Application of POCOP Pincer Nickel Complexes to the Catalytic Hydroboration of Carbon Dioxide


Abstract
:1. Introduction
2. Results and Discussion
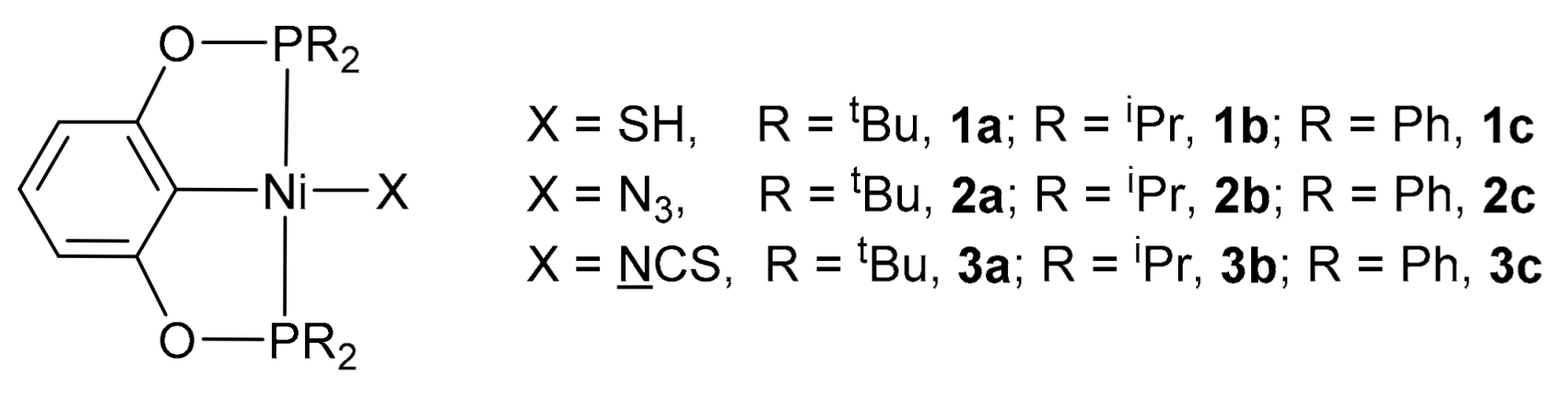
2.1. Synthesis and Characterization of the Ni Catalysts
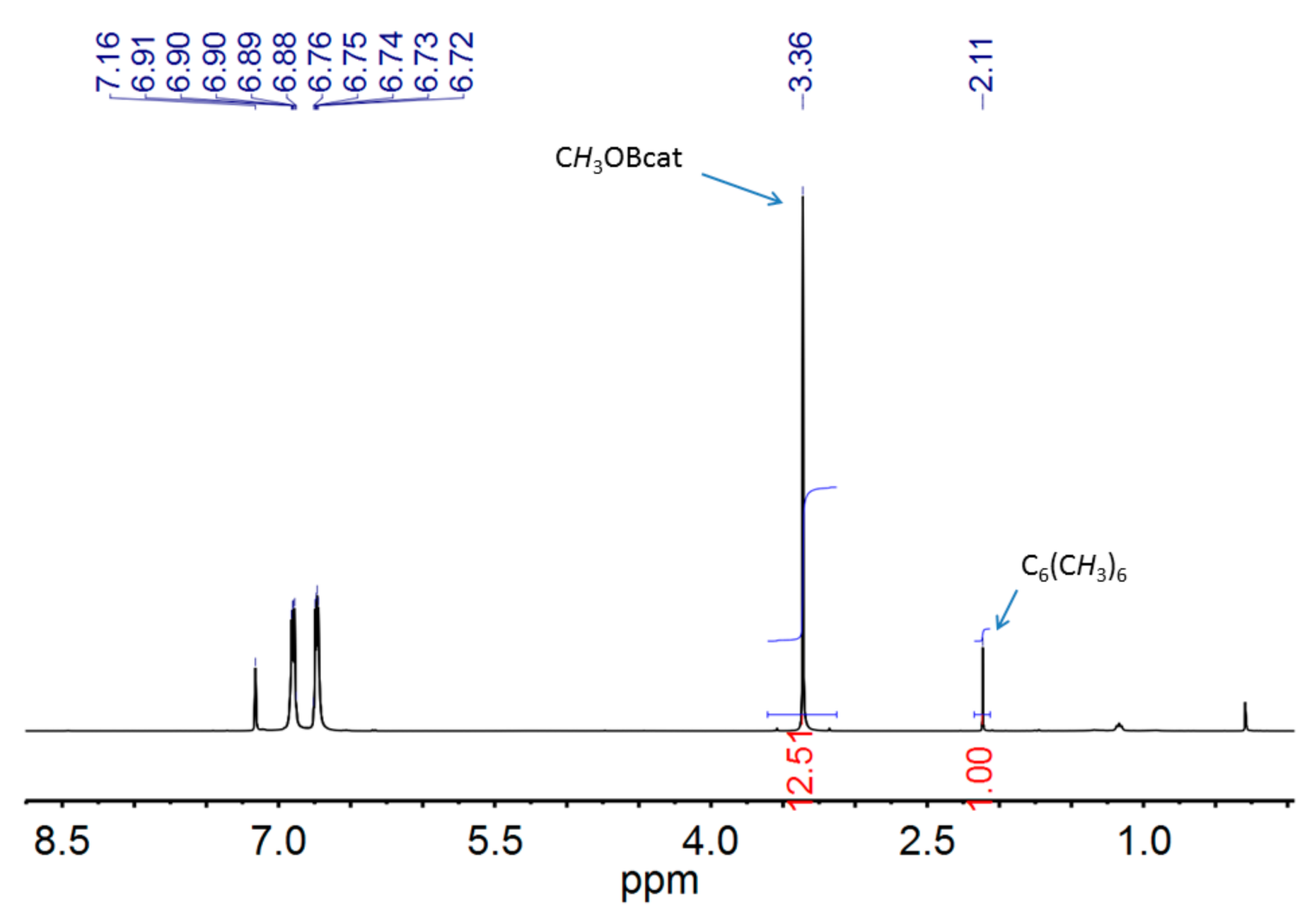
2.2. Catalytic Hydroboration of CO2 with HBcat
3. Materials and Methods
3.1. General Information
3.2. Synthesis of [2,6-(Ph2PO)2C6H3]NiSH (1c)
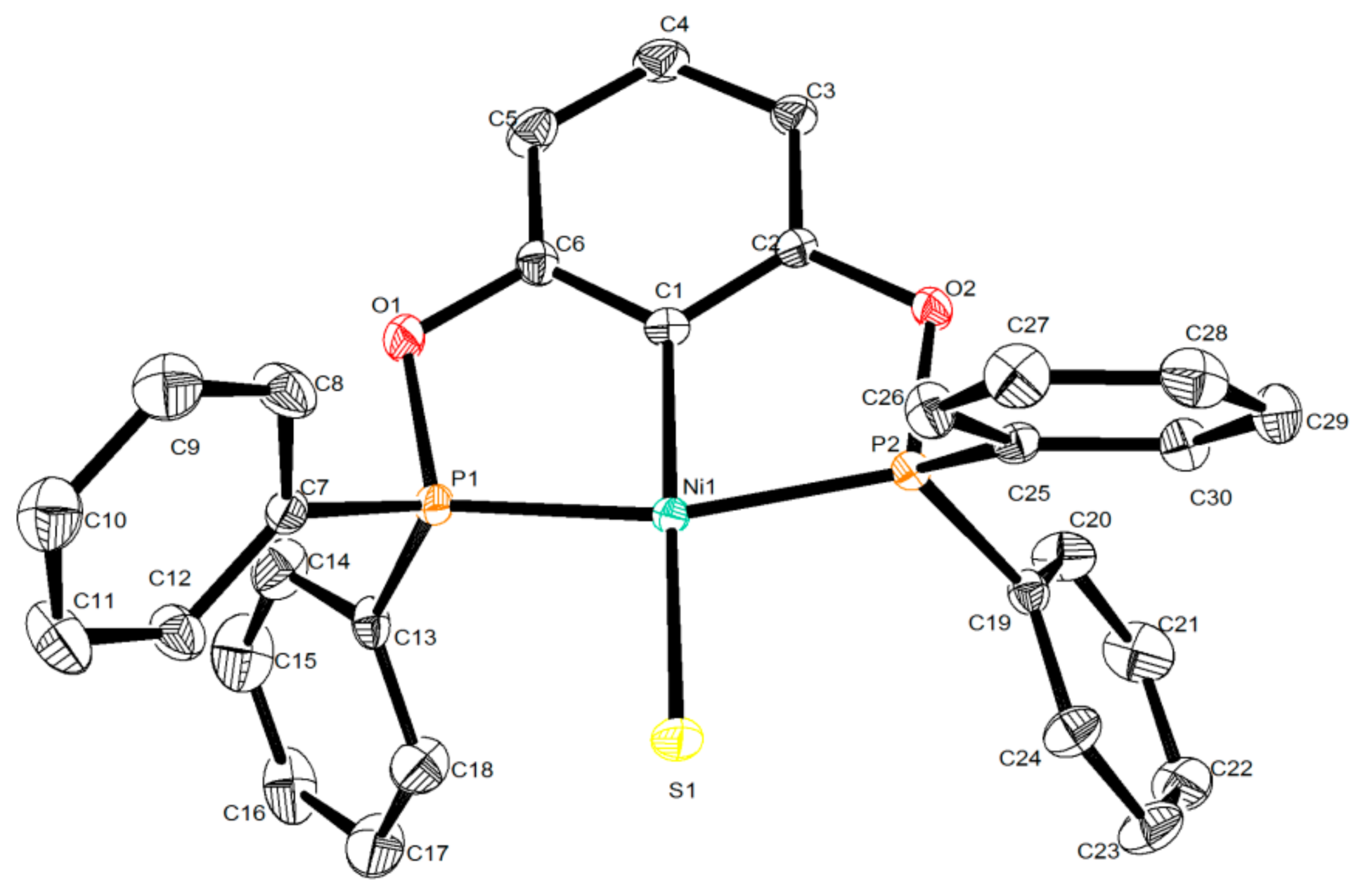
3.3. X-ray Structure Determination of [2,6-(Ph2PO)2C6H3]NiSH (1c)
3.4. Procedure for the Catalytic Hydroboration of CO2
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kortlever, R.; Shen, J.; Schouten, K.J.P.; Calle-Vallejo, F.; Koper, M.T.M. Catalysts and reaction pathways for the electrochemical reduction of carbon dioxide. J. Phys. Chem. Lett. 2015, 6, 4073–4082. [Google Scholar] [CrossRef] [PubMed]
- Costentin, C.; Roberta, M.; Savéant, J.-M. Catalysis of the electrochemical reduction of carbon dioxide. Chem. Soc. Rev. 2013, 42, 2423–2436. [Google Scholar] [CrossRef] [PubMed]
- Takeda, H.; Ishitani, O. Development of efficient photocatalytic systems for CO2 reduction using mononuclear and multinuclear metal complexes based on mechanistic studies. Coord. Chem. Rev. 2010, 254, 346–354. [Google Scholar] [CrossRef]
- Chong, C.C.; Kinjo, R. Catalytic hydroboration of carbonyl derivatives, imines, and carbon dioxide. ACS Catal. 2015, 5, 3238–3259. [Google Scholar] [CrossRef]
- Benson, E.E.; Kubiak, C.P.; Sathrum, A.J.; Smieja, J.M. Electrocatalytic and homogeneous approaches to conversion of CO2 to liquid fuels. Chem. Soc. Rev. 2009, 38, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-H.; Himeda, Y.; Muckerman, J.T.; Manbeck, G.F.; Fujita, E. CO2 hydrogenation to formate and methanol as an alternative to photo- and electrochemical CO2 reduction. Chem. Rev. 2015, 115, 12936–12973. [Google Scholar] [CrossRef] [PubMed]
- Ali, K.A.; Abdullah, A.Z.; Mohamed, A.R. Recent development in catalytic technologies for methanol synthesis from renewable sources: A critical review. Renew. Sustain. Energy Rev. 2015, 44, 508–518. [Google Scholar] [CrossRef]
- Ganesh, I. Conversion of carbon dioxide into methanol—A potential liquid fuel: Fundamental challenges and opportunities (a review). Renew. Sustain. Energy Rev. 2014, 31, 221–257. [Google Scholar] [CrossRef]
- Fontaine, F.-G.; Courtemanche, M.-A.; Légaré, M.-A. Transition-metal-free catalytic reduction of carbon dioxide. Chem. Eur. J. 2014, 20, 2990–2996. [Google Scholar] [CrossRef] [PubMed]
- Habisreu-tinger, S.N.; Schmidt-Mende, L.; Stolarczyk, J.K. Photocatalytic reduction of CO2 on TiO2 and other semiconductors. Angew. Chem. Int. Ed. 2013, 52, 7372–7408. [Google Scholar] [CrossRef] [PubMed]
- Appel, A.M.; Bercaw, J.E.; Bocarsly, A.B.; Dobbek, H.; DuBois, D.L.; Dupuis, M.; Ferry, J.G.; Fujita, E.; Hille, R.; Kenis, P.J.A. Frontiers, opportunities, and challenges in biochemical and chemical catalysis of CO2 fixation. Chem. Rev. 2013, 113, 6621–6658. [Google Scholar] [CrossRef] [PubMed]
- Aresta, M.; Dibenedetto, A.; Angelini, A. Catalysis for the valorization of exhaust carbon: From CO2 to chemicals, materials, and fuels. Technological use of CO2. Chem. Rev. 2014, 114, 1709–1742. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, H.; Aresta, M.; Armor, J.N.; Barteau, M.A.; Beckman, E.J.; Bell, A.T.; Bercaw, J.E.; Creutz, C.; Dinjus, E.; Dixon, D.A.; et al. Catalysis research of relevance to carbon management: Progress, challenges, and opportunities. Chem. Rev. 2001, 101, 953–996. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Zhang, J.; Krause, J.A.; Guan, H. An efficient nickel catalyst for the reduction of carbon dioxide with a borane. J. Am. Chem. Soc. 2010, 132, 8872–8873. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Zhang, J.; Patel, Y.J.; Krause, J.A.; Guan, H. Pincer-ligated nickel hydridoborate complexes: The dormant species in catalytic reduction of carbon dioxide with boranes. Inorg. Chem. 2013, 52, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Patel, Y.J.; Krause, J.A.; Guan, H. Catalytic properties of nickel bis(phosphinite) pincer complexes in the reduction of CO2 to methanol derivatives. Polyhedron 2012, 32, 30–34. [Google Scholar] [CrossRef]
- Wesselbaum, S.; vom Stein, T.; Klankermayer, J.; Leitner, W. Hydrogenation of carbon dioxide to methanol by using a homogeneous ruthenium–phosphine catalyst. Angew. Chem. Int. Ed. 2012, 51, 7499–7502. [Google Scholar] [CrossRef] [PubMed]
- Huff, C.A.; Sanford, M.S. Cascade catalysis for the homogeneous hydrogenation of CO2 to methanol. J. Am. Chem. Soc. 2011, 133, 18122–18125. [Google Scholar] [CrossRef] [PubMed]
- Rezayee, N.M.; Huff, C.A.; Sanford, M.S. Tandem amine and ruthenium-catalyzed hydrogenation of CO2 to methanol. J. Am. Chem. Soc. 2015, 137, 1028–1031. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, K.; Sasaki, Y.; Kawai, M.; Watanabe, T.; Saito, M. Ruthenium complex catalysed hydrogenation of carbon dioxide to carbon monoxide, methanol and methane. J. Chem. Soc. Chem. Commun. 1993, 0, 629–631. [Google Scholar] [CrossRef]
- Wesselbaum, S.; Moha, V.; Meuresch, M.; Brosinski, S.; Thenert, K.M.; Kothe, J.; vom Stein, T.; Englert, U.; Hölscher, M.; Klankermayer, J.; et al. Hydrogenation of carbon dioxide to methanol using a homogeneous ruthenium–triphos catalyst: From mechanistic investigations to multiphase catalysis. Chem. Sci. 2015, 6, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Kothandaraman, J.; Goeppert, A.; Czaun, M.; Olah, G.A.; Prakash, G.K.S. Conversion of CO2 from air into methanol using a polyamine and a homogeneous ruthenium catalyst. J. Am. Chem. Soc. 2016, 138, 778–781. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Williams, T.J. Di(carbene)-supported nickel systems for CO2 reduction under ambient conditions. ACS Catal. 2016, 6, 6670–6673. [Google Scholar] [CrossRef]
- Bontemps, S.; Vendier, L.; Sabo-Etienne, S. Borane-mediated carbon dioxide reduction at ruthenium: Formation of C1 and C2 compounds. Angew. Chem. Int. Ed. 2012, 51, 1671–1674. [Google Scholar] [CrossRef] [PubMed]
- Hadlington, T.J.; Kefalidis, C.E.; Maron, L.; Jones, C. Efficient reduction of carbon dioxide to methanol equivalents catalyzed by two-coordinate amido–germanium(II) and−tin(II) hydride complexes. ACS Catal. 2017, 7, 1853–1859. [Google Scholar] [CrossRef]
- Ma, Q.-Q.; Liu, T.; Li, S.; Zhang, J.; Chen, X.; Guan, H. Highly efficient reduction of carbon dioxide with a borane catalyzed by bis(phosphinite) pincer ligated palladium thiolate complexes. Chem. Commun. 2016, 52, 14262–14265. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Meng, W.; Ma, Q.-Q.; Zhang, J.; Li, H.; Li, S.; Zhao, Q.; Chen, X. Hydroboration of CO2 catalyzed by bis(phosphinite) pincer ligated nickel thiolate complexes. Dalton Trans. 2017, 46, 4504–4509. [Google Scholar] [CrossRef] [PubMed]
- Tamang, S.R.; Findlater, M. Cobalt catalysed reduction of CO2 via hydroboration. Dalton Trans. 2018, 47, 8199–8203. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Gonçalves, T.P.; Zhao, Q.; Gong, D.; Lai, Z.; Wang, Z.; Zheng, J.; Huang, K.-W. Diverse catalytic reactivity of a dearomatized PN3P*–nickel hydride pincer complex towards CO2 reduction. Chem. Commun. 2018, 54, 11395–11398. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.-G.; Li, B.-J.; Shi, Z.-J. Exploration of new C−O electrophiles in cross-coupling reactions. Acc. Chem. Res. 2010, 43, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Tasker, S.Z.; Standley, E.A.; Jamison, T.F. Recent advances in homogeneous nickel catalysis. Nature 2014, 509, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Cao, Z.-C.; Shi, Z.-J. Exploration of earth-abundant transition metals (Fe, Co, and Ni) as catalysts in unreactive chemical bond activations. Acc. Chem. Res. 2015, 48, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Medley, C.M.; Krause, J.A.; Guan, H. Mechanistic insights into C−S cross-coupling reactions catalyzed by nickel bis(phosphinite) pincer complexes. Organometallics 2010, 29, 6393–6401. [Google Scholar] [CrossRef]
- Zhang, J.; Adhikary, A.; King, K.M.; Krause, J.A.; Guan, H. Substituent effects on Ni–S bond dissociation energies and kinetic stability of nickel arylthiolate complexes supported by a bis(phosphinite)-based pincer ligand. Dalton Trans. 2012, 41, 7959–7968. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Meng, W.; Adhikary, A.; Li, S.; Ma, N.; Zhao, Q.; Yang, Q.; Eberhardt, N.A.; Leahy, K.M.; Krause, J.A.; et al. Metathesis reactivity of bis(phosphinite) pincer ligated nickel chloride, isothiocyanate and azide complexes. J. Organomet. Chem. 2016, 804, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.-Q.; Liu, T.; Adhikary, A.; Zhang, J.; Krause, J.A.; Guan, H. Using CS2 to probe the mechanistic details of decarboxylation of bis(phosphinite)-ligated nickel pincer formate complexes. Organometallics 2016, 35, 4077–4082. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, T.; Ma, Q.-Q.; Li, S.; Chen, X. A reaction of [2,6-(tBu2PO)2C6H3]NiSCH2Ph with BH3·THF: Borane mediated C–S bond cleavage. Dalton Trans. 2018, 47, 6018–6024. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, T.; Wei, C.; Chang, J.; Ma, Q.-Q.; Li, S.; Ma, N.; Chen, X. The reactivity of mercapto groups against boron hydrides in pincer ligated nickel mercapto complexes. Chem. Asian J. 2018, 13. [Google Scholar] [CrossRef] [PubMed]
- Vabre, B.; Spasyuk, M.D.; Zargarian, D. Impact of backbone substituents on POCOP-Ni pincer complexes: A structural, spectroscopic, and electrochemical study. Organometallics 2012, 31, 8561–8570. [Google Scholar] [CrossRef]
- Vabre, B.; Petiot, P.; Declercq, R.; Zargarian, D. Fluoro and trifluoromethyl derivatives of POCOP-type pincer complexes of nickel: Preparation and reactivities in SN2 fluorination and direct benzylation of unactivated arenes. Organometallics 2014, 33, 5173–5184. [Google Scholar] [CrossRef]
- Hao, J.; Vabre, B.; Zargarian, D. POCOP-ligated nickel siloxide complexes: Syntheses, characterization, and reactivities. Organometallics 2014, 33, 6568–6576. [Google Scholar] [CrossRef]
- Lapointe, S.; Vabre, B.; Zargarian, D. POCOP-type pincer complexes of nickel: Synthesis, characterization, and ligand exchange reactivities of new cationic acetonitrile adducts. Organometallics 2015, 34, 3520–3531. [Google Scholar] [CrossRef]
- Buil, M.L.; Elipe, S.; Esteruelas, M.A.; Oñate, E.; Peinado, E.; Ruiz, N. Five-coordinate complexes MHCl(CO)(PiPr3)2 (M = Os, Ru) as precursors for the preparation of new hydrido- and alkenyl-metallothiol and monothio−β-diketonato derivatives. Organometallics 1997, 16, 5748–5755. [Google Scholar] [CrossRef]
- Vicic, D.A.; Jones, W.D. Room-temperature desulfurization of dibenzothiophene mediated by [(i-Pr2PCH2)2NiH]2. J. Am. Chem. Soc. 1997, 119, 10855–10856. [Google Scholar] [CrossRef]
- Di Vaira, M.; Peruzzini, M.; Stoppioni, P. Hydrochalcogenide and hydride hydrochalcogenide derivatives of rhodium. Inorg. Chem. 1991, 30, 1001–1007. [Google Scholar] [CrossRef]
- Courtemanche, M.A.; Legare, M.A.; Maron, L.; Fontaine, F.G. A highly active phosphine–borane organocatalyst for the reduction of CO2 to methanol using hydroboranes. J. Am. Chem. Soc. 2013, 135, 9326–9329. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.; Knizek, J.; Nöth, H.; Schur, S.; Thomann, M. Beiträge zur chemie des bors. 237. Bis(benzo-l,3,2-dioxaborolanyl)oxid und 2-(o-hydroxyphenoxy)-benzo-l,3,2-dioxaborolan. Vorstufen zur synthese von catecholboran (benzo-l,3,2-dioxaborolan). Z. Anorg. Allg. Chem. 1997, 623, 901–907. [Google Scholar] [CrossRef]
- Chakraborty, S.; Krause, J.A.; Guan, H. Hydrosilylation of aldehydes and ketones catalyzed by nickel PCP-pincer hydride complexes. Organometallics 2009, 28, 582–586. [Google Scholar] [CrossRef]
- Castonguay, A.; Spasyuk, D.M.; Madern, N.; Beauchamp, A.L.; Zargarian, D. Regioselective hydroamination of acrylonitrile catalyzed by cationic pincer complexes of Nickel(II). Organometallics 2009, 28, 2134–2141. [Google Scholar] [CrossRef]
- Salah, A.B.; Zargarian, D. The impact of P-substituents on the structures, spectroscopic properties, and reactivities of POCOP-type pincer complexes of Nickel(II). Dalton Trans. 2011, 40, 8977–8985. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Benítez, V.; Baldovino-Pantaleón, O.; Herrera-Álvarez, C.; Toscano, R.A.; Morales-Morales, D. High yield thiolation of iodobenzene catalyzed by the phosphinite nickel PCP pincer complex: [NiCl{C6H3-2,6-(OPPh2)2}]. Tetrahedron Lett. 2006, 47, 5059–5062. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | X | R | Time (min) | Conversion (%) | TON | TOF (h−1) |
|---|---|---|---|---|---|---|
| 1a | SH | tBu | 30 | 100 | 450 ± 19 | 900 ± 38 |
| 1b | SH | iPr | 15 | 100 | 465 ± 15 | 1860 ± 60 |
| 2a | N3 | tBu | 30 | 100 | 480 ± 11 | 960 ± 22 |
| 2b | N3 | iPr | 15 | 100 | 477 ± 12 | 1908 ± 48 |
| 3a | NCS | tBu | 120 | 0 | 0 | 0 |
| 3b | NCS | iPr | 120 | 68 | 325 ± 17 | 163 ± 9 |
| Empirical formula | C30H24NiO2P2S | Volume, Å3 | 2582.0(3) |
| Formula weight | 569.20 | Z | 4 |
| Temp, K | 103(2) | dcalc, g cm−3 | 1.464 |
| Crystal system | Monoclinic | λ, Å | 0.71073 |
| Space group | P 1 21/c 1 | μ, mm−1 | 0.983 |
| a, Å | 14.9995(10) | No. of data collected | 29,194 |
| b, Å | 9.9798(5) | No. of unique data | 10,374 |
| c, Å | 17.2488(12) | Rint | 0.0895 |
| α (°) | 90 | Goodness-of-fit on F2 | 1.003 |
| β (°) | 90.195(3) | R1, wR2 (I > 2σ(I)) | 0.0583, 0.1182 |
| γ (°) | 90 | R1, wR2 (all data) | 0.1111, 0.1411 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Chang, J.; Liu, T.; Cao, B.; Ding, Y.; Chen, X. Application of POCOP Pincer Nickel Complexes to the Catalytic Hydroboration of Carbon Dioxide. Catalysts 2018, 8, 508. https://doi.org/10.3390/catal8110508
Zhang J, Chang J, Liu T, Cao B, Ding Y, Chen X. Application of POCOP Pincer Nickel Complexes to the Catalytic Hydroboration of Carbon Dioxide. Catalysts. 2018; 8(11):508. https://doi.org/10.3390/catal8110508
Chicago/Turabian StyleZhang, Jie, Jiarui Chang, Ting Liu, Bula Cao, Yazhou Ding, and Xuenian Chen. 2018. "Application of POCOP Pincer Nickel Complexes to the Catalytic Hydroboration of Carbon Dioxide" Catalysts 8, no. 11: 508. https://doi.org/10.3390/catal8110508