characterisation and stereochemical changes induced by heat treatment. J. Chem. Soc. Dalton Trans. 1981, 2376–2384. [Google Scholar] [CrossRef]
- Mani, B.; de Neufville, J.P. Dehydration of chemically and electrochemically impregnated (CI and EI) nickel hydroxide electrodes. J. Electrochem. Soc. 1988, 135, 800–803. [Google Scholar] [CrossRef]
- Miessler, G.L.; Fischer, P.J.; Tarr, D.A. Inorganic Chemistry, 5th ed.; Pearson Education: London, UK, 2014; pp. 374–377. [Google Scholar]
- Casañas-Montes, B.; Díaz, A.; Barbosa, C.; Ramos, C.; Collazo, C.; Meléndez, E.; Queffelec, C.; Fayon, F.; Clearfield, A.; Bujoli, B.; et al. Molybdocene dichloride intercalation into zirconium phosphate nanoparticles. J. Organomet. Chem. 2015, 791, 34–40. [Google Scholar] [CrossRef]
- Sun, L.; Boo, W.J.; Sue, H.-J.; Clearfield, A. Preparation of α-zirconium phosphate nanoplatelets with wide variations in aspect ratios. New J. Chem. 2007, 31, 39–43. [Google Scholar] [CrossRef]
- Costantino, U.; Szirtes, L.; Kuzmann, E.; Megyeri, J.; Lázár, K. Exchange of iron ions into layers of α-zirconium phosphate. Solid State Ion. 2001, 141, 359–364. [Google Scholar] [CrossRef]
- Yamashita, T.; Hayes, P. Analysis of XPS spectra of Fe2+ and Fe3+ ions in oxide materials. Appl. Surf. Sci. 2008, 254, 2441–2449. [Google Scholar] [CrossRef]
- Nemoshkalenko, V.V.; Didyk, V.V.; Krivitskii, V.P.; Senekevich, A.I. Investigation of the atomic charges in iron, cobalt and nickel phosphides. Zhurnal Neorganicheskoi Khimii 1983, 28, 2182–2192. [Google Scholar]
- Hernández-Huesca, R.; Braos-Garcı́a, P.; Mérida-Robles, J.; Maireles-Torres, P.; Rodrı́guez-Castellón, E.; Jiménez-López, A. Cobalt-based alumina pillared zirconium phosphate catalysts for the selective catalytic reduction of NO by propane. Chemosphere 2002, 48, 467–474. [Google Scholar] [CrossRef]
- Nardi, K.L.; Yang, N.; Dickens, C.F.; Strickler, A.L.; Bent, S.F. Creating highly active atomic layer deposited NiO electrocatalysts for the oxygen evolution reaction. Adv. Energy Mater. 2015, 5, 1500412. [Google Scholar] [CrossRef]
- Cheikhi, N.; Ziyad, M. Methane conversion to higher alkanes over ruthenium and cobalt loaded Zr3(PO4)4. J. Phys. IV 2005, 123, 301–305. [Google Scholar] [CrossRef]
- Zhan, Y.; Lu, M.; Yang, S.; Liu, Z.; Lee, J.Y. The origin of catalytic activity of nickel phosphate for oxygen evolution in alkaline solution and its further enhancement by iron substitution. ChemElectroChem 2016, 3, 615–621. [Google Scholar] [CrossRef]
- Colón, J.L.; Thakur, D.S.; Yang, C.-Y.; Clearfield, A.; Martin, C.R. X-ray photoelectron spectroscopy and catalytic activity of α-zirconium phosphate and zirconium phosphate sulfophenylphosphonate. J. Catal. 1990, 124, 148–159. [Google Scholar] [CrossRef]
- Arfelli, M.; Mattogno, G.; Ferragina, C.; Massucci, M.A. XPS characterization of γ-zirconium phosphate and of some of its intercalation compounds. A comparison with the α-zirconium phosphate analogues. J. Incl. Phenom. Mol. Recognit. Chem. 1991, 11, 15–27. [Google Scholar] [CrossRef]
- Xie, J.; Wang, R.; Bao, J.; Zhang, X.; Zhang, H.; Li, S.; Xie, Y. Zirconium trisulfide ultrathin nanosheets as efficient catalysts for water oxidation in both alkaline and neutral solutions. Inorg. Chem. Front. 2014, 1, 751–756. [Google Scholar] [CrossRef]
- Szirtes, L.; Megyeri, J.; Kuzmann, E.; Klencsár, Z. Electrical conductivity of transition metal containing crystalline zirconium phosphate materials. Solid State Ion. 2001, 145, 257–260. [Google Scholar] [CrossRef]
- Kijima, T. Direct preparation of θ-zirconium phosphate. Bull. Chem. Soc. Jpn. 1982, 55, 3031–3032. [Google Scholar] [CrossRef]







© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanchez, J.; Ramos-Garcés, M.V.; Narkeviciute, I.; Colón, J.L.; Jaramillo, T.F. Transition Metal-Modified Zirconium Phosphate Electrocatalysts for the Oxygen Evolution Reaction. Catalysts 2017, 7, 132. https://doi.org/10.3390/catal7050132
Sanchez J, Ramos-Garcés MV, Narkeviciute I, Colón JL, Jaramillo TF. Transition Metal-Modified Zirconium Phosphate Electrocatalysts for the Oxygen Evolution Reaction. Catalysts. 2017; 7(5):132. https://doi.org/10.3390/catal7050132
Chicago/Turabian StyleSanchez, Joel, Mario V. Ramos-Garcés, Ieva Narkeviciute, Jorge L. Colón, and Thomas F. Jaramillo. 2017. "Transition Metal-Modified Zirconium Phosphate Electrocatalysts for the Oxygen Evolution Reaction" Catalysts 7, no. 5: 132. https://doi.org/10.3390/catal7050132
APA StyleSanchez, J., Ramos-Garcés, M. V., Narkeviciute, I., Colón, J. L., & Jaramillo, T. F. (2017). Transition Metal-Modified Zirconium Phosphate Electrocatalysts for the Oxygen Evolution Reaction. Catalysts, 7(5), 132. https://doi.org/10.3390/catal7050132