BF3·Et2O-Promoted Decomposition of Cyclic α-Diazo-β-Hydroxy Ketones: Novel Insights into Mechanistic Aspects


 ,
,  , ,
, ,  and
and
Abstract
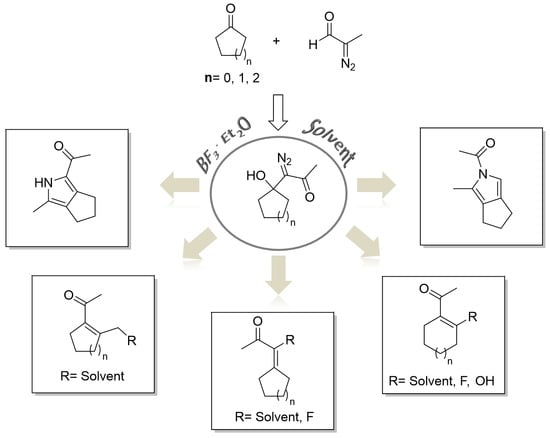
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
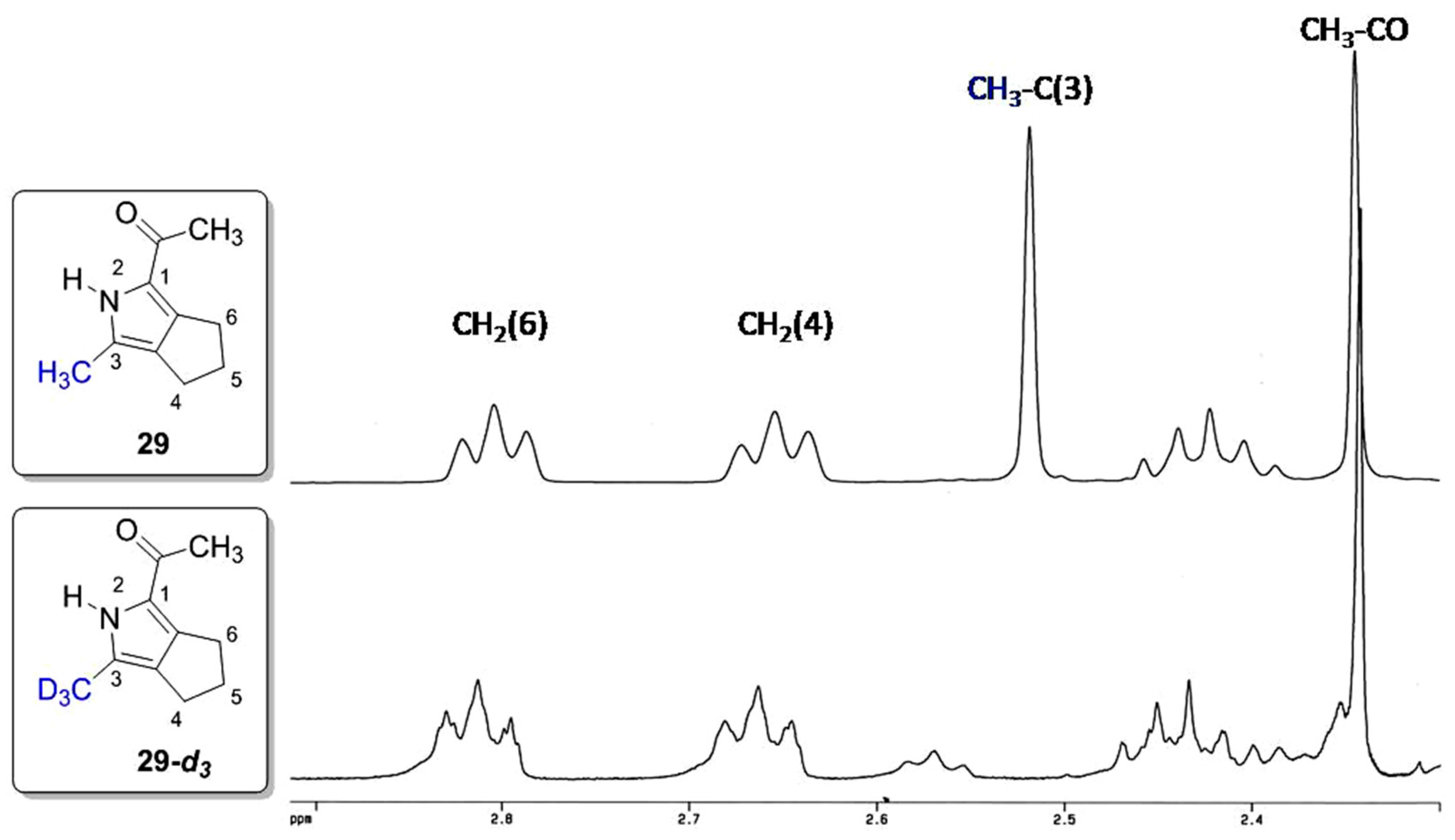
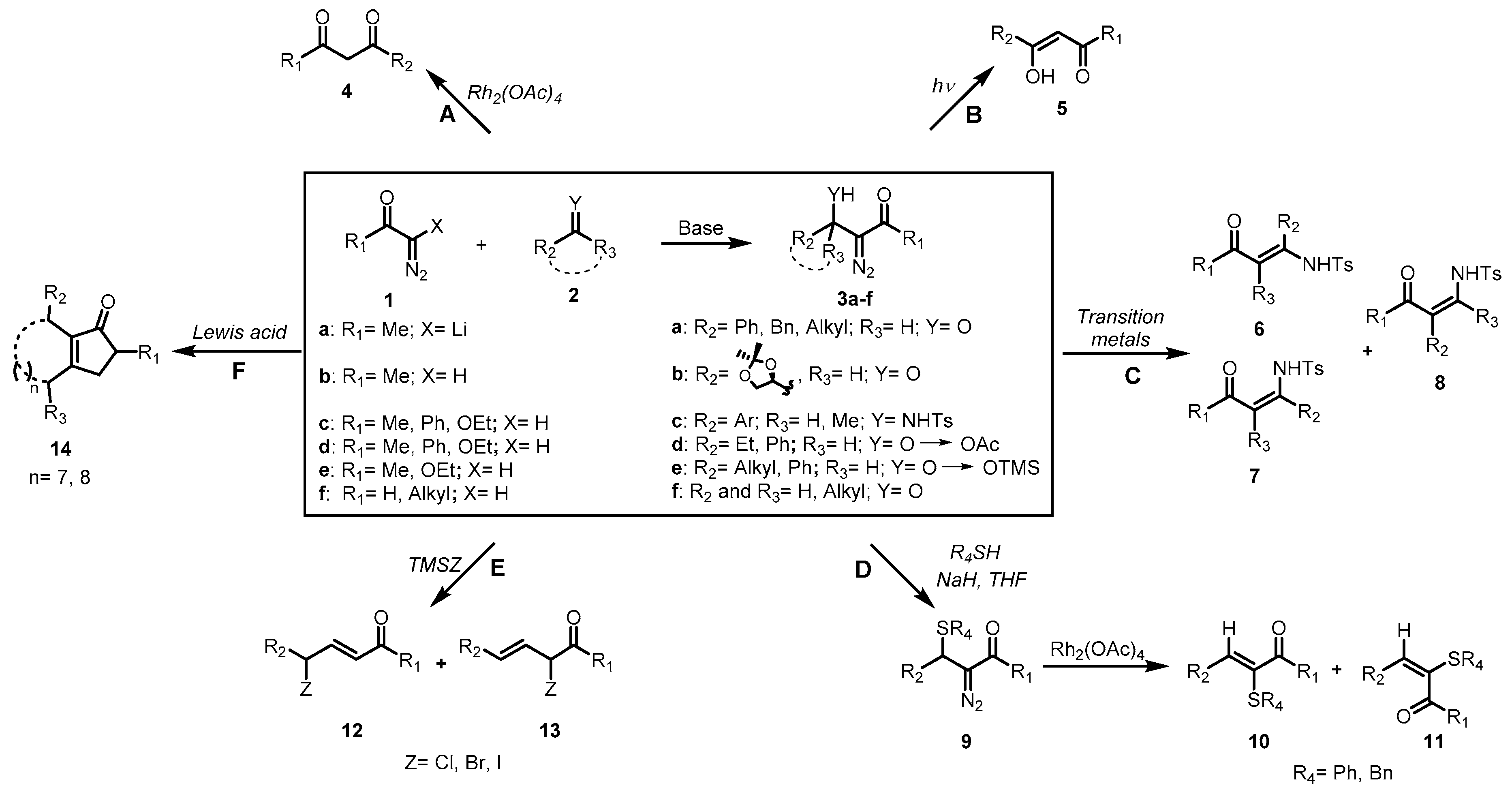
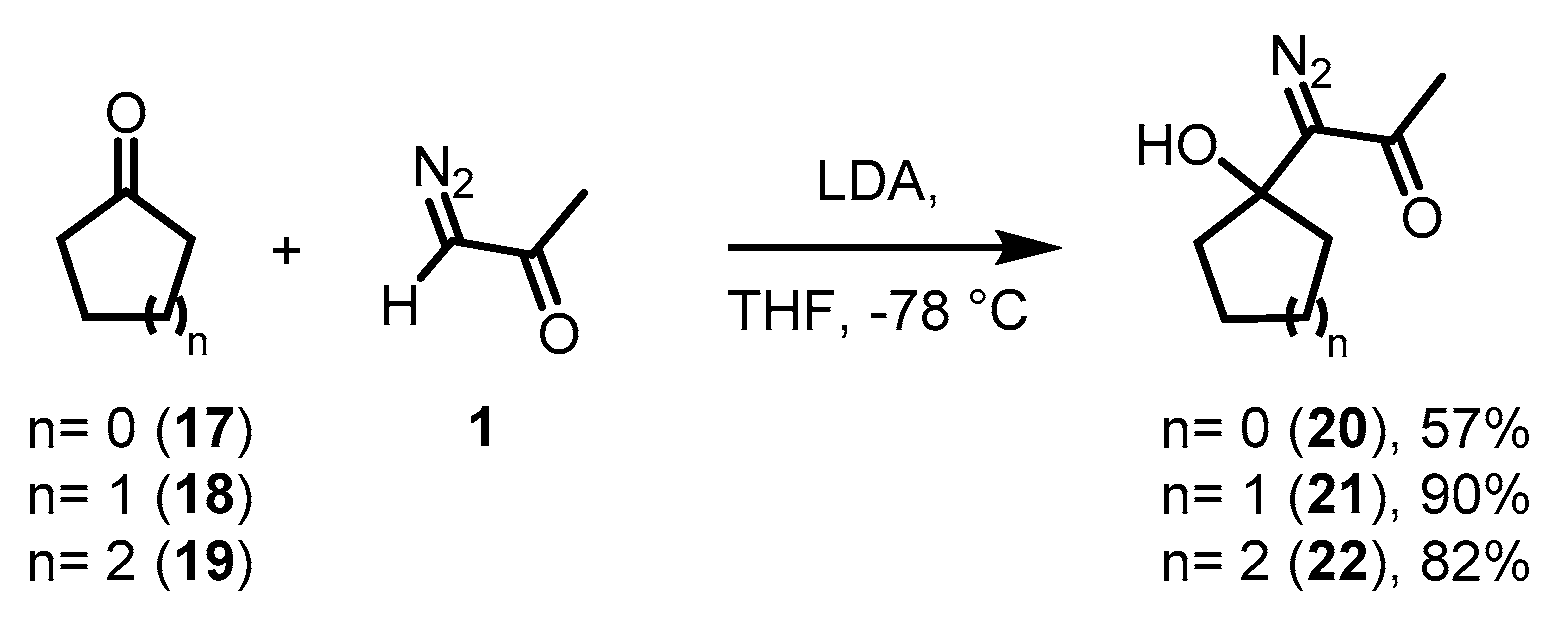
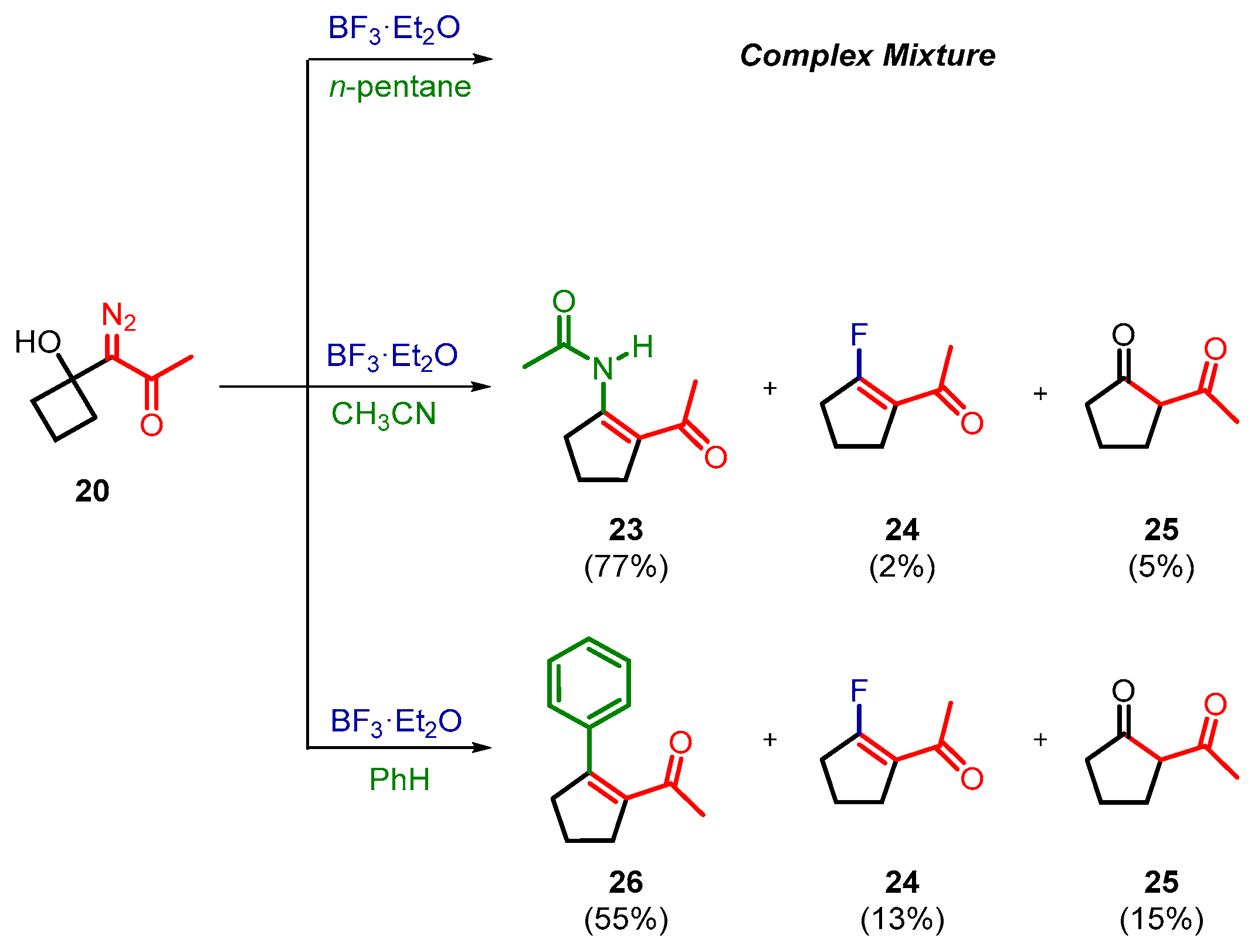
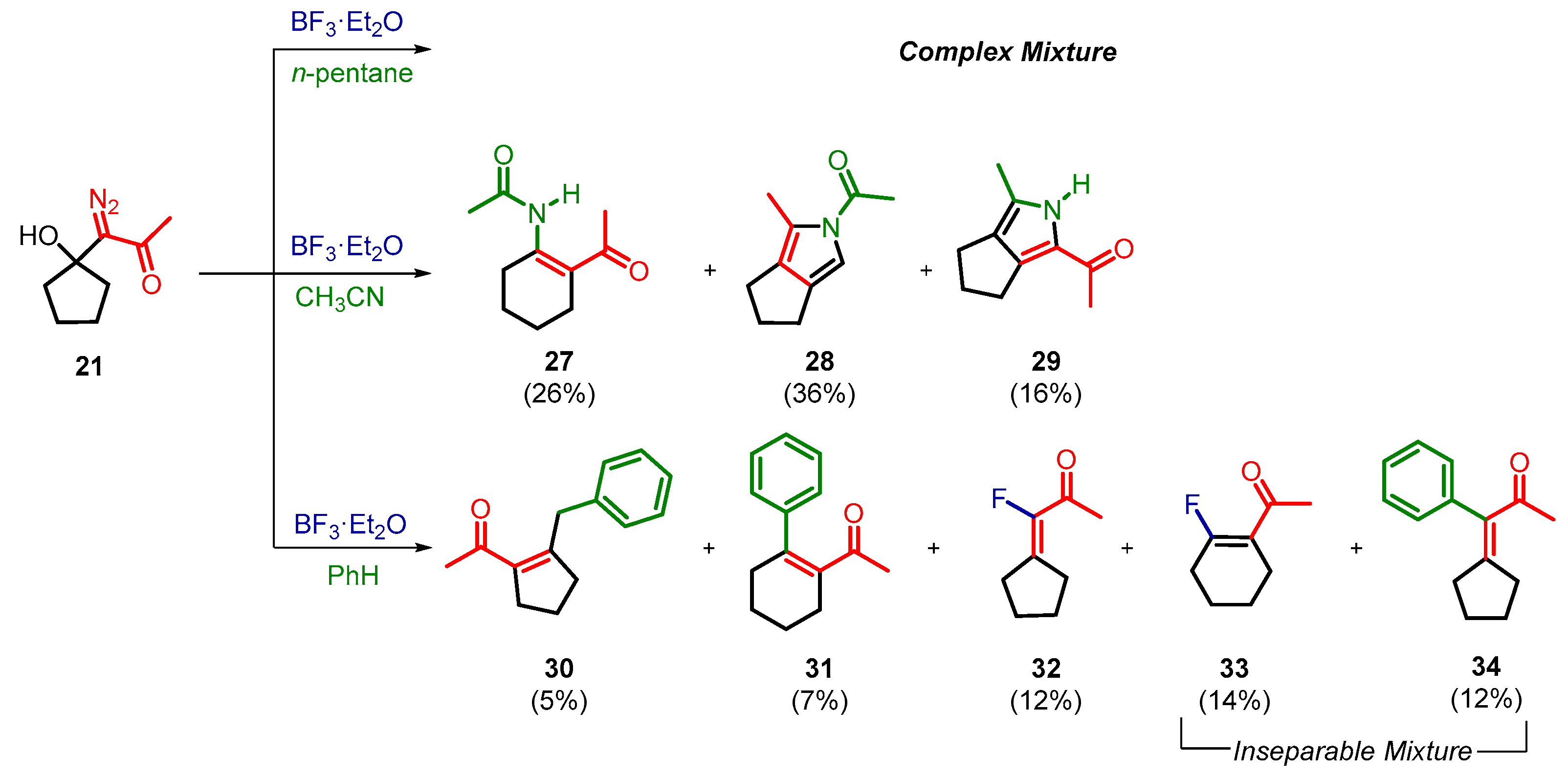
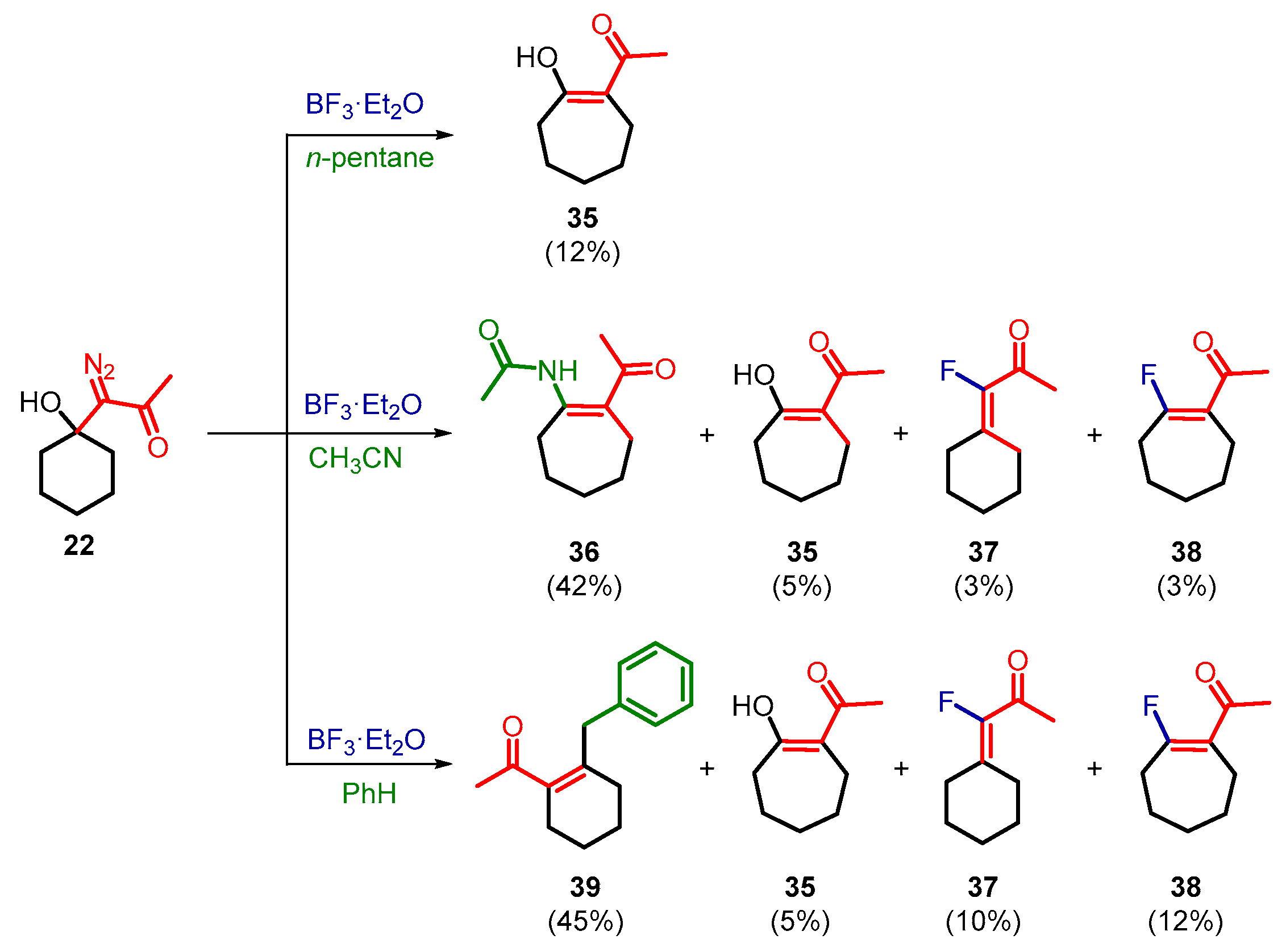
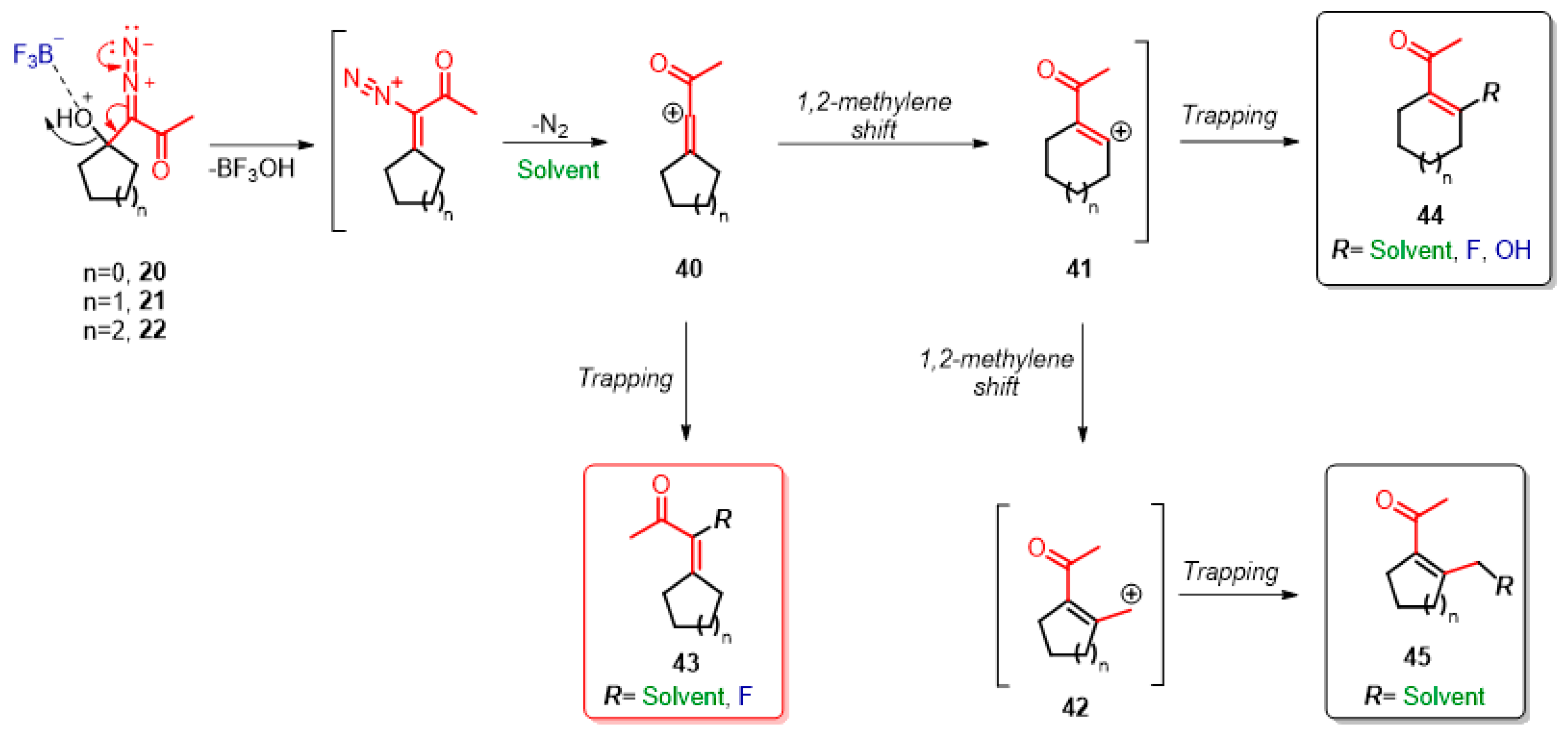
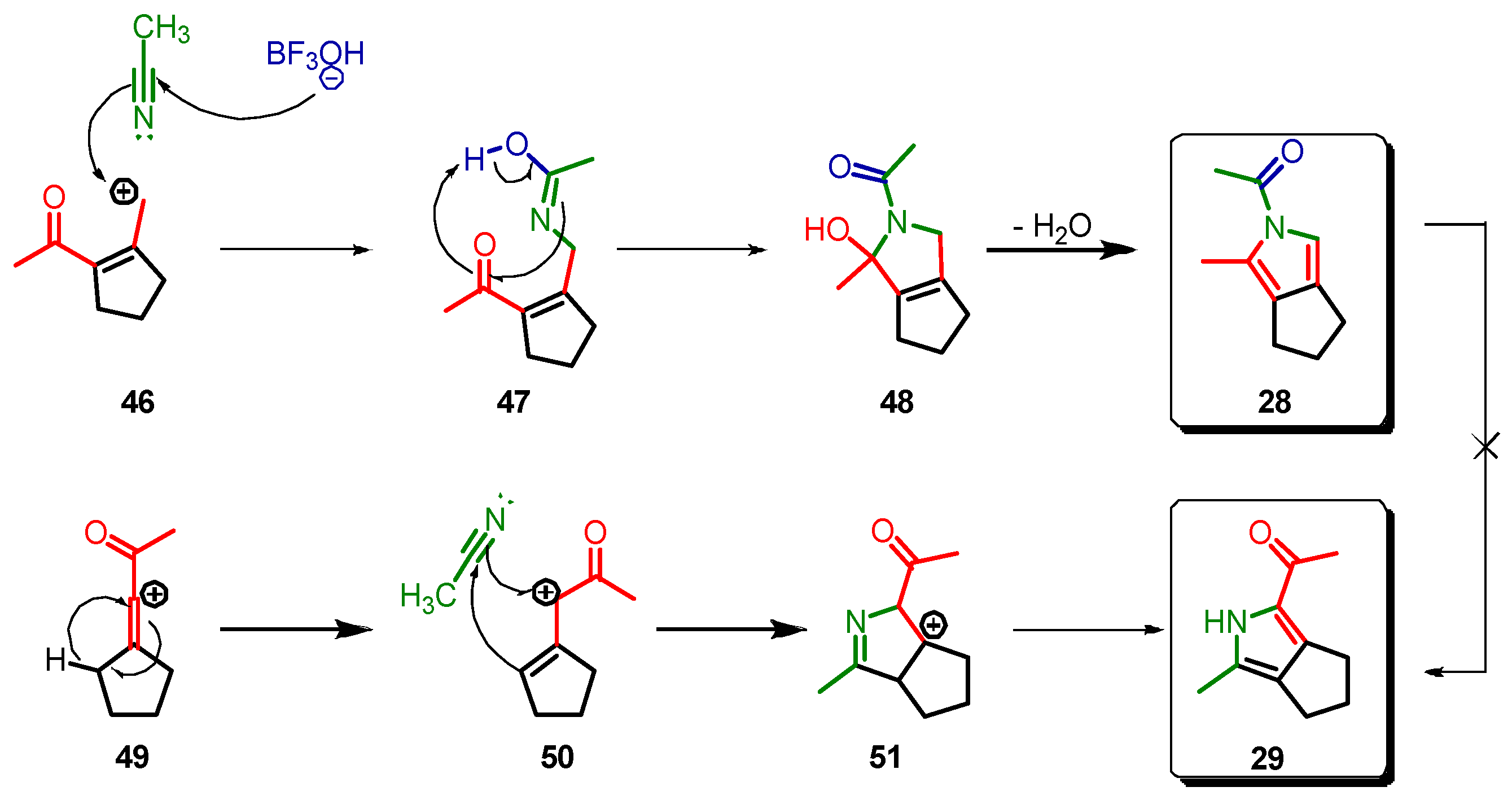
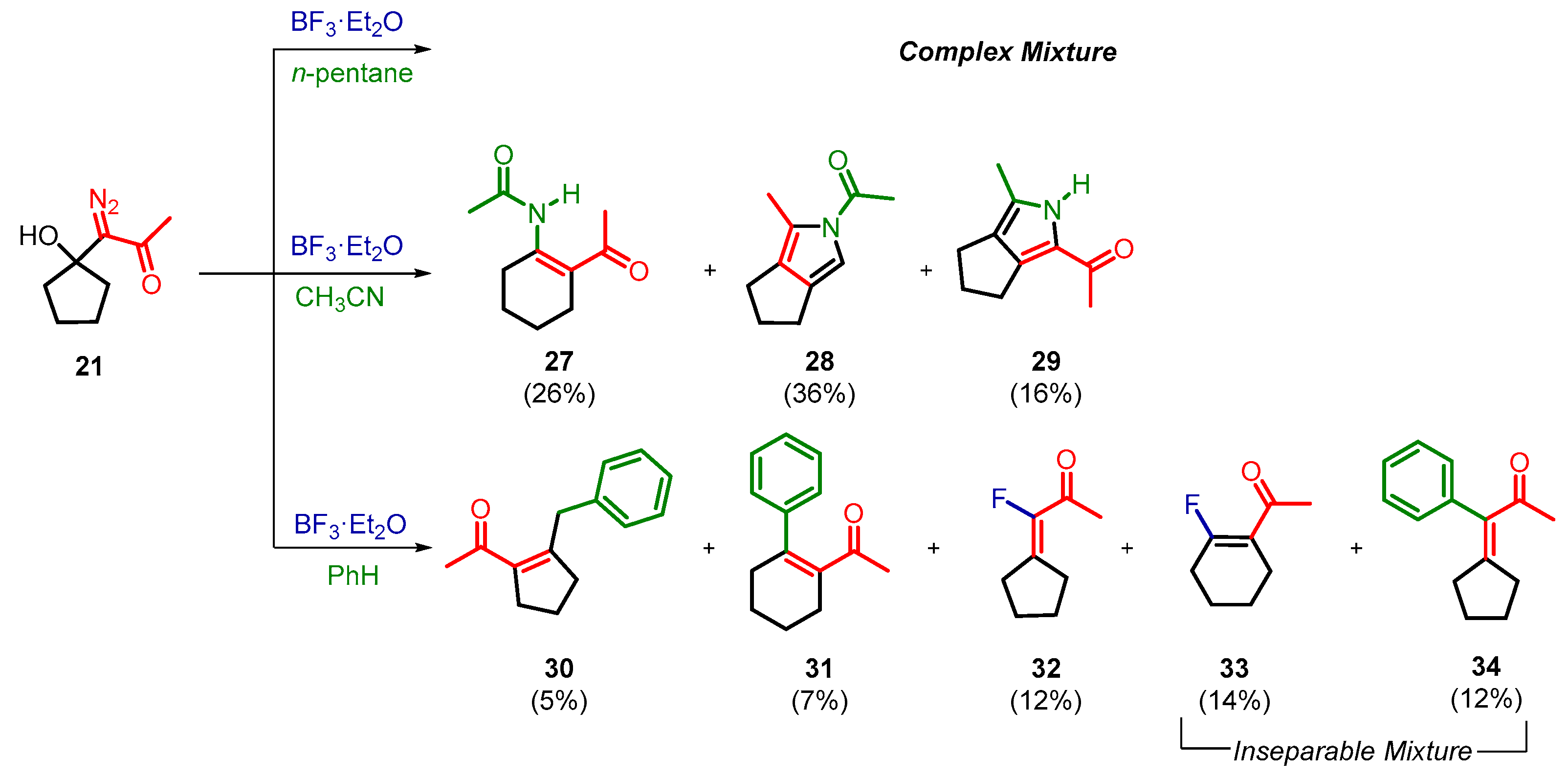
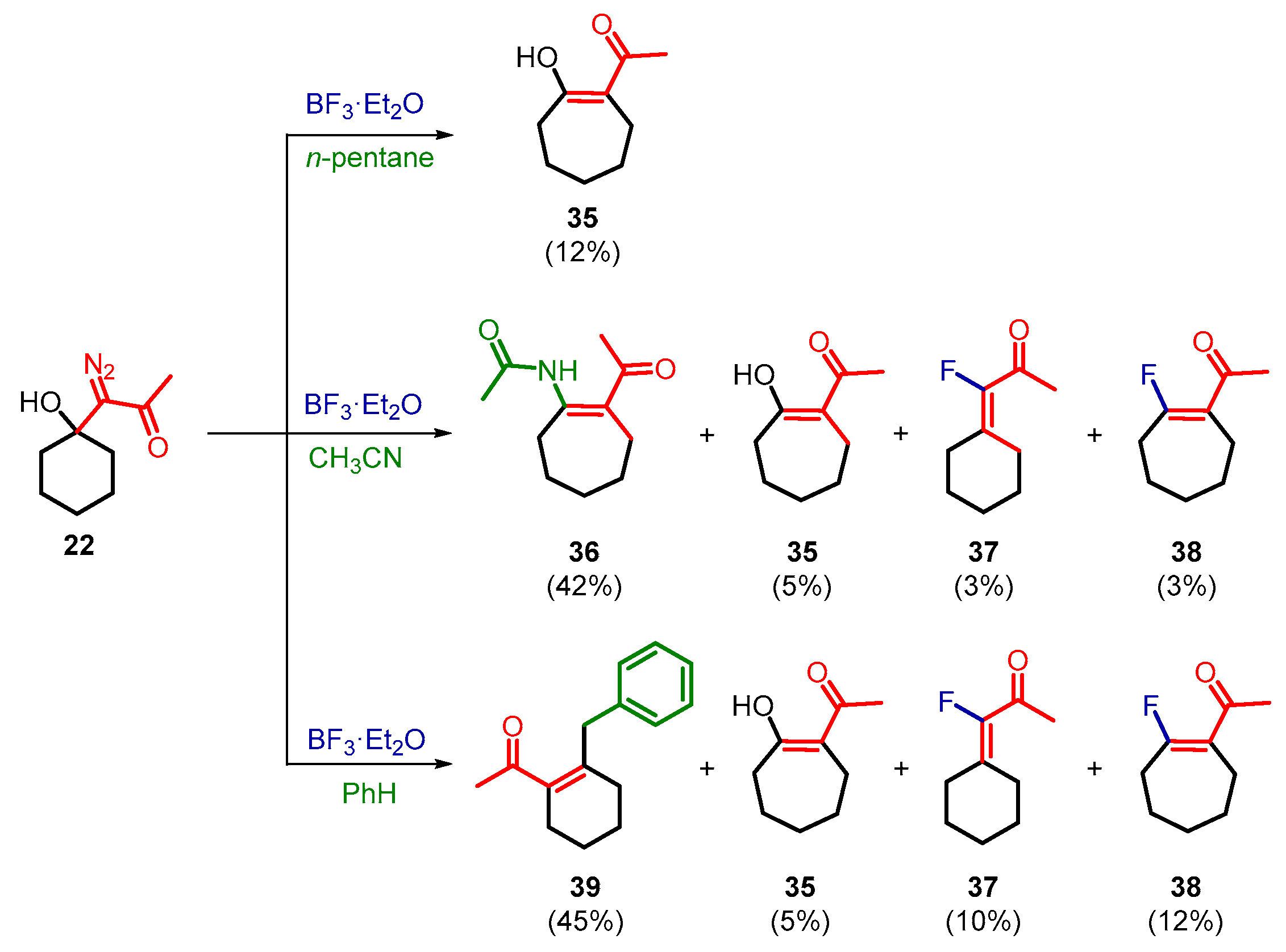
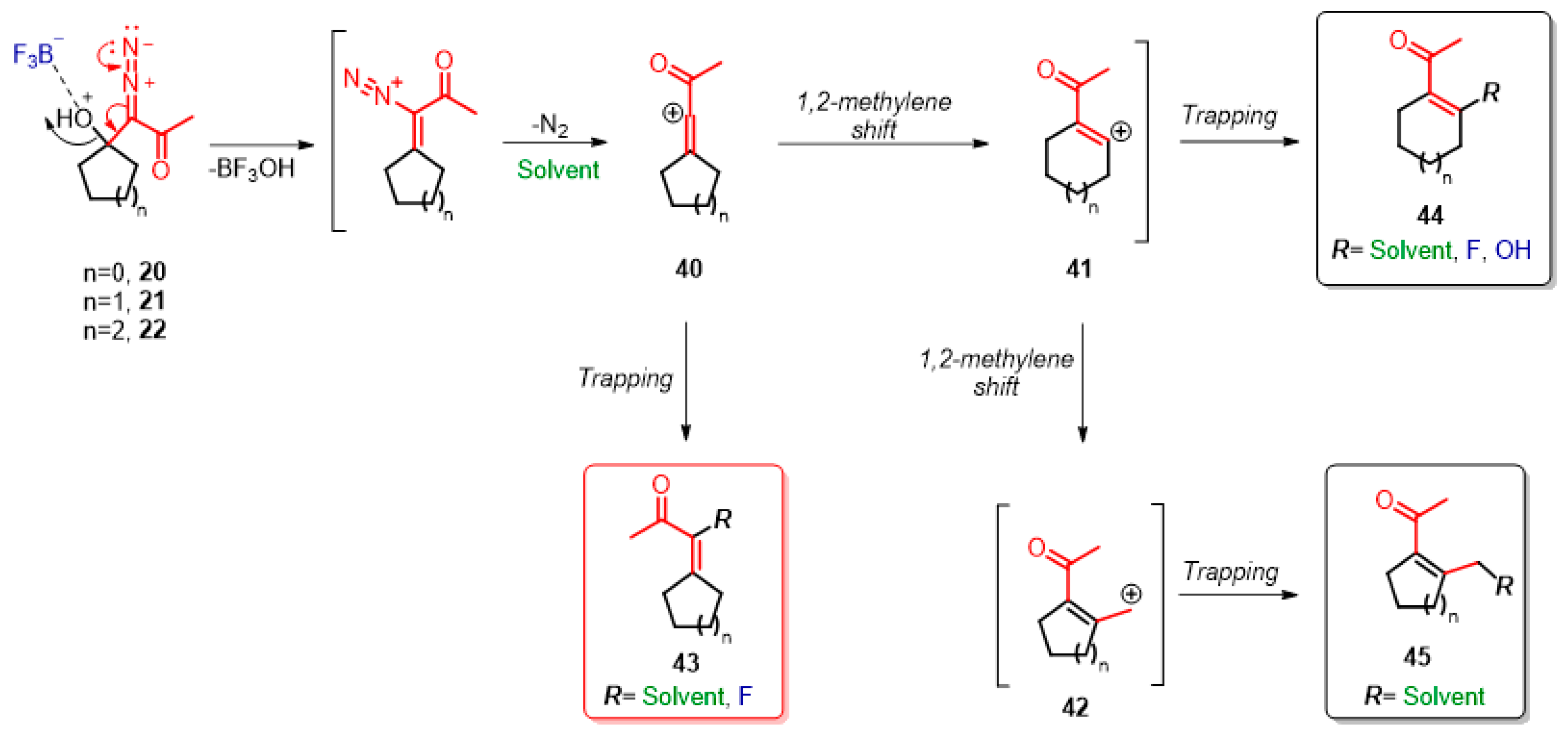
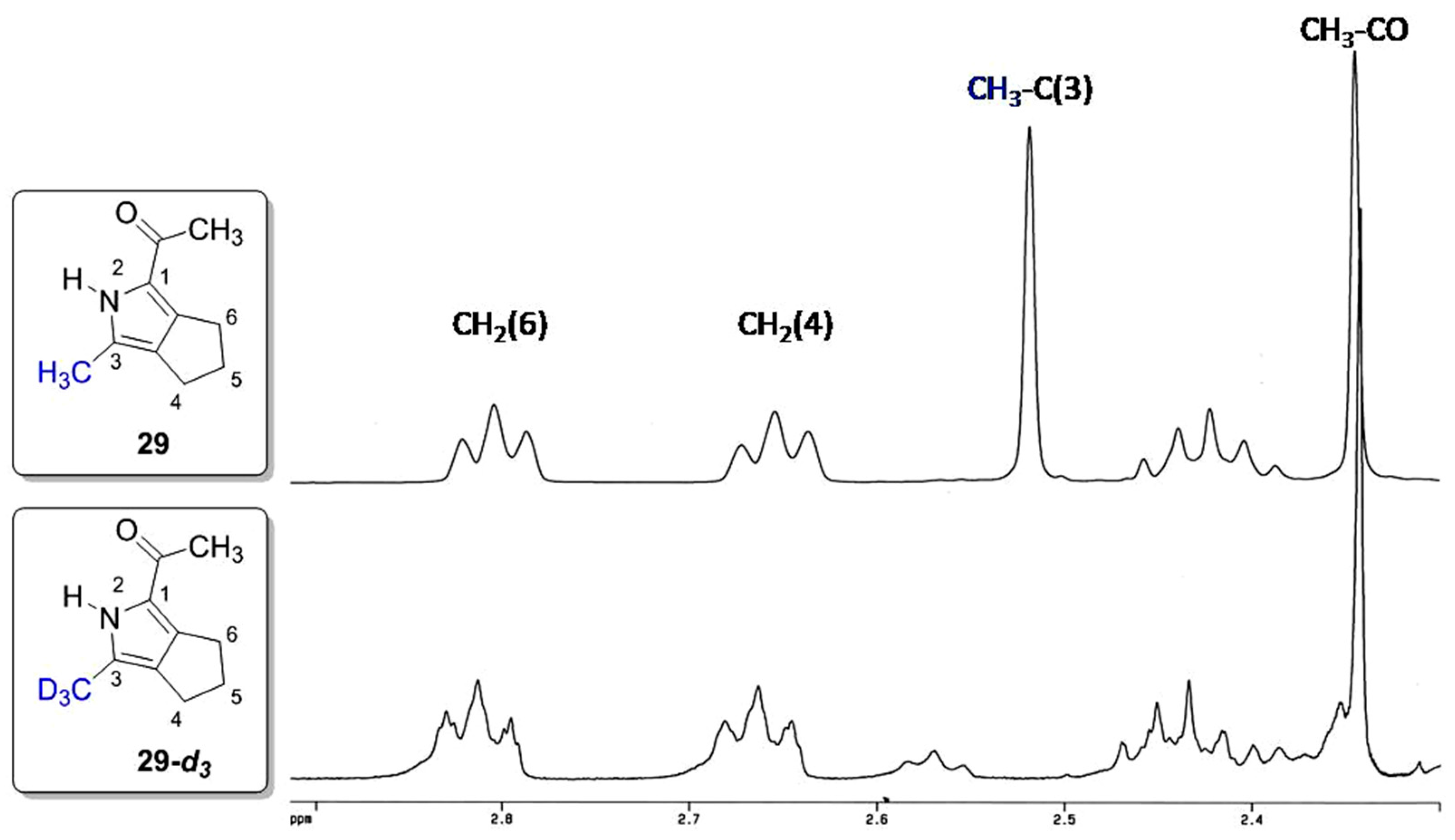
2. Results and Discussion
3. Materials and Methods
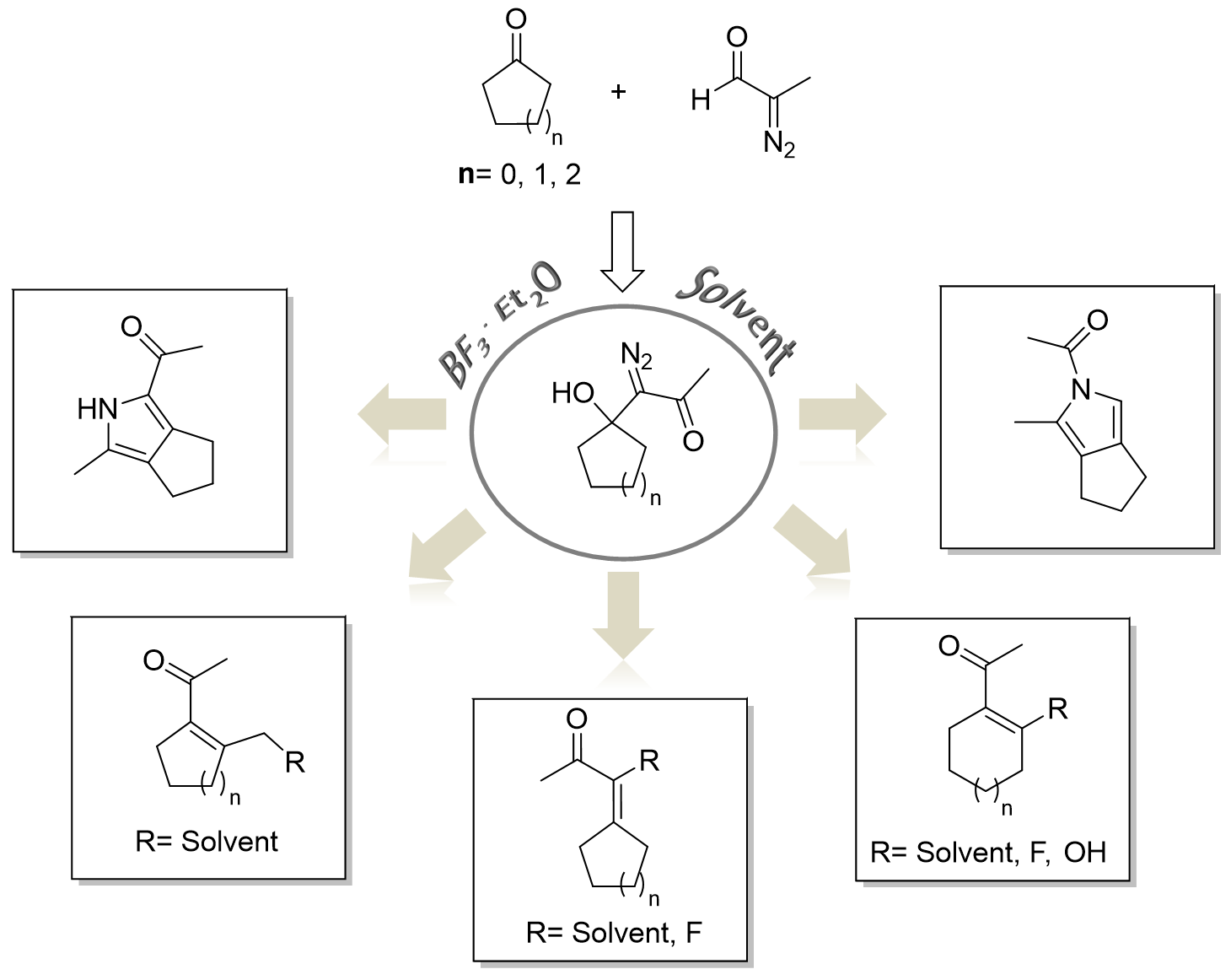
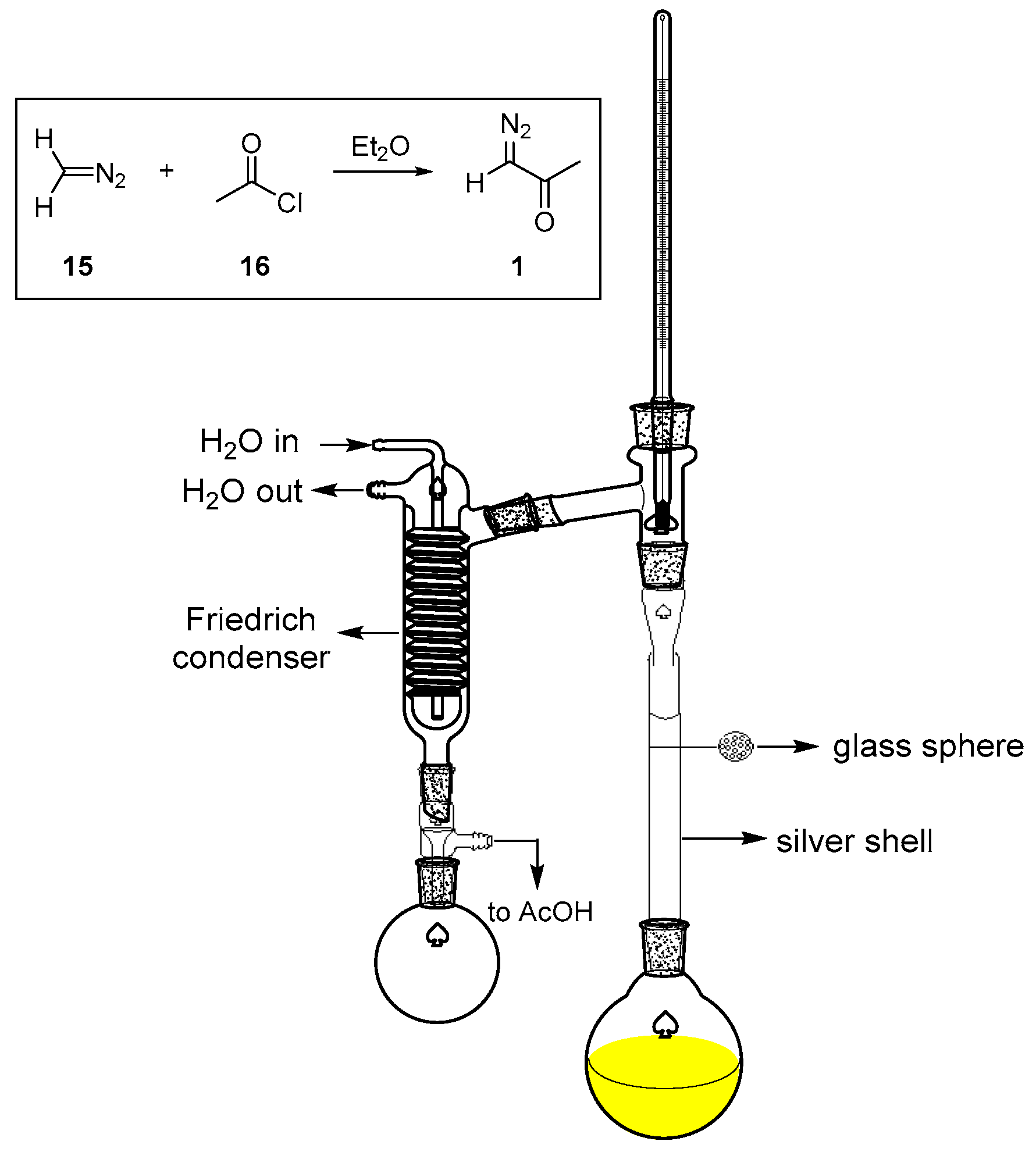
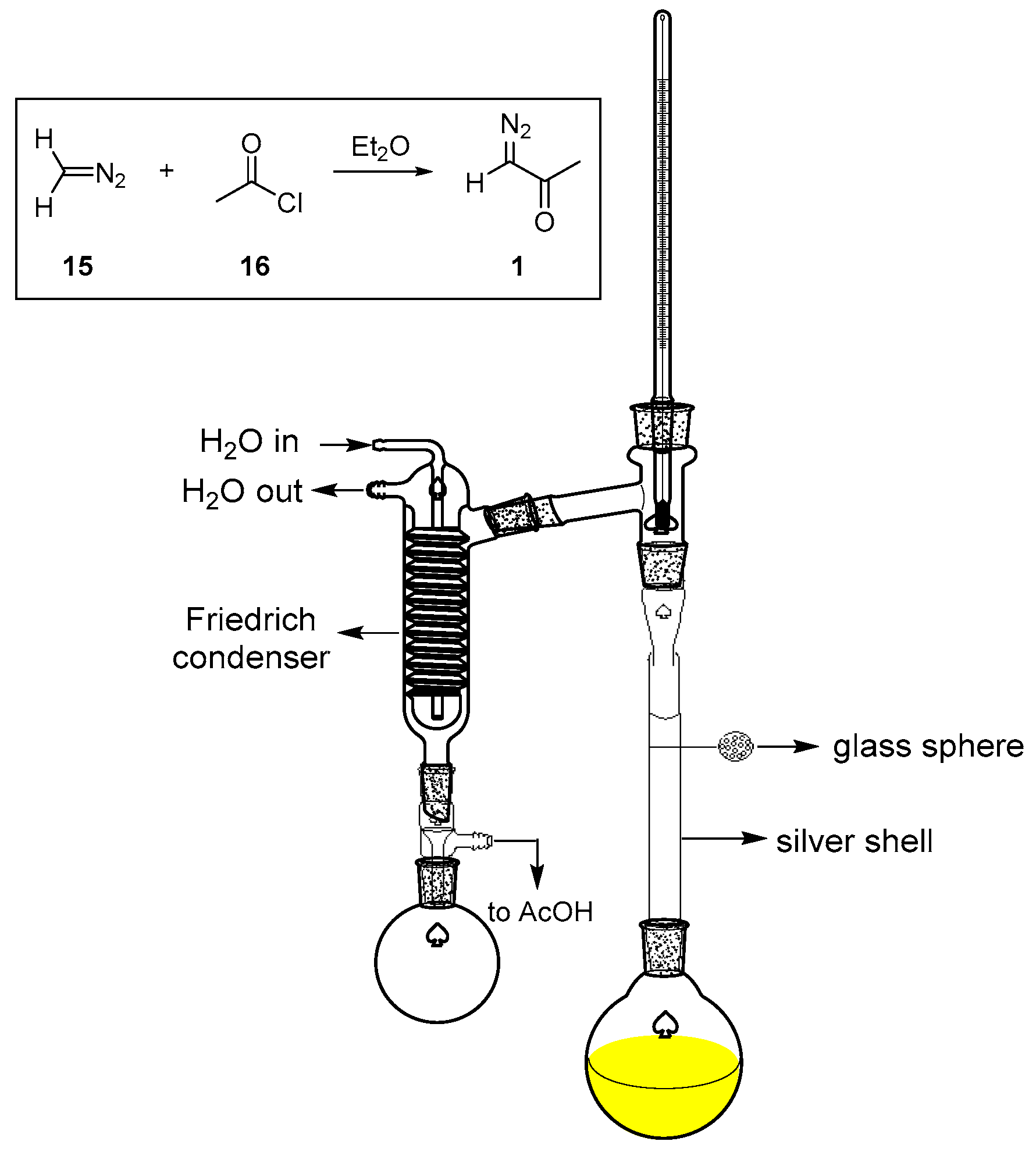
3.1. Preparation of DAA (1)
3.2. General Procedures for the Preparation of 1-Diazo-1-(1-hydroxycycloalkyl)acetone Derivatives 20–22
3.3. General Procedure for BF3·Et2O-Induced Decomposition of 1-Diazo-1-(1-hydroxycycloalkyl)acetone 20–22
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Curtius, T. Ueber die einwirkung von salpetriger säure auf salzasauren glycocollälther. Ber. Dtsch. Chem. Ges. 1883, 16, 2230–2231. [Google Scholar] [CrossRef]
- Candeias, N.R.; Paterna, R.; Gois, P.M.P. Homologation reaction of ketones with diazo compounds. Chem. Rev. 2016, 116, 2937–2981. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, J. Recent development of reactions with α-diazocarbonyl compounds as nucleophiles. Chem. Commun. 2009, 5350–5361. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, J. Recent studies on the reactions of α-diazocarbonyl compounds. Tetrahedron 2008, 64, 6577–6605. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Beckwith, R.E.J. Catalytic enantioselective C–H activation by means of metal−carbenoid-induced C–H insertion. Chem. Rev. 2003, 103, 2861–2904. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M.P.; McKervey, M.A.; Ye, T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds; Wiley-Interscience: New York, NY, USA, 1998; ISBN 978-0-471-13556-2. [Google Scholar]
- Ye, T.; McKervey, M.A. Organic synthesis with α-diazo carbonyl compounds. Chem. Rev. 1994, 94, 1091–1160. [Google Scholar] [CrossRef]
- Bug, T.; Hartnagel, M.; Schlierf, C.; Mayr, H. How nucleophilic are diazo compounds? Chem. Eur. J. 2003, 9, 4068–4076. [Google Scholar] [CrossRef] [PubMed]
- Pellicciari, R.; Fringuelli, R.; Sisani, E.; Curini, M. An improved two-step route for the preparation of β-diketones from aldehydes and its application to the synthesis of β-damascone. J. Chem. Soc. Perkin Trans. 1981, 9, 2566–2569. [Google Scholar] [CrossRef]
- Padwa, A.; Hornbuckle, S.F.; Zhang, Z.; Zhi, L. Synthesis of 1,3-diketones using α-diazo ketones and aldehydes in the presence of tin(II) chloride. J. Org. Chem. 1990, 55, 5297–5299. [Google Scholar] [CrossRef]
- Ye, T.; McKewey, M.A. Synthesis of chiral N-protected α-amino-β-diketones from α-diazoketones derived from natural amino acids. Tetrahedron 1992, 48, 8007–8022. [Google Scholar] [CrossRef]
- Sarabia-García, F.; Cebrián, G.M.P.; López, A.H.; López-Herrera, F.J. Reactions of monosaccharide derivatives with diazocarbonyl compounds. Reactivity and synthetic applications. Tetrahedron 1998, 54, 6867–6896. [Google Scholar] [CrossRef]
- López-Herrera, F.J.; Sarabia-García, F. β-Oxy-α-diazo carbonyl compounds. I. Photochemistry of chiral β-oxy-α-diazo methyl ketones. Stereoselective synthesis of chiral macrolide synthons. Tetrahedron Lett. 1993, 34, 3467–3470. [Google Scholar] [CrossRef]
- López-Herrera, F.J.; Sarabia-García, F. β-Oxy-α-diazo carbonyl compounds. II. conversion to chiral α-oxy-α′-diazo ketones and photochemical reaction. Tetrahedron Lett. 1994, 35, 2929–2935. [Google Scholar] [CrossRef]
- Jiang, N.; Qu, Z.; Wang, J. 1,2-Aryl and 1,2-hydride migration in transition metal complex catalyzed diazo decomposition: a novel approach to α-aryl-β-enamino esters. Org. Lett. 2001, 3, 2989–2992. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Ma, Z.; Qu, Z.; Xing, X.; Xie, L.; Wang, J. Investigation of the transition-metal- and acid-catalyzed reactions of β-(N-tosyl)amino diazo carbonyl compounds. J. Org. Chem. 2003, 68, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Liu, Y.; Wang, J. DBU-catalyzed condensation of acyldiazomethanes to aldehydes in water and a new approach to ethyl β-hydroxy α-arylacrylates. Tetrahedron Lett. 2007, 48, 1147–1149. [Google Scholar] [CrossRef]
- Xu, F.; Shi, W.; Wang, J. 1,2-Thio group migration in Rh(II) carbene reactions. J. Org. Chem. 2005, 70, 4191–4194. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Zhang, Z.; Zhang, J.; Wang, J. Reaction of β-trimethylsiloxy α-diazocarbonyl compounds with trimethylsilyl halides: A novel diazo decomposition process. Tetrahedron Lett. 2005, 46, 8873–8875. [Google Scholar] [CrossRef]
- Miyauchi, K.; Hori, K.; Hirai, T.; Takebayashi, M.; Ibata, T. The acid-catalyzed decomposition of α-diazo β-hydroxy ketones. Bull. Chem. Soc. Jpn. 1981, 54, 2142–2146. [Google Scholar] [CrossRef]
- Cleary, S.E.; Hensinger, M.J.; Brewer, M. Remote C–H insertion of vinyl cations leading to cyclopentenones. Chem. Sci. 2017, 8, 6810–6814. [Google Scholar] [CrossRef] [Green Version]
- Pellicciari, R.; Natalini, B. Ring expansion of thiochroman-4-one and isothiochroman-4-one with ethyl diazo(lithio)acetate to tetrahydrobenzothiepin β-oxoesters. J. Chem. Soc. Perkin Trans. 1977, 1822–1824. [Google Scholar] [CrossRef]
- Pellicciari, R.; Fringuelli, R.; Ceccherelli, P.; Sisani, E.J. β-Keto esters from the rhodium(II) acetate catalysed conversion of α-diazo-β-hydroxy esters. J. Chem. Soc. Chem. Commun. 1979, 21, 959–960. [Google Scholar] [CrossRef]
- Pellicciari, R.; Sisani, E.; Fringuelli, R. A new synthesis of β-damascone. Tetrahedron Lett. 1980, 21, 4039–4042. [Google Scholar] [CrossRef]
- Pellicciari, R.; Natalini, B.; Cecchetti, S.; Fringuelli, R. Reduction of α-diazo-β-hydroxy esters to β-hydroxy esters: Application in one of two convergent syntheses of a (22S)-22-hydroxy bile acid from fish bile and its (22R)-epimer. J. Chem. Soc. Perkin Trans. 1985, 3, 493–497. [Google Scholar] [CrossRef]
- Pellicciari, R.; Natalini, B.; Fringnelli, R. An efficient procedure for the regiospecific preparation of d-homo-steroid derivatives. Steroids 1987, 49, 433–441. [Google Scholar] [CrossRef]
- Pellicciari, R.; Natalini, B.; Sadeghpour, B.M.; Rosato, G.C.; Ursini, A. The reaction of α-diazo-β-hydroxy esters with boron trifluoride. J. Chem. Soc. Chem. Commun. 1993, 24, 1798–1800. [Google Scholar] [CrossRef]
- Pellicciari, R.; Natalini, B.; Sadeghpour, B.M.; Marinozzi, M.; Snyder, J.P.; Williamson, B.L.; Kuethe, J.T.; Padwa, A. The reaction of α-diazo-β-hydroxy esters with boron trifluoride etherate: Generation and rearrangement of destabilized vinyl cations. A detailed experimental and theoretical study. J. Am. Chem. Soc. 1996, 118, 1–12. [Google Scholar] [CrossRef]
- Gioiello, A.; Venturoni, F.; Natalini, B.; Pellicciari, R. BF3·Et2O-Induced decomposition of ethyl 2-diazo-3-hydroxy-3,3-diarylpropanoates in acetonitrile: A novel approach to 2,3-diaryl β-enamino ester derivatives. J. Org. Chem. 2009, 74, 3520–3523. [Google Scholar] [CrossRef]
- Gioiello, A.; Venturoni, F.; Marinozzi, M.; Natalini, B.; Pellicciari, R. Exploring the synthetic versatility of the Lewis acid induced decomposition reaction of α-diazo-β-hydroxy esters. The case of ethyl diazo(3-hydroxy-2-oxo-2,3-dihydro-1H-indol-3-yl)acetate. J. Org. Chem. 2011, 76, 7431–7437. [Google Scholar] [CrossRef]
- Marinozzi, M.; Pertusati, F.; Serpi, M. λ5-Phosphorus-containing α-diazo compounds: A valuable tool for accessing phosphorus-functionalized molecules. Chem. Rev. 2016, 116, 13991–14055. [Google Scholar] [CrossRef]
- Arndt, F.; Amende, J. Synthesen mit diazo-methan, V.: Über die reaktion der aäurechloride mit diazo-methan. J. Ber. Dtsch. Chem. Ges. 1928, 61, 1122–1124. [Google Scholar] [CrossRef]
- Diazomethane should be treated with extreme caution. It is very toxic, and the pure gas and liquid explode readily even from contact with sharp edges of glass. For more safety information see Black, T.H. Aldrichimica 1983, 16, 1–22.
- Schollkopf, U.; Brinhidai, B.; Frasnelli, H.; Meyer, R.; Beckhaus, H. Metallsubstituierte carbene und C-metallierte diazoalkane, VI1)α-diazo-β-hydroxy-carbonsäureester und -ketone aus carbonyl- und diazolithioverbindungen sowie ihre umlagerung zu β-ketocarbonsäureestern und β-diketonen. Liebigs Ann. Chem. 1974, 1767–1783. [Google Scholar] [CrossRef]
- Jones, R.A.; Stokes, M.J. Phase-transfer catalysis-3: Acetylation of 2-acylcycloalkanones. Tetrahedron 1984, 40, 1051–1060. [Google Scholar] [CrossRef]
- Miyahara, Y.; Ito, Y.N. AlCl3-mediated aldol cyclocondensation of 1,6- and 1,7-diones to cyclopentene and cyclohexene derivatives. J. Org. Chem. 2014, 79, 6801–6807. [Google Scholar] [CrossRef] [PubMed]
- Ostercamp, D.L. Vinylogous imides. II. Ultraviolet spectra and the application of Woodward’s rules. J. Org. Chem. 1970, 35, 1632–1641. [Google Scholar] [CrossRef]
- Cai, Y.; Jalan, A.; Kubosumi, A.R.; Castle, S.L. Microwave-promoted tin-free iminyl radical cyclization with TEMPO trapping: A practical synthesis of 2-acylpyrroles. Org. Lett. 2015, 17, 488–491. [Google Scholar] [CrossRef]
- Hettrick, C.M.; Kling, J.K.; Scott, W.J. Palladium-catalyzed cross-coupling of β-(methanesulfonyl)oxyenones with organostannanes. J. Org. Chem. 1991, 56, 1489–1492. [Google Scholar] [CrossRef]
- Normant, J.F.; Foulon, J.P.; Masure, D.; Sauvetre, R.; Villieras, J. A new preparation of trifluorovinyllithium. Applications to the syntheses of trifluorovinylalcohols and of α-fluoro-α-ethylenic acids and derivatives. Synthesis 1975, 2, 122–125. [Google Scholar] [CrossRef]
- Colonge, J.; Bonnard, L. Synthesis of anthracene hydrocarbons. Bull. Soc. Chim. Fr. 1958, 742–746. [Google Scholar]
- Sessler, J.L.; Roznyatovskiy, V.V.; Lynch, V.M. Novel β-substituted calix[4]pyrroles. J. Porphyr. Phthalocyanines 2009, 13, 322–325. [Google Scholar] [CrossRef]
- Benniston, A.C. Photo- and redox-active [2]rotaxanes and [2]catenanes. Chem. Soc. Rev. 1996, 25, 427–435. [Google Scholar] [CrossRef]
- Lopez, F.J.; Jett, M.-F.; Muchowski, J.M.; Nitzan, D.; O’Yang, C. Synthesis and biological evaluation of ketorolac analogs. Heterocycles 2002, 56, 91–95. [Google Scholar] [CrossRef]
- Kinnel, R.B.; Gehrken, H.P.; Scheuer, P.J. Palau’amine: A cytotoxic and immunosuppressive hexacyclic bisguanidine antibiotic from the sponge Stylotella agminata. J. Am. Chem. Soc. 1993, 115, 3376–3377. [Google Scholar] [CrossRef]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Venturoni, F.; Cerra, B.; Marinozzi, M.; Camaioni, E.; Gioiello, A.; Pellicciari, R. BF3·Et2O-Promoted Decomposition of Cyclic α-Diazo-β-Hydroxy Ketones: Novel Insights into Mechanistic Aspects. Catalysts 2018, 8, 600. https://doi.org/10.3390/catal8120600
Venturoni F, Cerra B, Marinozzi M, Camaioni E, Gioiello A, Pellicciari R. BF3·Et2O-Promoted Decomposition of Cyclic α-Diazo-β-Hydroxy Ketones: Novel Insights into Mechanistic Aspects. Catalysts. 2018; 8(12):600. https://doi.org/10.3390/catal8120600
Chicago/Turabian StyleVenturoni, Francesco, Bruno Cerra, Maura Marinozzi, Emidio Camaioni, Antimo Gioiello, and Roberto Pellicciari. 2018. "BF3·Et2O-Promoted Decomposition of Cyclic α-Diazo-β-Hydroxy Ketones: Novel Insights into Mechanistic Aspects" Catalysts 8, no. 12: 600. https://doi.org/10.3390/catal8120600
APA StyleVenturoni, F., Cerra, B., Marinozzi, M., Camaioni, E., Gioiello, A., & Pellicciari, R. (2018). BF3·Et2O-Promoted Decomposition of Cyclic α-Diazo-β-Hydroxy Ketones: Novel Insights into Mechanistic Aspects. Catalysts, 8(12), 600. https://doi.org/10.3390/catal8120600