1. Introduction
Biodiesel is made from renewable stock including vegetable oils and animal fats via a transesterification reaction. Glycerol is produced in huge quantities from the process of biodiesel production. Hence, much effort is made to convert glycerol into useful products by a variety of catalytic reactions [
1,
2,
3]. Glycerol can be utilized for the synthesis of biodiesel additives as well as industrial solvents such as propylene glycols, glycerol ethers, esters, glycerol carbonate, glyceric acids, and other products. Glycerol as oxygenated additive and cannot be added directly to fuel because it can be decomposed and polymerized at high temperatures in the engine. Therefore, glycerol is converted to the compounds that can be used as fuel additives [
4]. For example, acetines, especially diacetin (DA) and triacetin (TA), are used as important additives to improve the cold properties and decrease the viscosity of biodiesel.
Acetines can be synthesized by reaction between glycerol and acetic acid over homogeneous mineral acids, such as H
2SO
4, HCl, and H
3PO
4 [
5], and heterogeneous solid acids including sulfonic acid groups supported mesoporous silica [
6], niobic acid, Starbon acid, niobic acid supported heteropolytungstate, dodecatungstophosphoric acid immobilized on to a silica matrix, supported ionic liquids, TPA supported on Cs-containing zirconia, Amberlysts [
7,
8], SO
42–/γ-Al
2O
3 and Cu-Ni/γ-Al
2O
3 catalyst [
5], zeolites, Lewis acid SnCl
2, heteropolyacids loaded materials, acid-treated montmorillonite clay, and some other catalysts [
8,
9,
10,
11]. These heterogeneous catalysts can prevent reactor corrosion, difficult catalyst separation, and extra neutralization of the homogeneous catalyst and production of waste and side products. However, the solid acid catalysts have some disadvantages such as the small amount of acidic sites, low thermal stability, quick loss of catalytic activity, and pore-size restriction (as zeolites with small pore size provide poor selectivity to DA and TA) [
5,
12]. Moreover, water could deactivate the acidic sites due to competition with the reactants or cause leaching of the hydrophilic support of the solid catalysts and inhibit the esterification reaction which results in low selectivity for the favored DA and TA [
9,
12]. Thus, a water-tolerant solid acid catalyst or water removal substances are necessary for the esterification of glycerol [
4,
8]. However, lower usability, poor hydrothermal stability, and low selectivity to TA has been presented due to the catalyst preparation methods. It was described that catalysts provided by a grafting method via reaction of the functional groups of the support with the metal precursor can prevent migration, agglomeration, and sintering throughout the following thermal treatments. Regarding the disadvantages, catalysts prepared by wet impregnation method, progressing through the chemi- or physisorption of metal groups onto the support surface, suffer from remarkable leaching and present low selectivity to TA. For solving these problems, mesoporous silicate and aluminosilicate samples can be considered for glycerol esterification with acetic acid. Despite the porosity and large pore diameter of these catalysts, some problems such as the low degree of effective acidic sites over these mesoporous solids exist. However, these drawbacks can be improved by acidifying and functionalizing mesoporous solids with sulfonic groups [
1,
9].
The modification of bentonite clays was studied by pillaring or intercalation of complexes, anions, and organic compounds between layers of clay minerals. These modifying processes caused different properties and reactivities that increased the requests for this group of clay minerals as catalysts [
13].
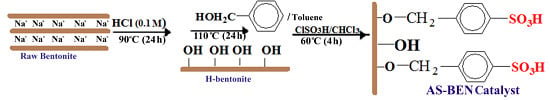
In this work, a reusable, environmentally friendly, and inexpensive catalyst with good catalytic performance and thermal stability was produced using bentonite as a support, grafted by benzylsulfonic acid groups. The catalyst performance was then examined for the esterification of glycerol with acetic acid. The reaction was studied under reaction parameters optimized in our previous work [
14] and a Dean-Stark apparatus was used to separate water from the products.
2. Results and Discussion
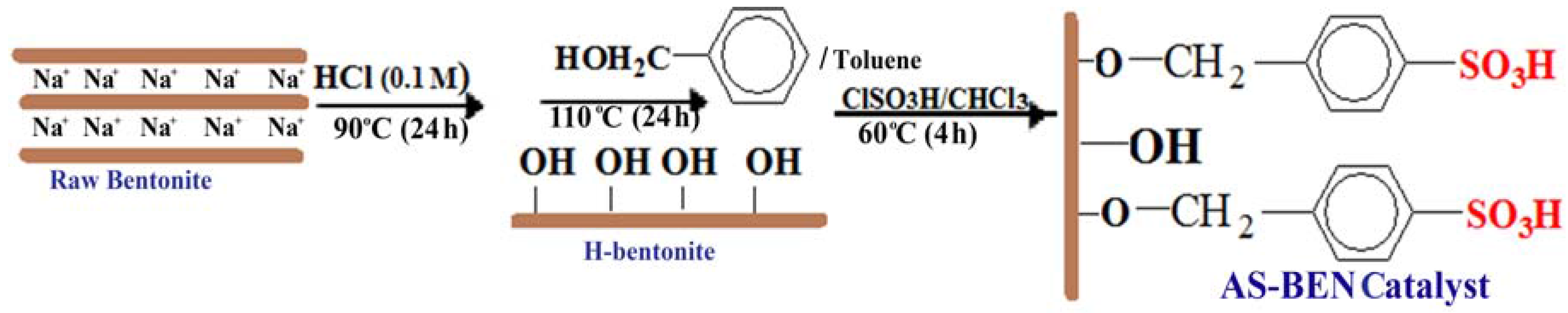
Heterogeneous catalysts have shown good activity for glycerol esterification but big challenges such as low reusability, poor thermal stability, and selectivity to TA still exist for the proper design of an appropriate solid catalyst in esterification reactions. In fact, the water produced in the esterification reaction may lead to deactivation of acidic sites. One of the methods for preparing stable catalyst involves grafting of a modifying agent into surface of inorganic supports and continuous removal of water from the reaction medium. Therefore, we decided to modify natural bentonite (BEN) as a support for manufacturing arenesulfonic acid-functionalized catalyst using the following procedure (
Scheme 1). Raw bentonite was activated by HCl and benzyl alcohol, respectively, to give arylbentonite (Ar-BEN) with covalent bonds between the hydroxyl groups of the bentonite layer and benzyl alcohol molecules. The benzyl groups then were reacted with ClSO
3H to produce arenesulfonic acid bentonite (AS-BEN) as catalyst.
Acid activation with 0.1 M HCl was performed to increase the number of hydroxyl sites on the clay mineral surfaces by formation of structural imperfections in the tetrahedral sheet and by substituting interlayer cations (Na
+, K
+ and Ca
2+) with hydronium ions without significant leaching of Al from the bentonite framework and without the loss of the layer pattern [
15].
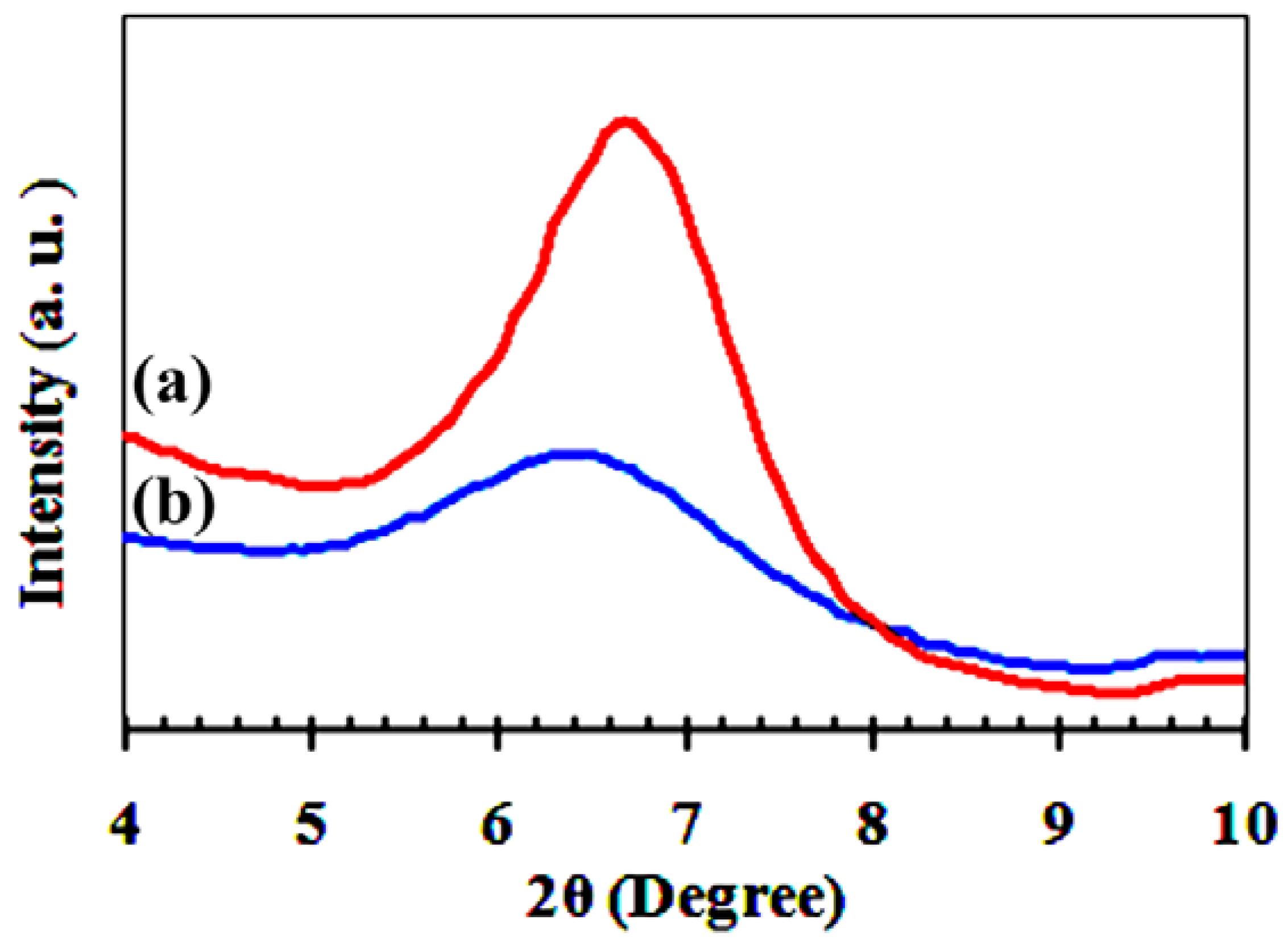
X-ray diffraction (XRD) patterns of acidified bentonite (H-BEN) and AS-BEN are presented as evidence for the demonstration of modified clays. According to the XRD patterns (
Figure 1), after grafting of arylsulfonic acid groups to H-BEN, the characteristic peaks corresponding to montmorillonite at 2θ = 6.8° (d
001 = 1.293 nm) shift to a lower diffraction angle (2θ = 6.4°, d
001 = 1.381 nm) [
15,
16]. This shift towards a lower angle confirms the intercalation and grafting of arylsulfonic acid groups in the internal and external surfaces of H-BEN [
16].
Field emission scanning electron microscopy (FE-SEM) was utilized to study the morphology of modified bentonite.
Figure 2 shows scanning electron microphotographs of AS-BEN. The characteristic layered structure of clay was retained after functionalization of raw bentonite.
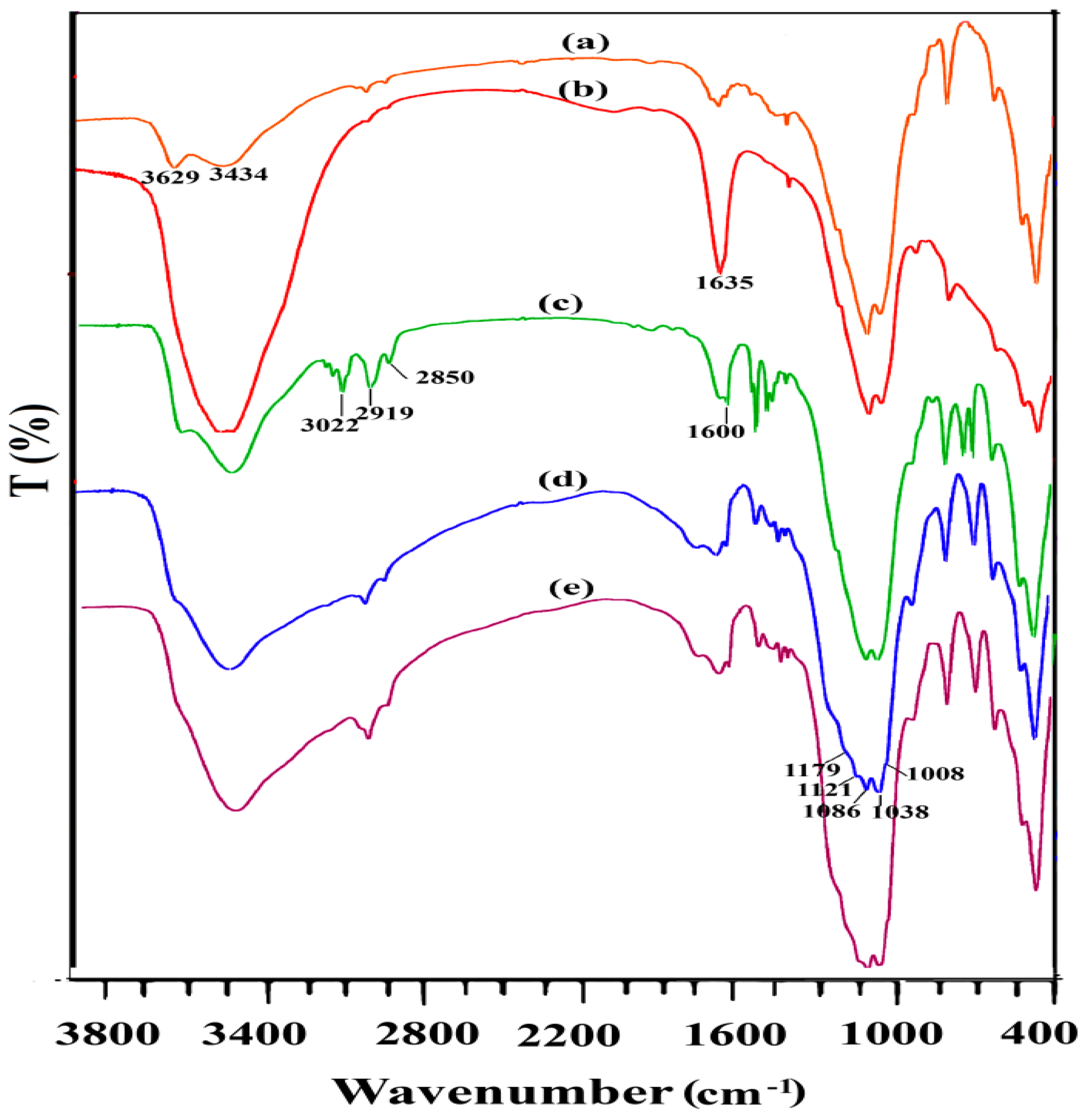
All spectra in
Figure 3 show vibrational bands at 1089 cm
−1, 519 cm
−1, and 469 cm
−1 attributed to the stretching vibrations of Si-O-Si groups and bending vibration of Si-O-Al and Si-O-Si, respectively. The bands at 3434 cm
−1 and 1635 cm
−1 attributed to the stretching vibration and corresponding bending vibration bands from the physically adsorbed water. The relatively strong IR absorption bands around 3629 cm
−1 are attributed to the stretching vibration of the Mg-OH, Al-OH, or Fe-OH structural hydroxyl groups. Bands at 2850–2935 cm
−1 were assigned to the -C-H asymmetric and symmetric stretching of the methylene groups and the several bands at 3020–3060 cm
−1 were attributed to the stretching vibrations -C-H of aromatic groups which confirmed incorporation of the aromatic ring on the surface [
13,
16,
17]. In addition, for the Ph-SO
3H group, vibrational bands around 1008, 1036, and 1178 cm
−1 were attributed to the O=S=O symmetric stretching, C-S stretching, and O=S=O asymmetric stretching modes of vibration, respectively. However, these bands may overlap the stretching bands of Si-O bonds [
18].
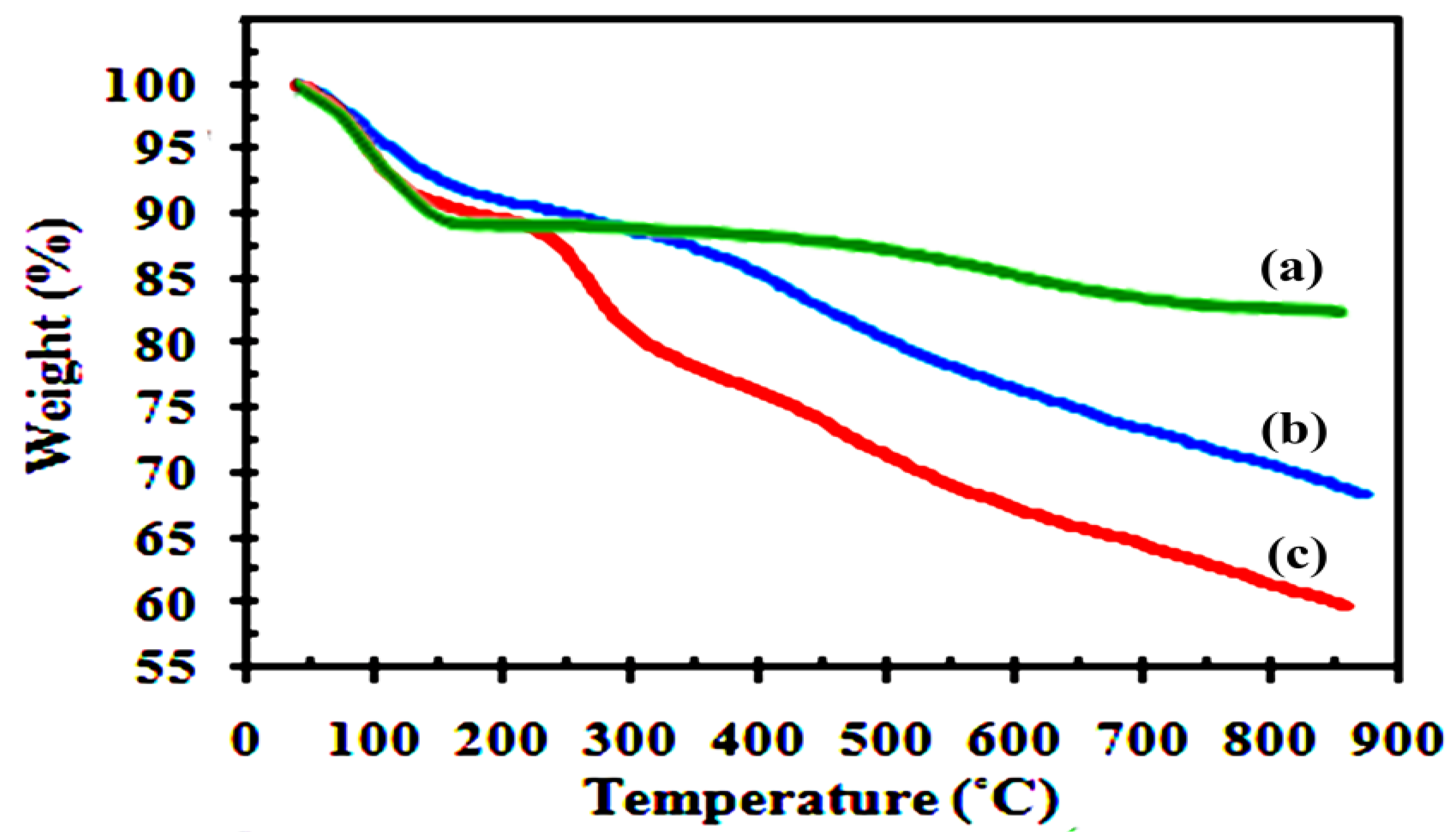
Figure 4 shows thermogravimetric curves for H-BEN, Ar-BEN, and AS-BEN catalyst prior to use in the esterification reaction. For unmodified H-BEN, the onset of 11.7% weight loss starts at 30 °C and ends at 150 °C, and is caused by desorption of physically adsorbed water from the outer surfaces and between bentonite layers. The second loss-stage (4.5%) at T > 500 °C can be due to the dehydroxylation of the silicate layers. The Ar-BEN displayed extra weight loss (15%) at about 200–600 °C assigned to the loss of bonded benzyl alcohol molecules. The first weight loss between 200–500 °C was assigned to the benzyl alcohol molecules grafted on the external surface and at the edge of the clay, while the weight loss at about 550 °C was attributed to the strongly grafted benzyl alcohol molecules in the interlayer regions. In AS-BEN, three stages of decomposition were observed. It is noteworthy that the weight loss of approximately 9% at 250–350 °C can be due to the desulfonation of the sulfonic acid groups [
15,
16,
18].
The BET surface area, pore volume, and average pore diameter of BEN, H-BEN, and AS-BEN, were summarized in
Table 1. The specific surface area was obtained from the linear part of the BET plot. The average pore diameter, pore volume, and pore size distribution were calculated from the N
2 adsorption-desorption isotherms using the Barrett-Joyner-Halenda formula. In the raw bentonite, dry platelets of bentonite which are composed of one octahedral alumina sheet placed between two tetrahedral silica sheets, are most commonly grouped together in face-to-face arrangement, with exchangeable cations and small amounts of water in an interlayer region between each platelet. The intercalation of aryl groups in an interlayer region and the subsequent sulfonation of the intercalated groups (shown in XRD patterns) result in the formation of hydrogen bonding between sulfonic acid functions in the interlayer space, and due to the occupation or even blocking of some solid pores by the modifying agents, the specific surface area and the total pore volume are smaller than that of the H-bentonite. For the AS-BEN catalyst, the average pore diameter was increased due to the reduction of microporous contribution [
13,
15,
19]. Nonetheless, some surface areas and porosities were kept in all the meso structures after modification of the pore layers. Hence, when the AS-BEN catalyst was utilized, the reaction takes place in the pores, inner layers, and on the outer surface of the particles.
The acid capacities of H-BEN and AS-BEN were measured by a neutralization back titration procedure (
Table 1). For both samples, 0.1 g of solid powder was dispersed in 10 mL of 0.1 M NaOH solution and slowly shaken at ambient temperature for 24 h. The mixture was finally centrifugedand then the separated alkali solution was titrated against a 0.1 M HCl standard solution. The concentration of acidic sites (C) was calculated as follow [
9]:
In order to investigate the catalytic performance of AS-BEN, we performed the esterification of glycerol with acetic acid as a model reaction. The synthesis of glycerol ester was performed in toluene at optimized conditions of reaction examined in our previous work, acetic acid:glycerol molar ratio of 7, 0.074 wt % of catalyst (based on the total weight of glycerol), and 100 °C. Since water produced as a byproduct could prevent the progress of the reaction, the Dean-Stark trap with toluene in the side arm was used to remove water to significantly improve the glycerol conversion [
4,
20]. The first reaction was performed in toluene as solvent without adding the solid catalyst (blank run) at 0.5 h and 3 h. As can be seen from
Table 2, for both cases conversion of glycerol reached 47% and 58%, respectively. In the first 0.5 h, the MA selectivity (85%) was higher than DA and TA; as the reaction extended, the DA selectivity increased and its maximum selectivity was achieved (36%) within 3 h. The glycerol conversion and particularly high monoacetin selectivity in the control reaction implied that the synthesis of MA was considerably easier than its acetylation to DA and TA because the Gibb’s free energy for synthesis of MA (19.2 kJ·mol
−1) is substantially lower than those of DA (37.0 kJ·mol
−1) and TA (92.5 kJ·mol
−1) [
21]. The increase in selectivity for DA and TA within the reaction time is mainly due to the further acetylation of MA.
The above results reflect the contribution of the acetic acid (as weak acidic catalyst), temperature (in endothermic esterification), and toluene as a water removing agent in the progress of glycerol esterification [
4,
22]. It is noted that the conversion and selectivity of glycerol esterification depends on the nature of the catalyst and the reaction parameters. To investigate the catalytic effect of H-bentonite in the present work, the same mole of H-bentonite was utilized. The results showed that H-BEN compared to an AS-BEN catalyst presented an insignificant catalytic effect in esterification of glycerol with acid acetic at an acetic acid/glycerol molar ratio of 7, as show in
Table 2. The comparison of H-BEN and AS-BEN catalysts in glycerol esterification reaction performed here implied that the sulfonic acid-functionalized catalyst has a higher catalytic activity for the esterification. The results proposed that configuration of the surface acid moieties may play an important role in the intrinsic kinetics of glycerol esterification, in addition to the number and the strength of the acidic sites [
21].
In similar reaction conditions, the AS-BEN catalyst was employed to promote the esterification reaction of glycerol with acetic acid. The typical glycerol conversion and product selectivity in different reaction times are summarized in
Table 2. Rapid glycerol conversion in the early stage of the reaction can be explained by a better approachability of active sites of AS-BEN as catalyst which promotes glycerol conversion to MA.
In addition to the increasing conversion, the application of the AS-BEN catalyst strongly affects the selectivity of dominant products. In the first 0.5 h, DA selectivity of 66% and MA selectivity of 25% were achieved. The high TA selectivity of 74% was obtained during 3 h and within this period, the formation of other oxygenated hydrocarbons as byproducts was not detected at all. The considerable selectivity for TA demonstrated a high activity of the catalyst for formation of TA rather than DA as the product.
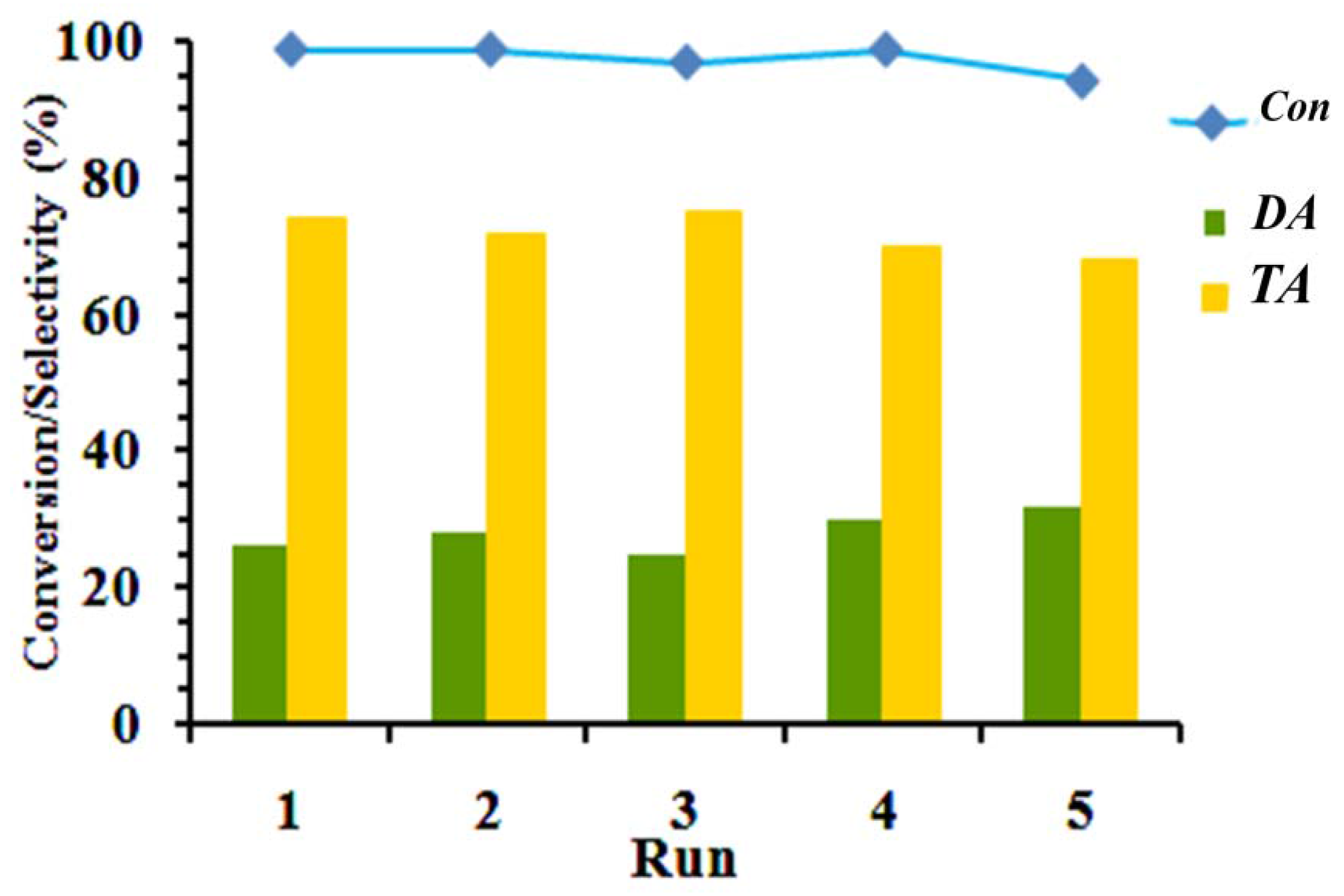
The critical factor for the application of any catalyst for industrial purposes is reusability of the catalyst after its consumption. The stability of the catalyst for glycerol conversion and product selectivity was investigated by performing consecutive batch reactions under toluene medium at an acetic acid:glycerol molar ratio of 7, 0.074 wt % of catalyst (based on the total weight of glycerol), and 100 °C. For using the catalyst in the next run, the solid catalyst was separated from the reaction products by centrifugation, washed in ethanol, and dried to remove deactivating components from the catalyst surface, each cycle was continued for 3 h.
As shown in
Figure 5, glycerol conversion and the selectivity to DA and TA do not change considerably, even after reutilization in 5 tests. Moreover, the IR spectrum of the fresh and recycled AS-BEN in
Figure 3 demonstrated a resistant catalyst structure after five tests.
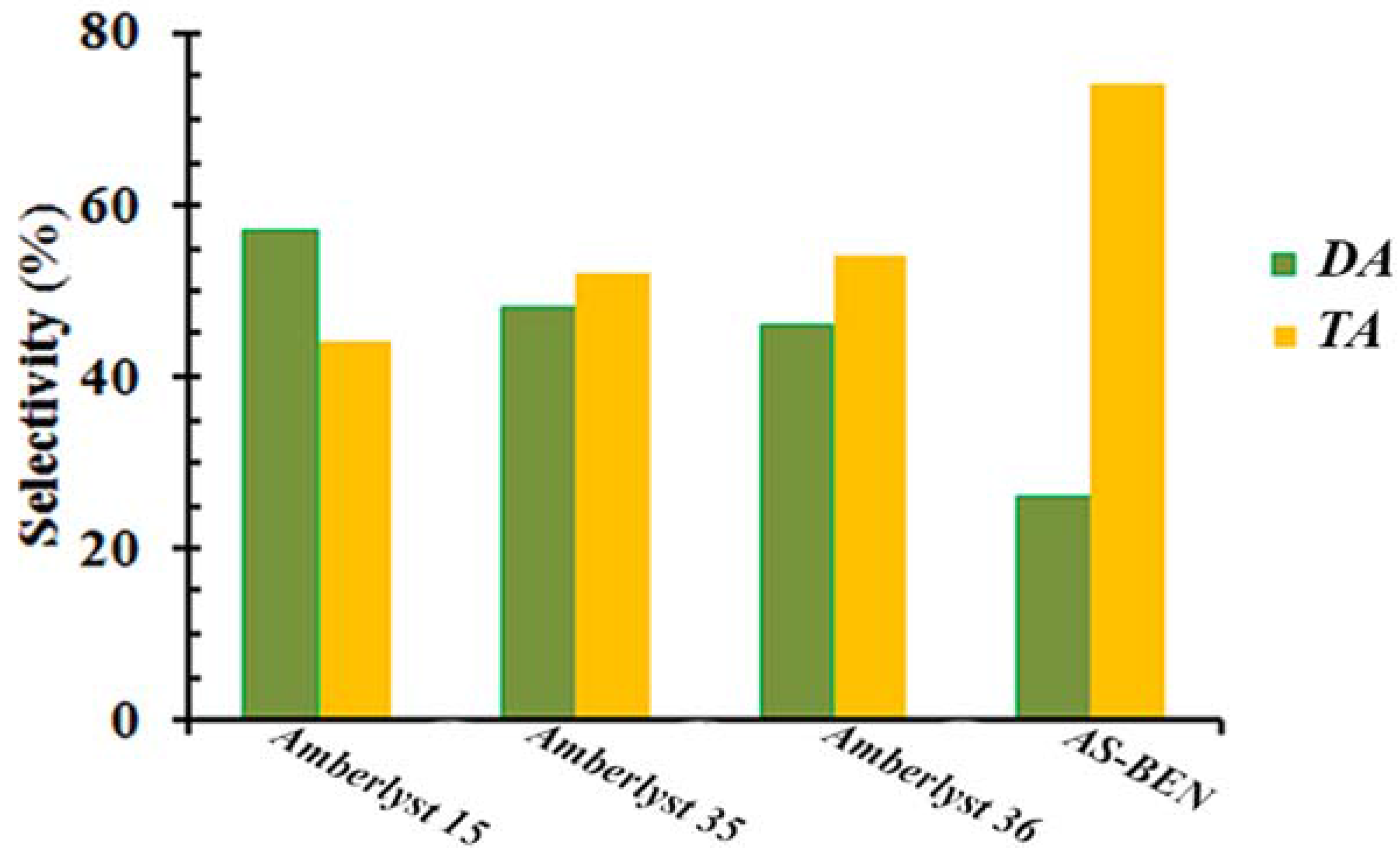
We also compared the catalytic activity of the synthesized catalyst with other solid acidic catalysts, some commercial sulfonic acids such as Amberlyst 15, 35, and 36 with the number of acid sites 4.7, 5.2, and 5.4 mmol·g
−1, respectively, in glycerol esterification with acetic acid.
Figure 6 shows the selectivity to acetins produced by each catalyst at the selected reaction time (3 h). All of the catalysts with the sulfonic acid groups used in the esterification displayed 100% glycerol conversion. The higher selectivity to the most desirable product (TA) over the AS-BEN was reached at 3 h, compared to Amberlyst 15, 35 and 36.
It is important to note that the Amberlysts 35 and 36 could not be recovered at the end of the reaction and these catalysts are partially disintegrated after 3 h and fully disintegrated and dissolved in the reaction medium after 8 h. These observations prove that the catalysts are physically/thermally instable in the reaction medium at 100 °C in the first hours of the reaction. Therefore, the stability of Amberlyst 36 was examined in only toluene that is used as an azeotropic agent in this work, at 100 °C. At this condition, the catalyst was completely stable even after 11 h. That is why our previous results in a continuous system with low product residence time over the catalyst [
14] are not repeated in a batch system. This work showed that the presence of the catalyst in the reaction medium in a batch system for a long time reduces the stability of Amberlysts 36 and dissolves the catalyst.
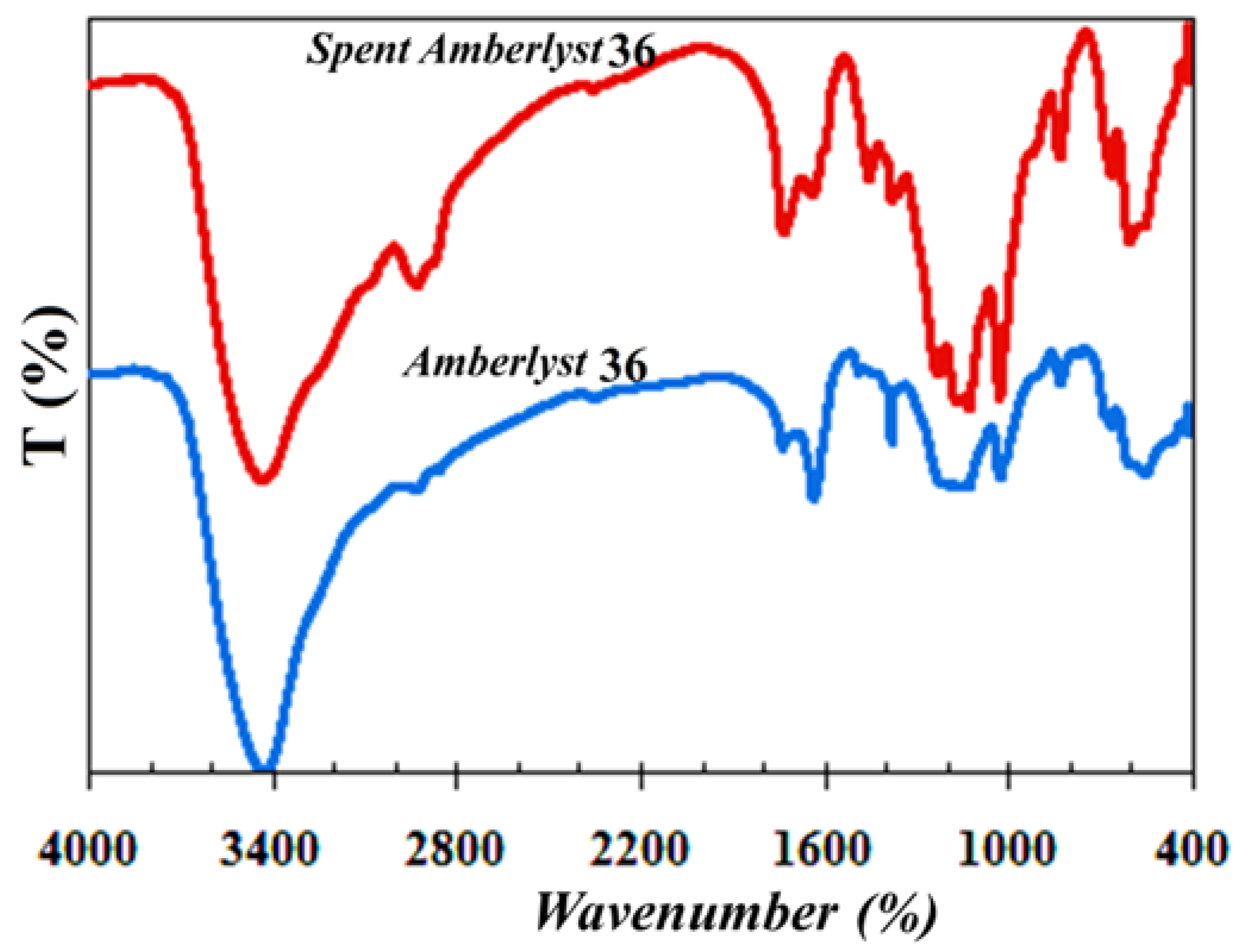
The FT-IR spectra of the Amberlyst 36 before and after (disintegrated spent catalyst) the reaction are presented in
Figure 7. The FT-IR spectrum of the disintegrated spent Amberlyst 36 after 3 h of reaction was distinct from the fresh one, demonstrating a partially disintegrated catalyst structure in the reaction medium.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
