Solvothermal Conversion of Lignosulfonate Assisted by Ni Catalyst: Investigation of the Role of Ethanol and Ethylene Glycol as Solvents

 ,
, 
Abstract
:1. Introduction
2. Results and Discussions
2.1. Lignosulfonate
2.2. Catalyst
2.3. Solvothermal Conversion of Lignosulfonate
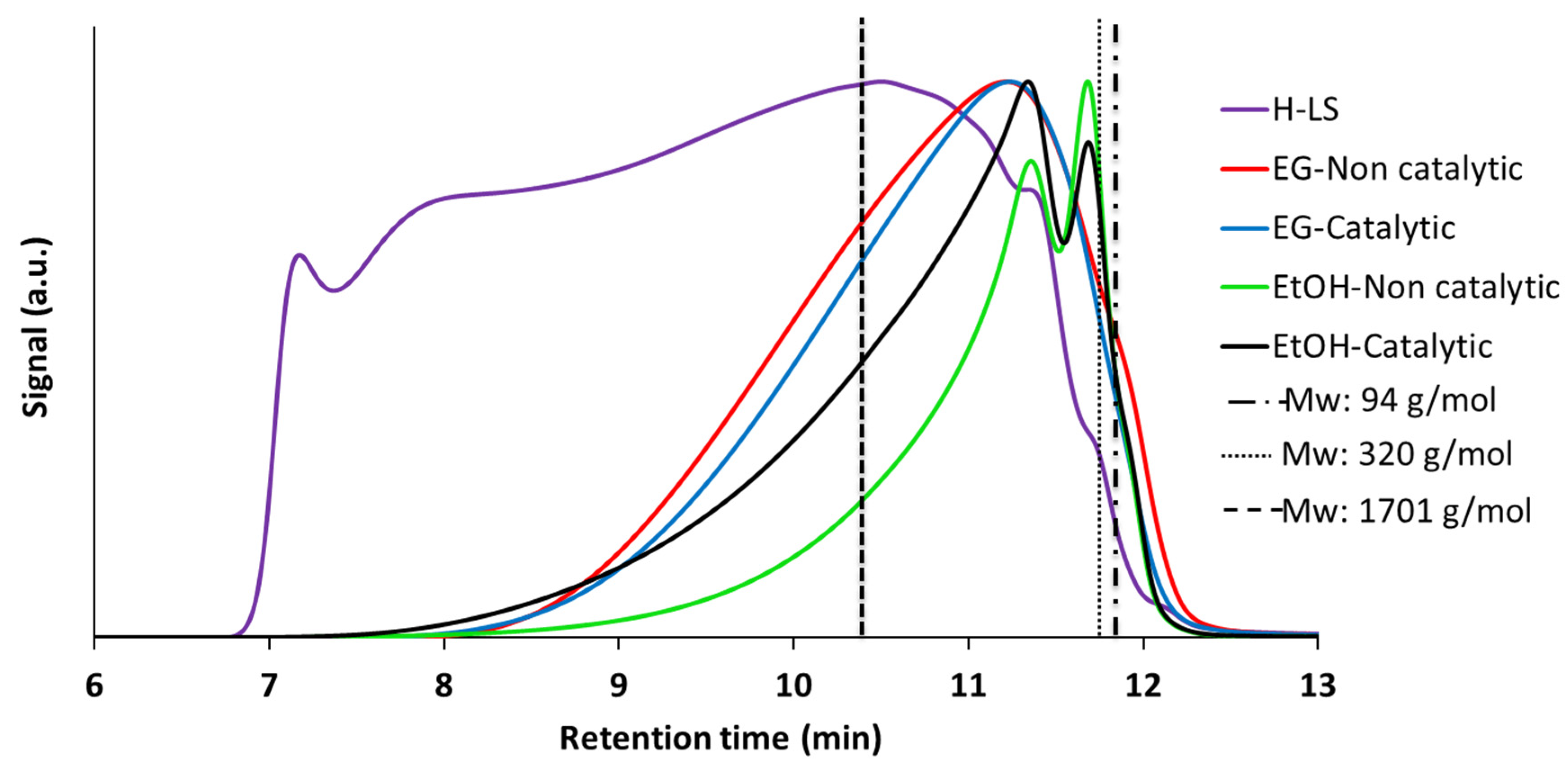
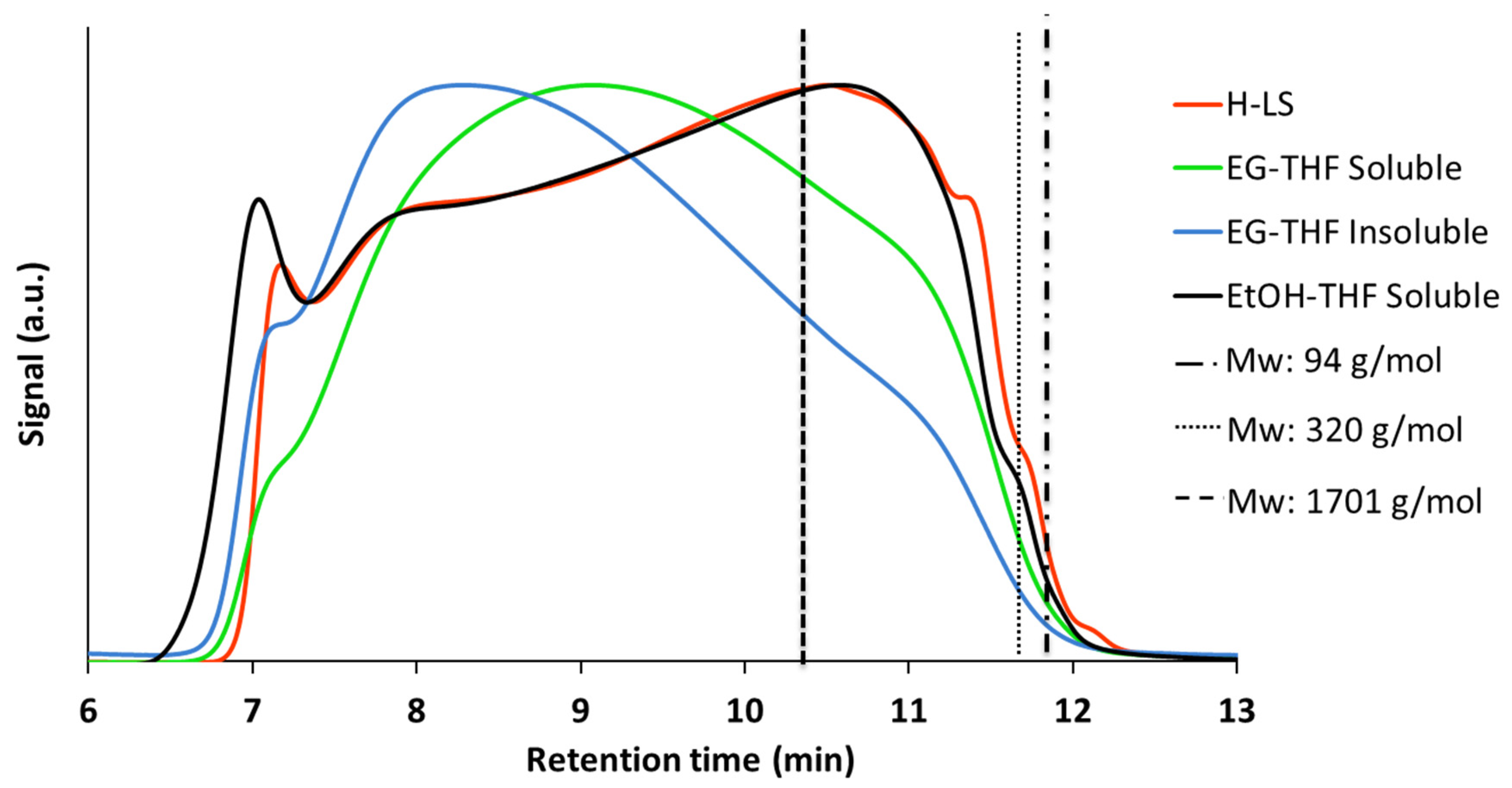
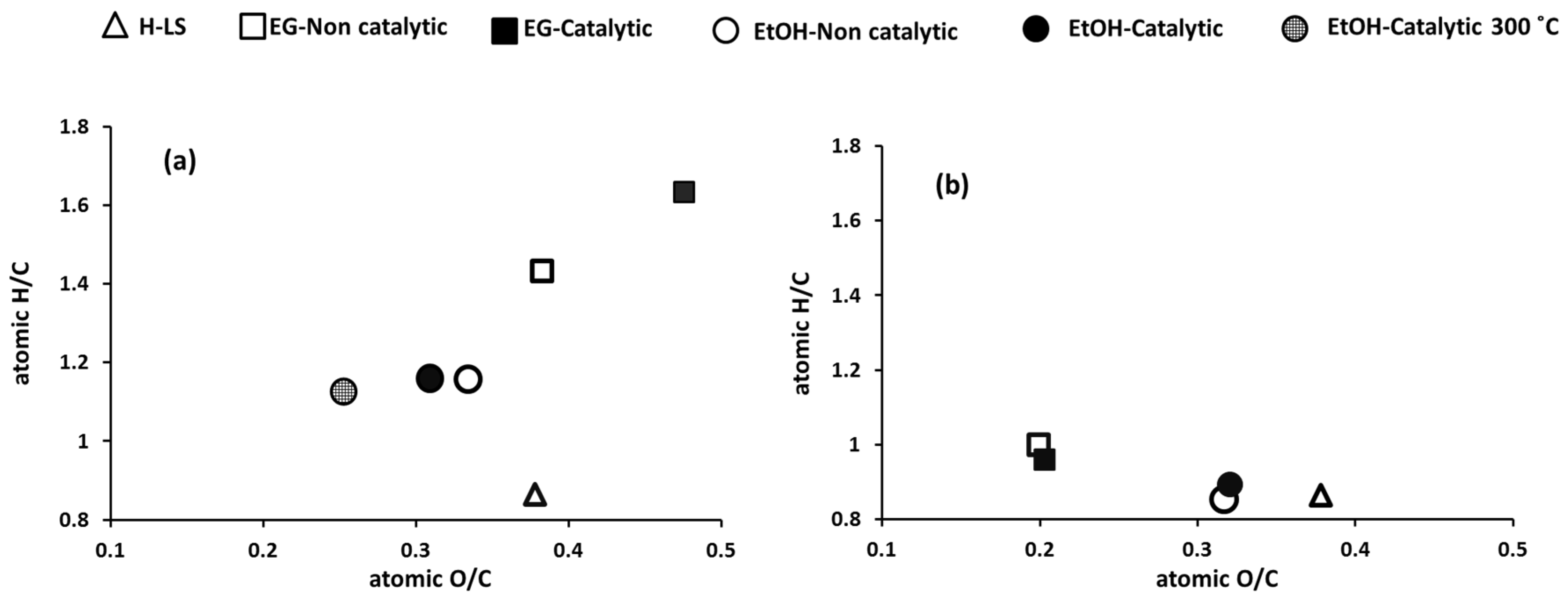
2.3.1. Characterization of the Products and Comparison between EtOH and EG as Reaction Media
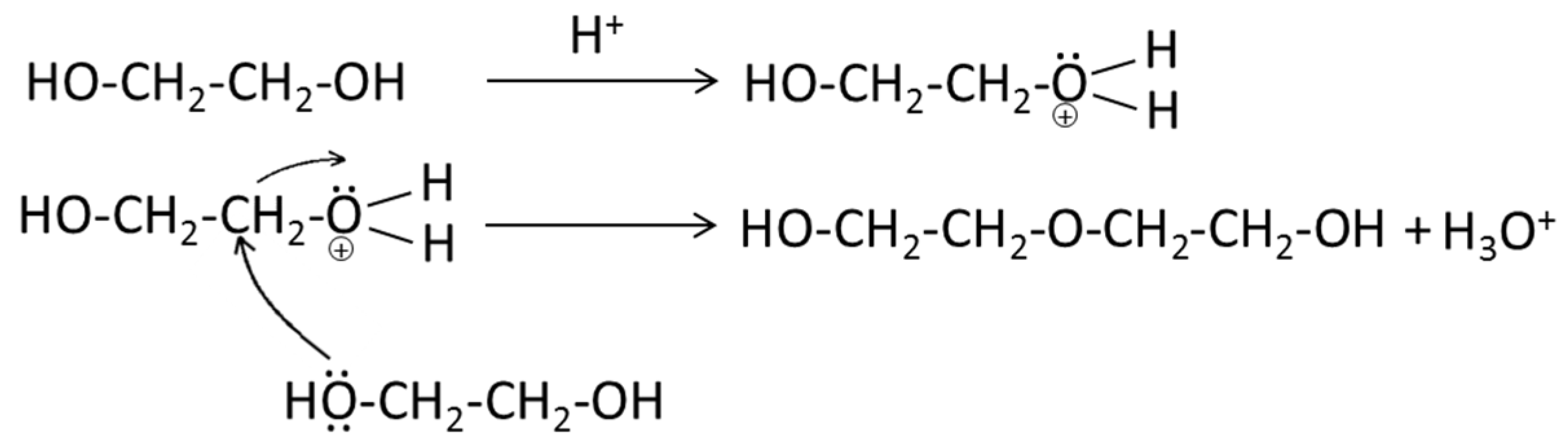
- EtOH remained almost intact at the reaction conditions whereas EG was, to a significant extent, converted via self-reaction. Depending on the value of the liquefied products from EG conversion, the conversion of EG may be unfavorable. However, it should be noted that conversion of EG was acid-catalyzed by H+ from sulfonate group. It is possible that the stability of EG can be improved by utilizing non-acid forms of lignosulfonate such as Na-LS. In support of this, although at lower temperature than applied here, Schutyser et al. [31] stated that EG remained stable during conversion of birch sawdust over Pd/C catalyst at 200 °C.
- The isolation of the oil from EtOH is simple and may be done by distillation. On the contrary, separation of reaction products in the EG medium is very challenging due to the high boiling point of EG and also partial solubility of EG in conventional solvents used for extraction of products.
- EG is a better end-capping agent in preventing formation of highly cross-linked solid fractions compared to EtOH. The solid residues left from reaction in EG can possibly be recycled in a continuous process and degraded to lower molecular weight compounds, while degradation of the solid char residue from EtOH medium could require severe reaction conditions or different processes such as pyrolysis or gasification. The char residue can also be burned for the energy supply.
- EtOH is in a supercritical condition at 250 °C. The operational pressure in EtOH tests rose up to 155 bar while the pressure in EG tests was up to 78 bar. Obviously, a lower pressure is favorable from industrial equipment design point of view. It is beneficial to optimize the process condition to lower process pressure by reduction of initial H2 pressure, reduction of EtOH to lignosulfonate ratio and operation at lower temperatures, though it may result in lower liquefied products yields.
2.3.2. Effect of the Catalyst Support on the Degradation of Lignosulfonate
2.3.3. Working State of the Ni-Based Catalyst
2.3.4. Catalyst Reusability
3. Materials and Methods
3.1. Feedstock and Chemicals
3.2. Catalyst
3.3. Depolymerization Reactions and Workup Procedure
3.4. Characterization and Analytical Techniques
3.4.1. Catalyst Characterization
3.4.2. Analysis of Feedstock and Depolymerization Products
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Calvo-Flores, F.G.; Dobado, J.A. Lignin as Renewable Raw Material. ChemSusChem 2010, 3, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Reknes, K. The Chemistry of Lignosulfonate and the Effect on Performance of Lignosulfonate Base Plasticizers and Superplasticizers. In Proceedings of the 29th OUR World in Concrete and Structures (OWICs), Singapore, 25–26 August 2004. [Google Scholar]
- Lora, J. Industrial Commercial Lignins: Sources, Properties and Applications. In Monomers, Polymers and Composites from Renewable Resources, 1st ed.; Belgacem, M.N., Gandini, A., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; pp. 225–241. ISBN 978-0-08-045316-3. [Google Scholar]
- Ma, R.; Hao, W.; Ma, X.; Tian, Y.; Li, Y. Catalytic ethanolysis of kraft lignin into high-value small-molecular chemicals over a nanostructured α-molybdenum carbide catalyst. Angew. Chem. 2014, 126, 7438–7443. [Google Scholar] [CrossRef]
- Huang, X.; Korányi, T.I.; Boot, M.D.; Hensen, E.J.M. Catalytic Depolymerization of Lignin in Supercritical Ethanol. ChemSusChem 2014, 7, 2276–2288. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Rinaldi, R. Corrigendum: Solvent effects on the hydrogenolysis of diphenyl ether with raney nickel and their implications for the conversion of lignin. ChemSusChem 2012, 5, 1455–1466. [Google Scholar] [CrossRef] [PubMed]
- Fredheim, G.E.; Braaten, S.M.; Christensen, B.E. Molecular weight determination of lignosulfonates by size-exclusion chromatography and multi-angle laser light scattering. J. Chromatogr. Coruña 2002, 942, 191–199. [Google Scholar] [CrossRef]
- Li, S.; Li, N.; Li, G.; Li, L.; Wang, A.; Cong, Y.; Wang, X.; Zhang, T. Lignosulfonate-based acidic resin for the synthesis of renewable diesel and jet fuel range alkanes with 2-methylfuran and furfural. Green Chem. 2015, 17, 3644–3652. [Google Scholar] [CrossRef]
- Megiatto, J.D.; Cerrutti, B.M.; Frollini, E. Sodium lignosulfonate as a renewable stabilizing agent for aqueous alumina suspensions. Int. J. Biol. Macromol. 2016, 82, 927–932. [Google Scholar] [CrossRef] [PubMed]
- Kilanowski, B. Ligninsulfonat. Available online: https://de.wikipedia.org/wiki/Ligninsulfonat (accessed on 25 September 2018).
- Product Functionalities. Available online: https://www.lignotech.com/Product-Functionalities (accessed on 25 September 2018).
- Vishtal, A.G.; Kraslawski, A. Challenges in Industrial Applications of Technical Lignins. BioResources 2011, 6, 3547–3568. [Google Scholar]
- Song, Q.; Wang, F.; Xu, J. Hydrogenolysis of lignosulfonate into phenols over heterogeneous nickel catalysts. Chem. Commun. 2012, 48, 7019–7021. [Google Scholar] [CrossRef] [PubMed]
- Horáček, J.; Mikkola, J.-P.; Samikannu, A.; Št’ávová, G.; Larsson, W.; Hora, L.; Kubička, D. Studies on Sodium Lignosulfonate Depolymerization over Al2O3 Supported Catalysts Loaded with Metals and Metal Oxides in a Continuous Flow Reactor. Top. Catal. 2013, 56, 794–799. [Google Scholar] [CrossRef]
- Shu, R.; Xu, Y.; Ma, L.; Zhang, Q.; Wang, T.; Chen, P.; Wu, Q. Hydrogenolysis process for lignosulfonate depolymerization using synergistic catalysts of noble metal and metal chloride. RSC Adv. 2016, 6, 88788–88796. [Google Scholar] [CrossRef]
- Bartholomew, C.H. Mechanisms of catalyst deactivation. Appl. Catal. A Gen. 2001, 212, 17–60. [Google Scholar] [CrossRef]
- Sun, J.; Dutta, T.; Parthasarathi, R.; Ho Kim, K.H.; Tolic, N.; Chu, R.K.; Isern, N.G.; Cort, J.R.; Simmons, B.A.; Singh, S. Rapid room temperature solubilization and depolymerization of polymeric lignin at high loadings. Green Chem. 2016, 18, 6012–6020. [Google Scholar] [CrossRef]
- Lahive, C.W.; Deuss, P.J.; Lancefield, C.S.; Sun, Z.; Cordes, D.B.; Young, C.M.; Tran, F.; Slawin, A.M.Z.; de Vries, J.G.; Kamer, P.C.J.; et al. Advanced Model Compounds for Understanding Acid-Catalyzed Lignin Depolymerization: Identification of Renewable Aromatics and a Lignin-Derived Solvent. J. Am. Chem. Soc. 2016, 138, 8900–8911. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Korányi, T.I.; Boot, M.D.; Hensen, E.J.M. Ethanol as capping agent and formaldehyde scavenger for efficient depolymerization of lignin to aromatics. Green Chem. 2015, 17, 4941–4950. [Google Scholar] [CrossRef] [Green Version]
- Rane, S.S.; Choi, P. Polydispersity Index: How Accurately Does It Measure the Breadth of the Molecular Weight Distribution? Chem. Mater. 2005, 17, 926. [Google Scholar] [CrossRef]
- Van den Bosch, S.; Renders, T.; Kennis, S.; Koelewijn, S.-F.; Van den Bossche, G.; Vangeel, T.; Deneyer, A.; Depuydt, D.; Courtin, C.M.; Thevelein, J.M.; et al. Integrating lignin valorization and bio-ethanol production: on the role of Ni-Al2O3 catalyst pellets during lignin-first fractionation. Green Chem. 2017, 19, 3313–3326. [Google Scholar] [CrossRef]
- Cheng, S.; Wilks, C.; Yuan, Z.; Leitch, M.; Xu, C.C. Hydrothermal degradation of alkali lignin to bio-phenolic compounds in sub/supercritical ethanol and water-ethanol co-solvent. Polym. Degrad. Stab. 2012, 97, 839–848. [Google Scholar] [CrossRef]
- Li, M.-F.; Sun, S.-N.; Xu, F.; Sun, R.-C. Organosolv fractionation of lignocelluloses for fuels, chemicals and materials: A biorefinery processing perspective. In Biomass Conversion—The Interface of Biotechnology, Chemistry and Materials Science, 1st ed.; Baskar, C., Baskar, S., Dhillon, R.S., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 341–379. ISBN 978-3-642-28417-5. [Google Scholar]
- Myrvold, B.O. Differences in solubility parameters and susceptibility to salting-out between softwood and hardwood lignosulfonates. Holzforschung 2016, 70. [Google Scholar] [CrossRef]
- Singh, R.; Prakash, A.; Dhiman, S.K.; Balagurumurthy, B.; Arora, A.K.; Puri, S.K.; Bhaskar, T. Hydrothermal conversion of lignin to substituted phenols and aromatic ethers. Bioresour. Technol. 2014, 165, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Tian, Q.; Li, N.; Liu, J.; Wang, M.; Deng, J.; Zhou, J.; Ma, Q. Catalytic hydrogenation of alkali lignin to bio-oil using fullerene-like vanadium sulfide. Energy Fuels 2015, 29, 255–261. [Google Scholar] [CrossRef]
- Caetano, C.S.; Guerreiro, L.; Fonseca, I.M.; Ramos, A.M.; Vital, J.; Castanheiro, J.E. Esterification of fatty acids to biodiesel over polymers with sulfonic acid groups. Appl. Catal. A Gen. 2009, 359, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Xu, Y.; Li, Y.; Wang, T.; Zhang, Q.; Ma, L.; He, M.; Li, K. Investigation on the esterification by using supercritical ethanol for bio-oil upgrading. Appl. Energy 2015, 160, 633–640. [Google Scholar] [CrossRef]
- Zhang, T.; Zhou, Y.; Liu, D.; Petrus, L. Qualitative analysis of products formed during the acid catalyzed liquefaction of bagasse in ethylene glycol. Bioresour. Technol. 2007, 98, 1454–1459. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, L.; Zhang, Z.; Wellard, R.M.; Bartley, J.P.; O’Hara, I.M.; Doherty, W.O.S. Characterisation of lignins isolated from sugarcane bagasse pretreated with acidified ethylene glycol and ionic liquids. Biomass Bioenergy 2014, 70, 498–512. [Google Scholar] [CrossRef]
- Schutyser, W.; Van den Bosch, S.; Renders, T.; De Boe, T.; Koelewijn, S.-F.; Dewaele, A.; Ennaert, T.; Verkinderen, O.; Goderis, B.; Courtin, C.M.; et al. Influence of bio-based solvents on the catalytic reductive fractionation of birch wood. Green Chem. 2015, 17, 5035–5045. [Google Scholar] [CrossRef]
- Mu, L.; Shi, Y.; Wang, H.; Zhu, J. Lignin in ethylene glycol and poly (ethylene glycol): Fortified lubricants with internal hydrogen bonding. ACS Sustain. Chem. Eng. 2016, 4, 1840–1849. [Google Scholar] [CrossRef]
- Kubo, S.; Yamada, T.; Hashida, K.; Ono, H. Grafting of ethylene glycol chains in lignin during the solvolysis for biomass conversion using ethylene carbonate/ethylene glycol system. Chem. Lett. 2007, 36, 502–503. [Google Scholar] [CrossRef]
- Rostrup-Nielsen, J.R. Some principles relating to the regeneration of sulfur-poisoned nickel catalyst. J. Catal. 1971, 21, 171–178. [Google Scholar] [CrossRef]
- Mortensen, P.M.; Gardini, D.; de Carvalho, H.W.P.; Damsgaard, C.D.; Grunwaldt, J.-D.; Jensen, P.A.; Wagner, J.B.; Jensen, A.D. Stability and resistance of nickel catalysts for hydrodeoxygenation: carbon deposition and effects of sulfur, potassium, and chlorine in the feed. Catal. Sci. Technol. 2014, 4, 3672–3686. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Xie, C.; Li, Y.; Song, C.; Bolin, T.B. Sulfur poisoning mechanism of steam reforming catalysts: an X-ray absorption near edge structure (XANES) spectroscopic study. Phys. Chem. Chem. Phys. 2010, 12, 5707. [Google Scholar] [CrossRef] [PubMed]
- Narani, A.; Chowdari, R.K.; Cannilla, C.; Bonura, G.; Frusteri, F.; Heeres, H.J.; Barta, K. Efficient catalytic hydrotreatment of Kraft lignin to alkylphenolics using supported NiW and NiMo catalysts in supercritical methanol. Green Chem. 2015, 17, 5046–5057. [Google Scholar] [CrossRef] [Green Version]
- Webb, P. Introduction to Chemical Adsorption Analytical Techniques and Their Applications to Catalysis; Micromeritics Instrument Corp.: Norcross, GA, USA, 2003. [Google Scholar]
- Katritzky, A.R.; Ignatchenko, E.S.; Barcock, R.A.; Lobanov, V.S.; Karelson, M. Prediction of gas chromatographic retention times and response factors using a general qualitative structure-property relationships treatment. Anal. Chem. 1994, 66, 1799–1807. [Google Scholar] [CrossRef]
- Scanlon, J.T.; Willis, D.E. Calculation of flame ionization detector relative response factors using the effective carbon number concept. J. Chromatogr. Sci. 1985, 23, 333–340. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular Weight g/mol | Composition wt.% | Atomic Ratio | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mw | Mn | C | H | O | S | Ash | Humidity | O/C | H/C | |
| H-LS | 9400 | 1900 | 61.1 | 4.4 | 30.8 | 3.1 | 0.6 | 2.5 | 0.38 | 0.86 |
| Catalyst | Particle Size [nm] | NH3 Desorbed [µmol/gcat] Weak | NH3 Desorbed [µmol/gcat] Strong |
|---|---|---|---|
| Ni/SiO2 | 7.5 | 55 (at 152 °C) | - |
| Ni/AC | 14.8 | N.A. * | N.A. |
| Ni/ZrO2 | N.A. | - | 373 (at 225 °C) |
| Ni/γ-Al2O3 | 32.5 | 602 (at 184 °C) | - |
| EtOH Medium | |||||
| Entry | Catalyst | Oil Yield wt.% | Solid Phase wt.% | ||
| THF Insoluble | THF Soluble | Total | |||
| 1 | Non-catalytic | 16 | 60 | 10 | 70 |
| 2 | Ni/SiO2 | 31 | 46 | 16 | 62 |
| 3 * | Ni/SiO2 | 47 | 30 | 9 | 39 |
| EG Medium | |||||
| Entry | Catalyst | Apparent/Estimated Oil Yield wt.% | Solid Phase wt.% | ||
| THF Insoluble | THF Soluble | Total | |||
| 4 | Non-catalytic | 89/20 | 6 | 74 | 80 |
| 5 | Ni/SiO2 | 93/32 | 23 | 45 | 68 |
| Experiment | Selectivity % | Monomer Yield wt.% | ||
|---|---|---|---|---|
| Guaiacol & Substituted Guaiacols | Aromatic Esters | Total | ||
| EtOH, Non-catalytic | 10.9 | 11.8 | 22.7 | 3.6 |
| EtOH, Ni/SiO2 | 6.6 | 6.3 | 12.9 | 4.0 |
| EG, Non-catalytic | 5.8 | - | 5.8 | 1.2 |
| EG, Ni/SiO2 | 2.6 | - | 2.6 | 0.8 |
| Entry | Catalyst | Oil Yield wt.% | Solid Yield wt.% |
|---|---|---|---|
| 1 | Ni/SiO2 | 31 | 62 |
| 2 | Ni/AC | 34 | 60 |
| 3 | Ni/ZrO2 | 34 | 69 |
| 4 | Ni/γ-Al2O3 | 33 | 67 |
| 5 | AC | 15 | 76 |
| 6 | SiO2 | 18 | 67 |
| 7 | Ni/SiO2 (sulfided) | 33 | 52 |
| 8 | Ni/SiO2 * | 30 | 60 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghafarnejad Parto, S.; Munkholt Christensen, J.; Saaby Pedersen, L.; Tjosås, F.; Degn Jensen, A. Solvothermal Conversion of Lignosulfonate Assisted by Ni Catalyst: Investigation of the Role of Ethanol and Ethylene Glycol as Solvents. Catalysts 2018, 8, 502. https://doi.org/10.3390/catal8110502
Ghafarnejad Parto S, Munkholt Christensen J, Saaby Pedersen L, Tjosås F, Degn Jensen A. Solvothermal Conversion of Lignosulfonate Assisted by Ni Catalyst: Investigation of the Role of Ethanol and Ethylene Glycol as Solvents. Catalysts. 2018; 8(11):502. https://doi.org/10.3390/catal8110502
Chicago/Turabian StyleGhafarnejad Parto, Soheila, Jakob Munkholt Christensen, Lars Saaby Pedersen, Freddy Tjosås, and Anker Degn Jensen. 2018. "Solvothermal Conversion of Lignosulfonate Assisted by Ni Catalyst: Investigation of the Role of Ethanol and Ethylene Glycol as Solvents" Catalysts 8, no. 11: 502. https://doi.org/10.3390/catal8110502
APA StyleGhafarnejad Parto, S., Munkholt Christensen, J., Saaby Pedersen, L., Tjosås, F., & Degn Jensen, A. (2018). Solvothermal Conversion of Lignosulfonate Assisted by Ni Catalyst: Investigation of the Role of Ethanol and Ethylene Glycol as Solvents. Catalysts, 8(11), 502. https://doi.org/10.3390/catal8110502