1. Introduction
Nucleoside analogues, NAs, form a very large family of compounds, with more than 40 presently used in medicine as antiviral and anticancer agents [
1,
2]. Actually, a variety of therapeutic nucleosides and nucleotides, which are in the WHO “List of essential medicines” are produced chemically, such as Abacavir (HIV therapeutic), Capecitabine (anticancer), Cytarabine (anticancer, antiviral), Vidarabine (antiviral), or Floxuridine (anticancer), among others. Due to the economic and social relevance of these kind of drugs, the availability of a stereo-selective and cheap synthetic method is relevant for the industry and developing countries.
Furthermore, nucleoside-5′-monophosphates, NMPs, are often used as food additives or intermediates by the pharmaceutical industry. For example, some dietary nucleotides, such as inosinic acid (inosine-5′-monophosphate, IMP) or guanosinic acid (guanosine-5′-monophosphate, GMP), are common additives used as flavour enhancers in foods, since they induce an umami taste sensation [
3]. As a result, current demand for nucleotides in the food additives market is increasing, and the production of nucleotides has been widely studied.
NAs and NMPs have been traditionally synthesized by chemical methods through multistep processes requiring protection and de-protection steps for the labile groups, and isolation in almost every step due to the poor regio- or stereoselectivity of the reactions [
4,
5,
6,
7,
8]. These drawbacks lead to a high price of these valuable compounds, limiting their application.
Nowadays, the application of bioprocesses that are catalyzed by whole cells or enzymes in industry is gaining ground against traditional chemical synthetic processes. In this context, the enzymatic synthesis of NAs and NMPs shows many advantages, such as one-pot reactions under mild conditions, high stereo-, and regioselectivity, and an environmentally friendly technology [
4,
5,
6,
7,
8,
9,
10,
11].
Purine metabolism is a metabolic route of vital importance in all the living organisms, since purines are essential for the synthesis of nucleic acids (DNA and RNA), proteins, and other metabolites. In the
de novo pathway cells use simple precursors like glycine, glutamine, or aspartate for the synthesis of the different purine nucleotides. On the contrary, the salvage pathway is composed by a group of reutilization routes by which the cell can satisfy its purine requirements from endogenous and/or exogenous sources of preformed purines. In this regard, numerous enzymes from purine salvage pathway have become valuable catalysts for mono or multi-enzymatic synthesis of nucleosides and nucleotides, such as nucleoside kinases (NKs) [
12,
13,
14,
15], phosphoribosyltransferases [
7,
9,
10,
11], nucleoside phosphorylases [
4,
8,
16,
17], 2′-deoxyribosyltransferases [
5,
18,
19,
20], among others.
The use of multi-enzymatic systems in organic synthesis offers several advantages, such as the realization of more complex synthetic schemes, the ability to make reversible processes irreversible, to shift the equilibrium reaction in desired way, and the partial or total elimination of product inhibition problems or the prevention of the shortage of substrates by dilution or degradation in the bulk media [
13]. In this regard, the aim of this work is the development of a novel multi-enzymatic system for the industrial production of different NAs and NMPs with application in pharmaceutical and food industry. To achieve this objective, an in vitro multienzymatic system composed by 2′-deoxyribosyltransferase from
Lactobacillus delbrueckii (
LdNDT) and the hypoxanthine-guanine-xanthine phosphoribosyltransferase from
Thermus themophilus HB8 (
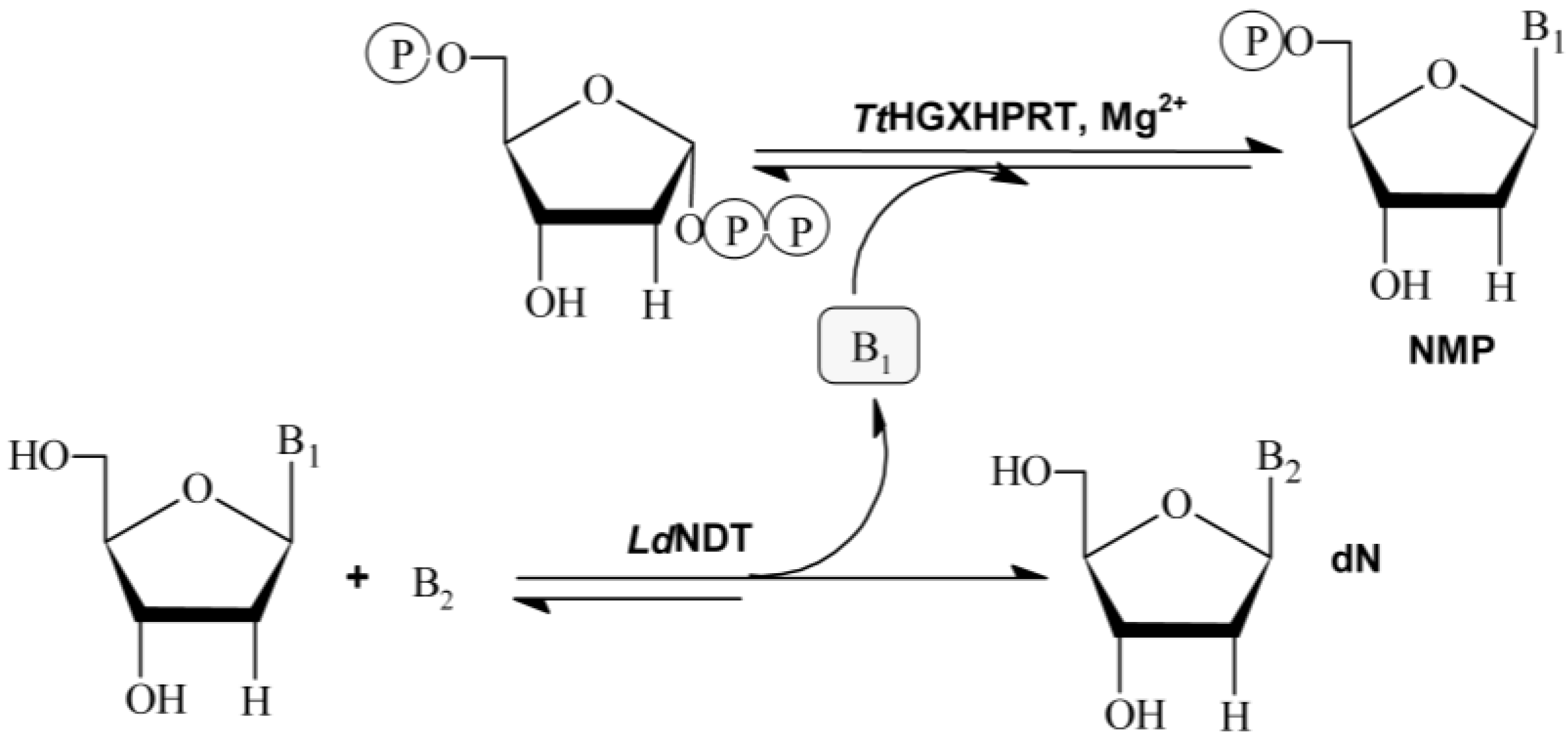
TtHGXPRT), is developed. As shown in
Figure 1, the sequential action of
LdNDT and hypoxanthine-guanine-xanthine
TtHGXPRT can efficiently catalyze the synthesis of NAs and NMPs in two steps.
Nucleoside 2′-deoxyribosyltransferases (NDTs, E.C. 2.4.2.6) catalyze the transglycosylation reaction between a 2′-deoxynucleoside donor and a nucleobase acceptor. According to their substrate specificity, NDTs can be classified into two classes: type I (PDT), specific for purines (Pur↔Pur), and type II (NDT), which catalyze the transfer between purines and/or pyrimidines (Pur↔Pur, Pur↔Pyr, Pyr↔Pyr) [
5,
19,
20] (
Figure 1).
Purine and pyrimidine phosphoribosyltransferases (PRTs) catalyze the reversible transfer of the 5-phosphoribosyl group from 5-phospho-α-
d-ribosyl-1-pyrophosphate (PRPP) to purine and pyrimidine nucleobases or derivatives in the presence of Mg
2+ (
Figure 1).
The coupled system LdNDT/TtHGXPRT allowed for the enzymatic production of different NAs by transglycosilation reaction using 2′-deoxynosine or 2′-deoxyguanosine as donors, and different modified purine nucleobases as acceptors. Interestingly, TtHGXPRT can slightly shift the equilibrium reaction of the NDT-catalyzed transglycosylation reaction in desired way, and partially eliminate undesired by-products (hypoxanthine or guanine) from bulk-media using PRTs to synthesize highly valuable nucleoside-5′-monophosphates (NMPs), commercially available as food additives (IMP or GMP).
3. Discussion
Nucleoside analogues are important molecules used as antiviral and antitumoral drugs [
1,
2,
21], because of their role as inhibitors of the replication of nucleic acids, and can also be employed as starting materials for antisense oligonucleotides. Modified nucleosides have been traditionally synthesized by different chemical methods, requiring the use of protection-deprotection steps, organic solvents and chemical reagents [
4,
5,
6,
7,
8,
9,
10,
11]. In this sense,
LdNDT has shown to be an interesting sustainable alternative to traditional chemical methods for the synthesis of many different purine nucleosides. However, the presence of by-products (nucleobases), or the difficulty to shift the equilibrium of the reaction in desired way, increases production costs, impeding the scale up these processes from laboratory to industry.
Cascades involving enzymes have recently become a thoroughly-investigated field, which offers efficient solutions to circumvent these problems, by removing by-products and leading to shift the reaction towards the formation of products. In this regard, the aim of this work is the development of a new efficient coupled system, which consists in two consecutive reactions catalyzed by LdNDT (Reaction 1) and TtHGXPRT (Reaction 2), which leads to: (i) a more efficient production of 2′-deoxynucleoside analogues due to the shift of the equilibrium of transglycosylation reaction, and (ii) the generation of NMPs with application in food industry.
For this purpose, we describe the production and biochemical characterization of 2′-deoxyribosyltransferase from
LdNDT. According substrate specificity studies,
LdNDT is a type II NDT, which recognizes both purine and pyrimidine bases and 2’-deoxynucleosides.
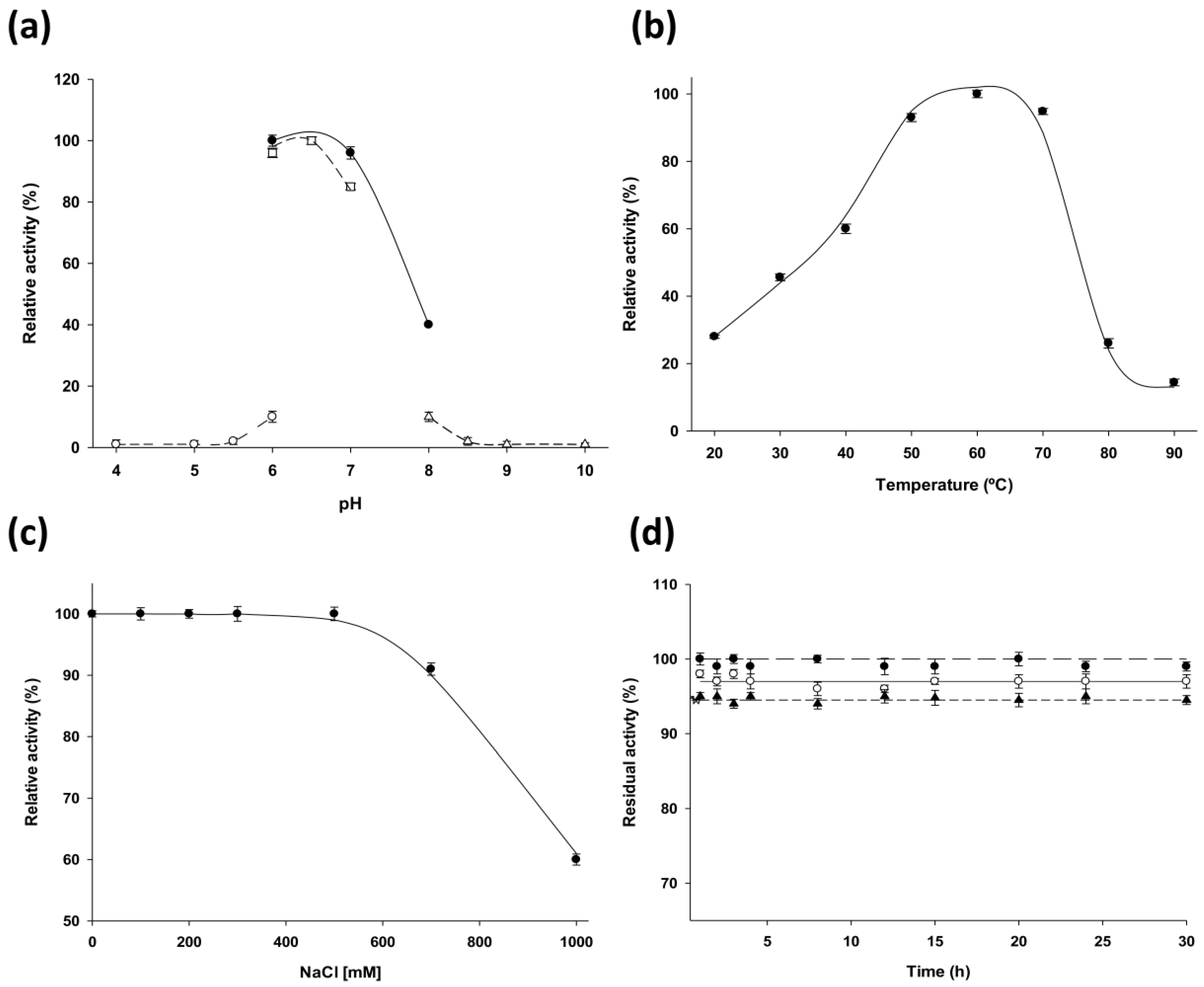
LdNDT present significant catalytic activity in a broad temperature range (from 40 °C to 70 °C) (
Figure 2b) and displays its optimal activity in the pH range 6–7 (
Figure 2a). According to experimental results,
LdNDT is also stable between pH 6–10 at 50 °C. These results suggest that alkaline environments (pH 8–10) could not inactivate the enzyme during this time period (
Figure 2d). In order to explore the potential of
LdNDT as biocatalyst, the enzymatic production of different purine 2′-deoxynucleoside analogues has been carried out.
Once operational conditions of
LdNDT were determined, a multienzymatic system composed by 2′-deoxyribosyltransferase from
Lactobacillus delbrueckii (
LdNDT) and the hypoxanthine-guanine-xanthine phosphoribosyltransferase from
Thermus themophilus HB8 (
TtHGXPRT) were developed. The use of coupled system
LdNDT/
TtHGXPRT allowed for the synthesis of different therapeutical purine nucleosides, such as 2-chloro-2′-deoxyadenosine (cladribine, approved FDA drug for the treatment of hairy-cell leukemia [
1], 2-fluro-2′-deoxyadenosine (a prodrug of 2-fluoroadenine, used in suicide gene therapy [
1]), among others. In addition, resulting by-products from transglycosylation reaction (hypoxanthine or guanine) were transformed to added value compounds (IMP or GMP). Moreover, due to
TtHXGPRT is a 6-oxopurine phosphoribosyltransferase (with a strict specificity for 6-oxopurines, such as guanine and hypoxanthine [
11]), the purine base derivatives used as acceptors in transglycosylation reaction (B
2) (
Figure 1) cannot be recognized by
TtHXGPRT. As a consequence of this, there will not undesired by-products in reaction medium.
In addition to this, the unusual tolerance to alkaline conditions of
LdNDT and
TtHGXPRT [
11], it is a very interesting advantage for covalent immobilization of the enzyme, an essential requisite for its industrial implementation. Covalent immobilization of proteins usually occurs via multipoint attachment through the region with higher density of primary amino groups, especially with ε-NH
2 from lysine residues [
22]. Due to this, covalent immobilization techniques usually employ long reaction times (2–24 h) and alkaline conditions (pH 8–10). In this way, the exceptional stability that is displayed by
LdNDT and
TtHGXPRT [
11] under alkaline conditions suggests that they could be efficiently immobilized.
However, this multienzymatic approach needs to improve several features in order to be applied as industrial biocatalyst, such as the possibility to shift totally the equilibrium or the high price of PRPP. One of the most significant limitations to the practical application of this multi-enzymatic system as industrial biocatalysts is the high cost and instability of PRPP. To address these issues, several attempts to produce PRPP from non-expensive substrates will tested in a future including different PRPP sources. In addition, high concentrations of PRPP and phosphate, buffer would chelate Mg2+ from reaction medium, and affect the efficiency of TtHGXPRT. In this sense, in order to scale up this process, the conversion of reaction 2 could be improved using higher concentrations of MgCl2.
4. Materials and Methods
4.1. Chemicals
Cell culture medium reagents were from Difco (St. Louis, MO, USA). Trimethyl ammonium acetate buffer was purchased from Sigma-Aldrich (Sigma-Aldrich, Madrid, Spain). All the other reagents and organic solvents were purchased to Scharlab (Scharlab, Barcelona, Spain) and Symta (Symta, Madrid, Spain). All natural and non-natural nucleosides and bases used in this work were provided by Carbosynth Ltd. (Carbosynth Ltd., Compton, UK).
4.2. Production and Purification of Recombinant LdNDT and TtHGXPRT
The encoding
ndt gene, which codifies 2′-deoxyribosyltransferase type II from
Lactobacillus delbrueckii subsp. lactis DSM 20072 (NCBI Reference Sequence: WP_002877839.1) was ordered and purchased from Genscript (Piscataway, NJ, USA). The coding sequence appeared as an
NdeI-
EcorI fragment was subcloned into the expression vector pET28b (+). The resultant, recombinant vector pET28b
LdNDT provided an N-terminal His6-tagged fusion protein with a thrombin cleavage site between the tag and the enzyme.
LdNDT was expressed in
E. coli BL21(DE3) and growth in LB medium at 37 °C with kanamycin 50 μg/mL. Protein overexpression was induced by adding 0.5 mM isopropyl-β-
d-1-thiogalactopyranoside and the cells were further grown for 3 h. These were harvested via centrifugation at 3500×
g. The resulting pellet was resuspended in 10 mM sodium phosphate buffer, pH 7.0. Crude extracts were prepared by French press lysis of cell suspensions. The lysate was centrifuged at 17,500×
g for 40 min and the supernatant was filtered through a 0.22 μm filter (Millipore). Cleared lysate was loaded onto a 5-mL HisTrap FF column (GE Healthcare) pre-equilibrated in a binding buffer (10 mM sodium phosphate buffer, pH 7.0, with 100 mM NaCl, and 10 mM imidazole) and the column was washed. Bound proteins were eluted using a linear gradient of imidazole (from 10 mM to 500 mM). After this process, fractions containing His6-tagged-
LdNDT were pooled and concentrated, and N-terminal His6-tagged was removed from
LdNDT using thrombin to avoid precipitation of the protein. In a second step, fractions containing
LdNDT were loaded onto a HiLoad 16/60 Superdex 200 prep grade column (GE Healthcare) pre-equilibrated in 25 mM sodium phosphate buffer, pH 7.0. Fractions with the protein of interest identified by SDS-PAGE were pooled and the protein was dialyzed against 10 mM sodium phosphate, pH 6.0, and concentrated and stored at 4 °C, until its use. Electrophoresis was carried out on 15% polyacrylamide slab gel with 25 mM Tris-HCl buffer, pH 8.6, 0.1% SDS [
23]. Protein concentration was determined spectrophotometrically by UV absorption at 280 nm using ε
280 = 34,380 M
−1cm
−1 [
24].
TtHGXPRT was produced and purified according to experimental protocols, as previously reported [
11].
4.3. N-deoxyribosyltransferase Assay
The standard activity assay was performed by incubating 0.3 μg of pure enzyme with 10 mM 2′-deoxyinosine (dIno) and 10 mM adenine (Ade) in 50 mM MES buffer pH 6.0 in a final volume of 40 μL. The reaction mixture was incubated at 40 °C for 10 min (300 rpm). Enzyme was inactivated by adding 40 μL of cold methanol in ice-bath and heating for 5 min at 100 °C. After centrifugation at 9000× g for 2 min, the samples were half-diluted with water and frozen at −20 °C. Nucleoside production was analysed using HPLC to measure quantitatively the reaction products, as described below. All of the determinations were carried out in triplicate and the maximum error was less than 5%. Under such conditions, one international activity unit (IU) was defined as the amount of enzyme producing 1 μmol/min of 2′-deoxyadenosine under the assay conditions.
4.4. Substrate Specificity
In order to explore the substrate specificity, 0.3 μg of LdNDT were incubated with 10 mM of purine and pyrimidine nucleosides and bases in 50 mM MES buffer, pH 6.5 in a final volume of 40 μL at 50 °C, and 300 rpm orbital shaking at different reaction times. At regular times, samples were taken, and enzymatic activity was evaluated, as described in analytical methods section.
4.5. Influence of pH, Temperature and Ionic Strength on Enzyme Activity
The pH profile of purified recombinant enzyme was initially determined using the standard assay described above with sodium citrate (pH 4–6), sodium phosphate (pH 6–8). MES (6–7) and sodium borate (pH 8–10) as reaction buffers (50 mM). The optimum temperature was determined using the standard assay across a 20–90 °C temperature range. The effect of ionic strength on 2′-deoxyribosyltransferase activity was studied by incubating 0.3 μg of enzyme with different concentrations of NaCl (0 to 1.0 M) in 50 mM MES buffer, pH 6.0 at 50 °C under standard conditions described for enzymatic assay.
4.6. Thermal and pH Stability of LdNDT
LdNDT was stored at −80 °C in 10 mM sodium phosphate, pH 7.0 for 300 days. Samples were taken periodically and enzymatic activity was evaluated. Storage stability was defined as the relative activity between the first and successive reactions. Moreover, thermal stability of LdNDT was assessed by incubating 0.3 μg of pure enzyme in a pH range from 6 to 10, at 50 °C for a period of 30 h. After this, the activity was measured using standard assay.
4.7. Enzymatic Production of Nucleoside Analogues by Soluble LdNDT
Enzymatic productions of non-natural nucleosides was carried out at variable amounts of enzyme, using different nucleoside and base analogues. Reactions were carried out incubating 0.3 μg of LdNDT with 1 mM nucleoside and base, in 50 mM sodium phosphate buffer pH 6.0 at 50 °C, in a final volume of 40 μL, with 300 rpm orbital shaking at different reaction times (20–120 min), and were stopped according experimental procedure described above. Enzymatic activity was evaluated, as described in analytical methods section.
4.8. Enzymatic Production of Purine NAs and Purine NMPs Catalyzed by Multi-Enzymatic System LdNDT/TtHGXPRT
Reaction mixtures contained 0.3 μg of electrophoretically pure LdNDT, 1 μg of TtHGXPRT, 3 mM 2′-deoxynucleoside, 1 mM nucleobase, 1 mM 5-phospho-α-d-ribosyl-1-pyrophosphate (5-PRPP) and 1.2 mM MgCl2 in 50 mM sodium phosphate buffer, pH 6.0, in a final volume of 40 μL. Reactions were conducted at 50 °C with shaking (300 rpm) at different reaction times (20–120 min), and were stopped according experimental procedure described above. Enzymatic activity was evaluated, as described in analytical methods section.
4.9. Analytical Methods
4.9.1. Analytical Methods for LdNDT
The production of nucleosides was quantitatively measured with an ACE 5 μm C18-PFP column 250 mm × 46 mm (Symta, Madrid, Spain) pre-equilibrated in 100% trimethyl ammonium acetate. Elution was carried out at a flow rate of 0.9 mL/min, by a discontinuous gradient: 0–10 min, 100% to 90% trimethyl ammonium acetate and 0% to 10% acetonitrile, and 10–20 min, 90% to 100% trimethyl ammonium acetate and 10% to 0% acetonitrile. The nucleoside product was eluted into the diode array detector for quantification at 230, 240, 254, and 260 nm.
Retention times for the reference natural and non-natural bases (hereafter abbreviated according to the recommendations of the IUPAC-IUB Commission on Biochemical Nomenclature) were as follows: adenine (Ade), 10.2 min; guanine (Gua), 7.3 min; hypoxanthine (Hyp), 7.5 min; benzimidazole (B), 29.6 min; 2,6-diaminopurine (2,6-DAP), 11.1 min; 6-mercaptopurine (6-M), 10.5 min; 6-methylpurine (6-MetPur): 13.8 min; 2-fluoroadenine (2-FAde), 14.4 min; 6-benzoyladenine (6-BenzoAde), 16.6; 2-chloroadenine (2-ClAde), 17.3 min; 6-chloropurine (6-ClPur), 16.7 min; 6-methoxyguanine (6-MeOGua), 16.4 min; 7-deaza-6-hydroxypurine (7-deaza6OHPur), 9.0 min; 7-deazaxanthine (7-deazaXan), 8.5 min; theophylline (The), 20.0 min; benzoyladenine (BenzoA); 22 min; and, xanthine (Xan), 8 min.
Retention times for the nucleosides (hereafter abbreviated according to the recommendations of the IUPAC-IUB Commission on Biochemical Nomenclature) were as follows: 2′-deoxyadenosine (dAdo), 15.5 min; 2′-deoxyguanosine (dGuo), 11.6 min; 2′-deoxyinosine (dIno), 12.0 min; 33.5; 2,6-diaminopurine-2′-deoxyribose (2,6-DAPdRib), 16.0 min; 6-mercaptopurine-2′-deoxyribose (6-MdRib), 13.5 min; 6-methylpurine-2′-deoxyribose (6-MetPurdRib), 17.7 min; 2-fluoro-2′-deoxyadenosine (2-F-dAdo), 19.0 min, 2-chloro-2′-deoxyadenosine (2-CldAdo), 23.2 min; 6-chloropurine-2′-deoxyribose (6-ClPurdRib), 22.9 min; 6-methoxy-2′-deoxyguanosine (6-MeOdGuo), 21.8 min; 7-deaza-6-hydroxypurine-2’-deoxyribose (7-deaza6OHPurdRib), 14.0 min; 7-deazaxanthine-2′-deoxyribose (7-deazadXao), 13.5 min; theophylline-2′-deoxyribose (ThedRib), 27.0 min; benzoyladenine-2’-deoxyribose (BenzoAdRib); 29.0 min; and, 2′-deoxyxanthosine (dXao), 13.0 min. The identification and quantification of most of reaction substrates and products have been performed in relation to external standards using the above, well characterized commercial products. In the case of 7-deaza-6-hydroxypurine-2′-deoxyribose, 7-deazaxanthine-2′-deoxyribose, theophylline-2′-deoxyribose, and benzoyladenine-2′-deoxyribose we use the decrease of corresponding starting nucleobases to quantify the formation of products.
4.9.2. Analytical Methods for Multi-Enzymatic System LdNDT/TtHGXPRT
The production of 2′-deoxynucleosides and nucleoside-5′-monophosphates was quantitatively measured with an ACE EXCEL 5 μm CN-ES column 250 mm × 4.6 mm (Symta, Madrid, Spain) equilibrated with 100% trimethyl ammonium acetate at a flow rate of 0.8 mL/min. The nucleoside product was eluted into the diode array detector for quantification at 230, 240, 254, and 260 nm.
Retention times for the reference natural and non-natural bases (hereafter abbreviated according to the recommendations of the IUPAC-IUB Commission on Biochemical Nomenclature) were as follows: adenine (Ade), 10.2 min; guanine (Gua), 5.8 min; hypoxanthine (Hyp), 5.3 min; 2,6-diaminopurine (2,6-DAP), 9.5 min; 6-mercaptopurine (6-M), 7.8 min; 6-methylpurine (6-MetPur): 14.5 min; 2-fluoroadenine (2-FAde), 14.4 min; 2-chloroadenine (2-ClAde), 25.0 min; and, 6-methoxyguanine (6-MeOGua), 19.4 min.
Retention times for the nucleosides (hereafter abbreviated according to the recommendations of the IUPAC-IUB Commission on Biochemical Nomenclature) were as follows: 2′-deoxyadenosine (dAdo), 19.5 min; 2′-deoxyguanosine (dGuo), 10.2 min; 2′-deoxyinosine (dIno), 9.0 min; 33.5; 2,6-diaminopurine-2’-deoxyribose (2,6-DAPdRib), 20.1 min; 6-mercaptopurine-2′-deoxyribose (6-MdRib), 11.5 min; 6-methylpurine-2′-deoxyribose (6-MetPurdRib), 33.4 min; 2-fluoro-2′-deoxyadenosine (2-F-dAdo), 34.0 min, 2-chloro-2′-deoxyadenosine (2-CldAdo), 65.0 min; and, 6-methoxy-2′-deoxyguanosine (6-MeOdGuo), 27.8 min.
Retention times for the nucleosides-5’-monophosphate (hereafter abbreviated according to the recommendations of the IUPAC-IUB Commission on Biochemical Nomenclature) were as follows: guanosine-5′-monophosphate (5′-GMP), 3.1 min; inosine-5′- monophosphate (5′-IMP), 3.4 min.
The identification and quantification of most of reaction substrates and products have been performed in relation to external standards using the above, well characterized commercial products.


 and
and 




{kind=link}
{kind=link}
{kind=link}