Non-Classical Anionic Naked N-Heterocyclic Carbenes: Fundamental Properties and Emerging Applications in Synthesis and Catalysis


Abstract
:
1. Introduction
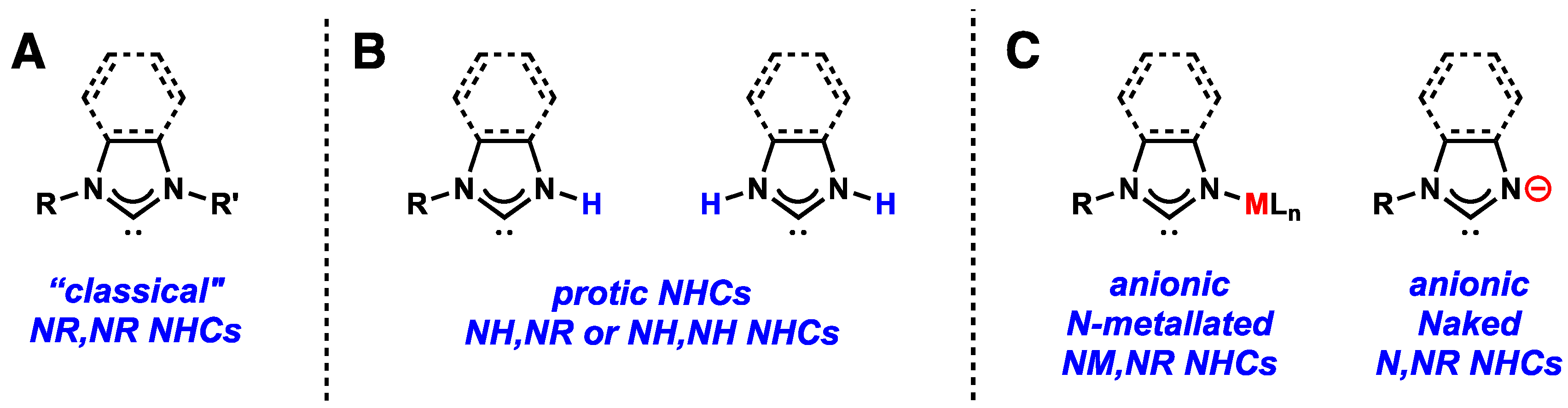
2. Non-Classical NHCs and Their Metal Complexes
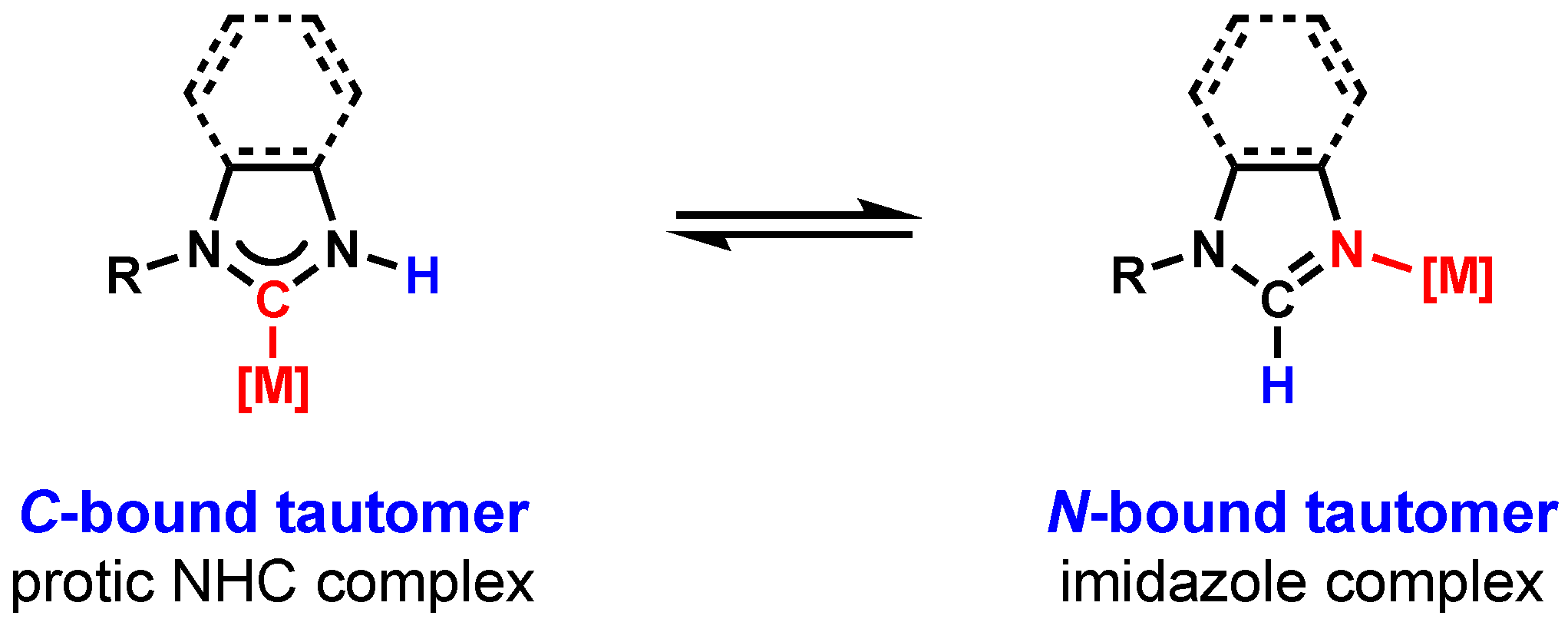
2.1. Protic NHC Metal Complexes
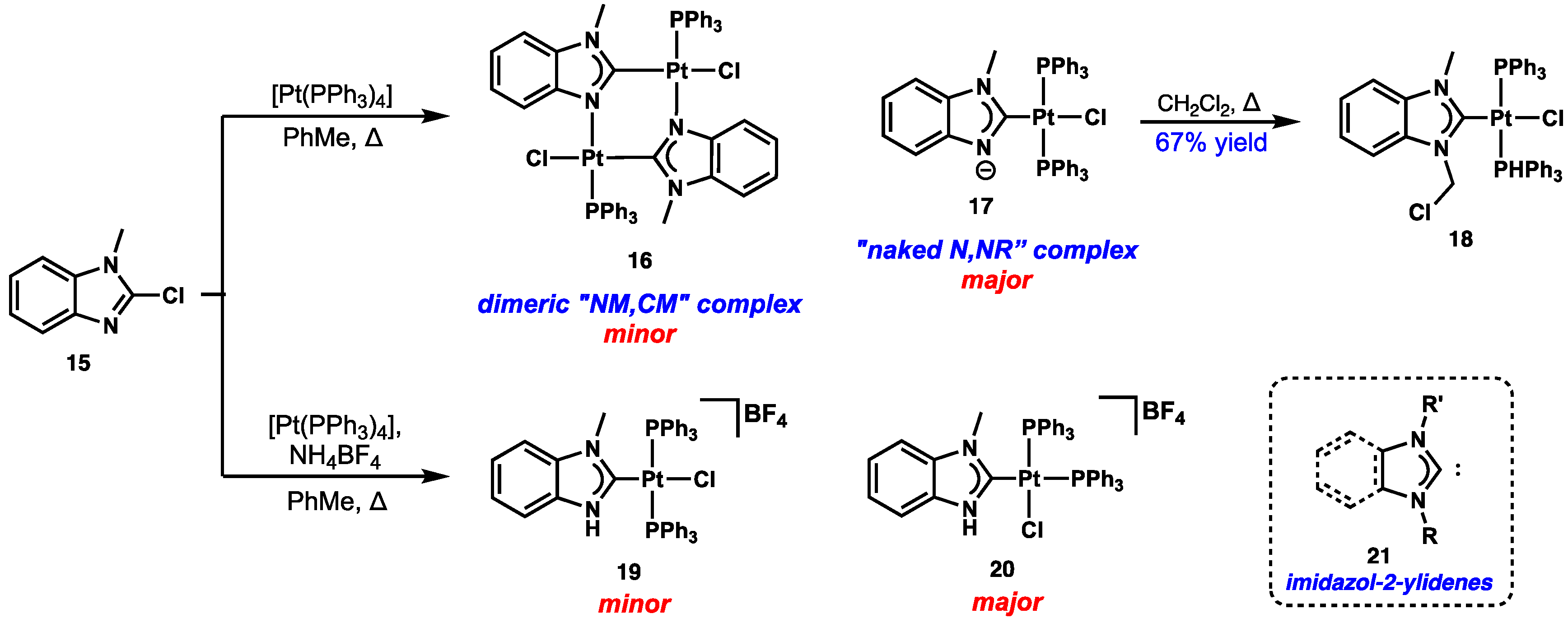
2.2. Anionic NM,NR and Naked N,NR NHC Metal Complexes
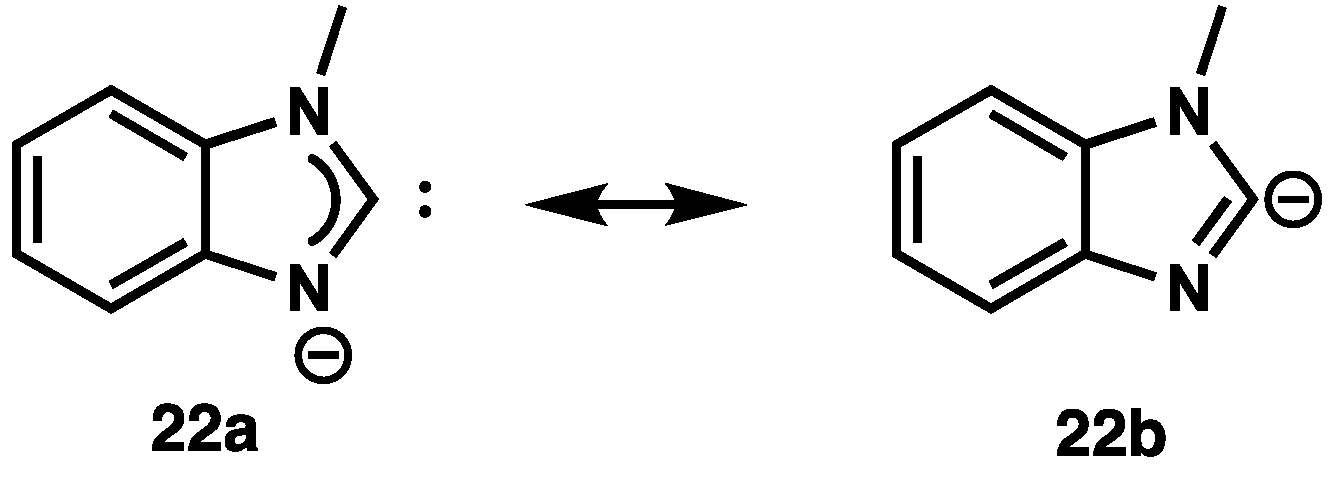
2.3. Carbene or Carbanion?
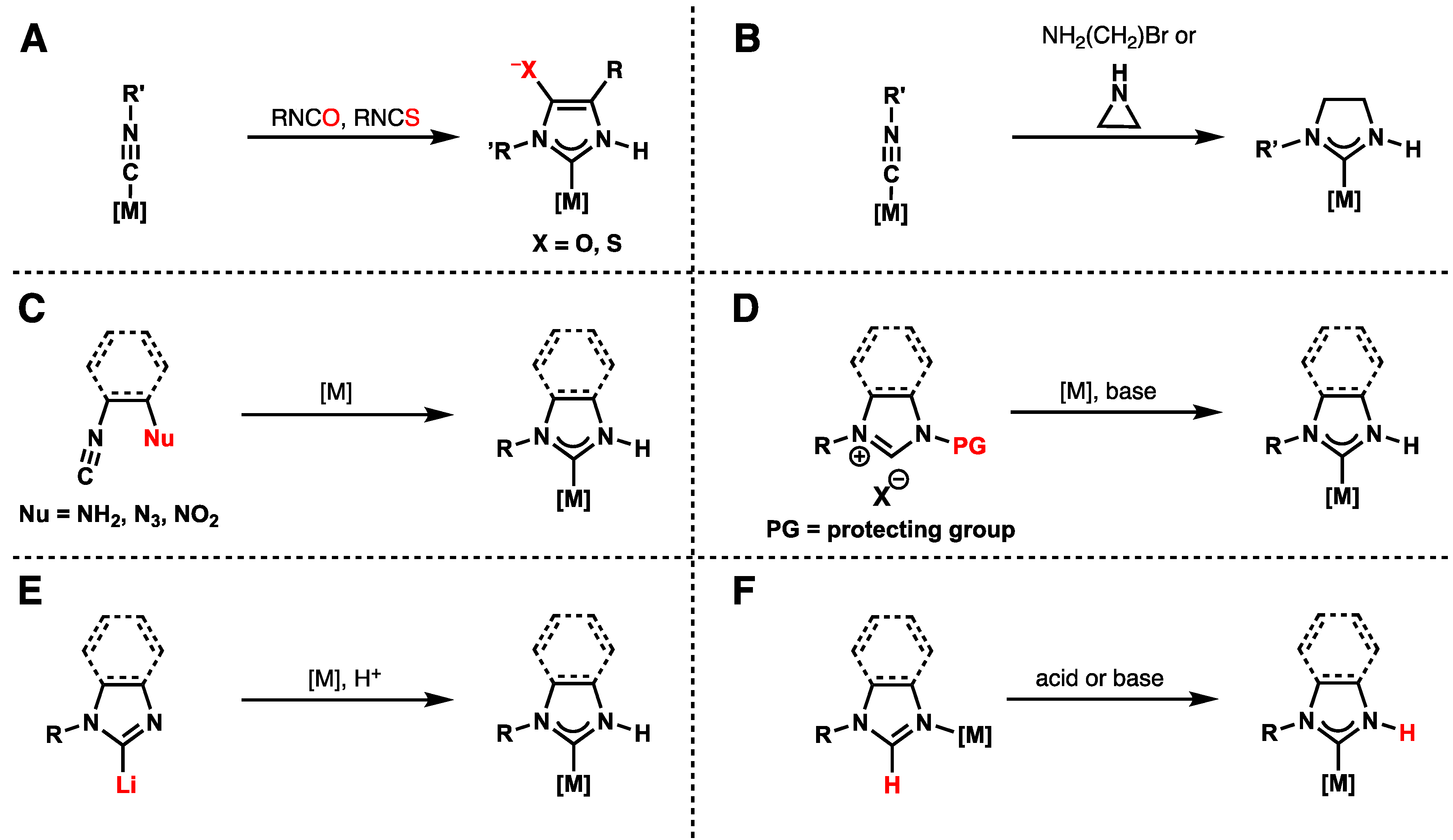
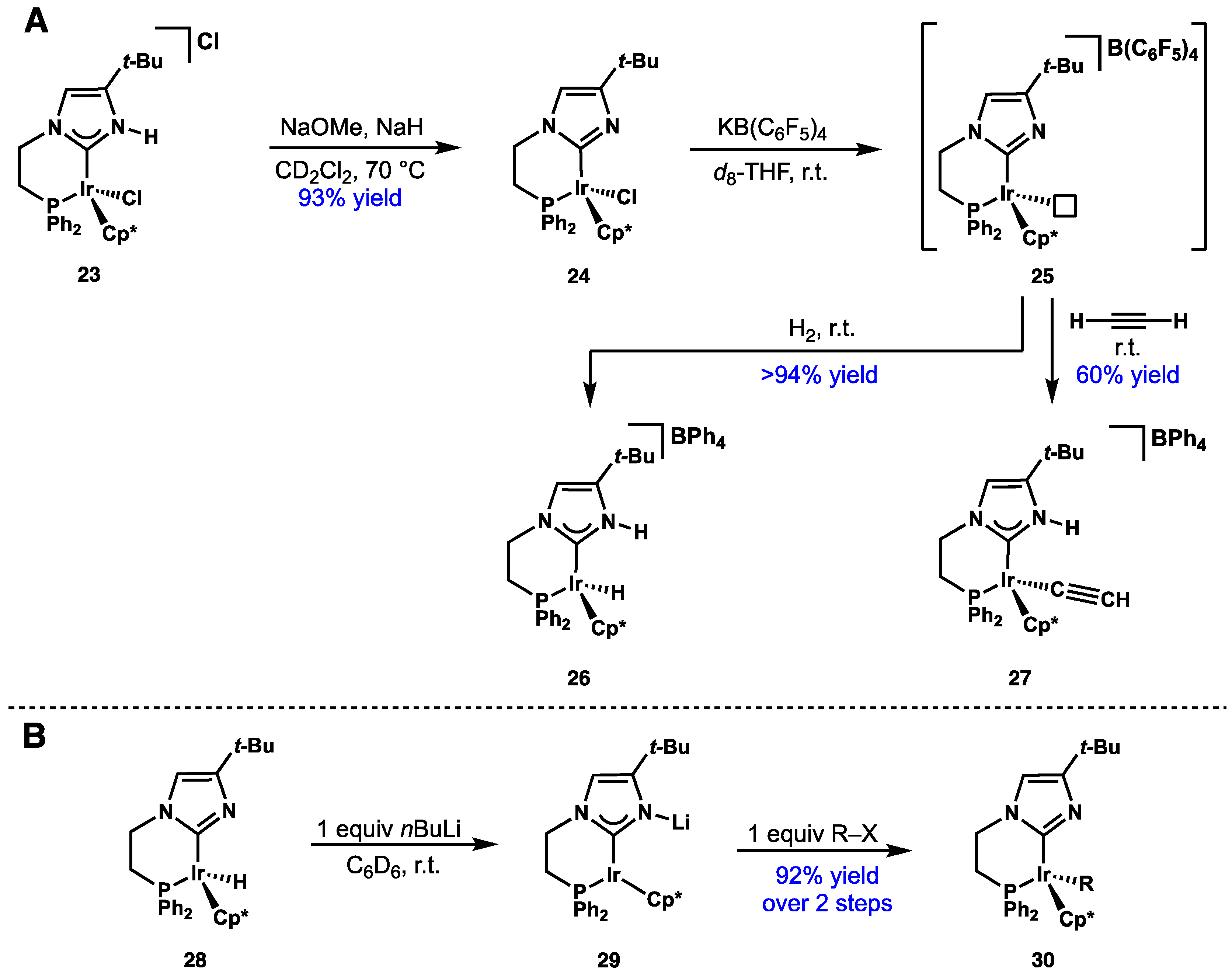
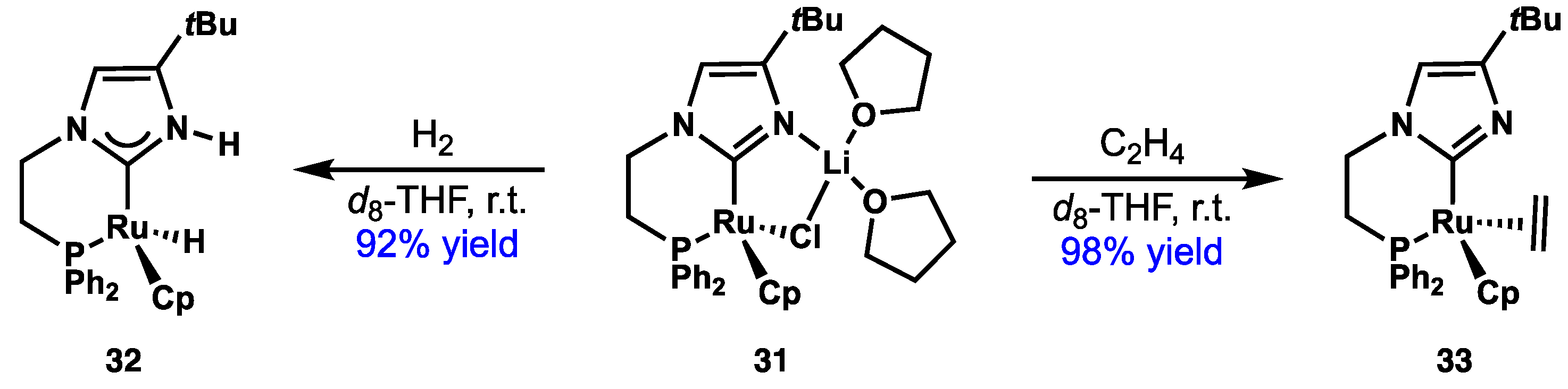
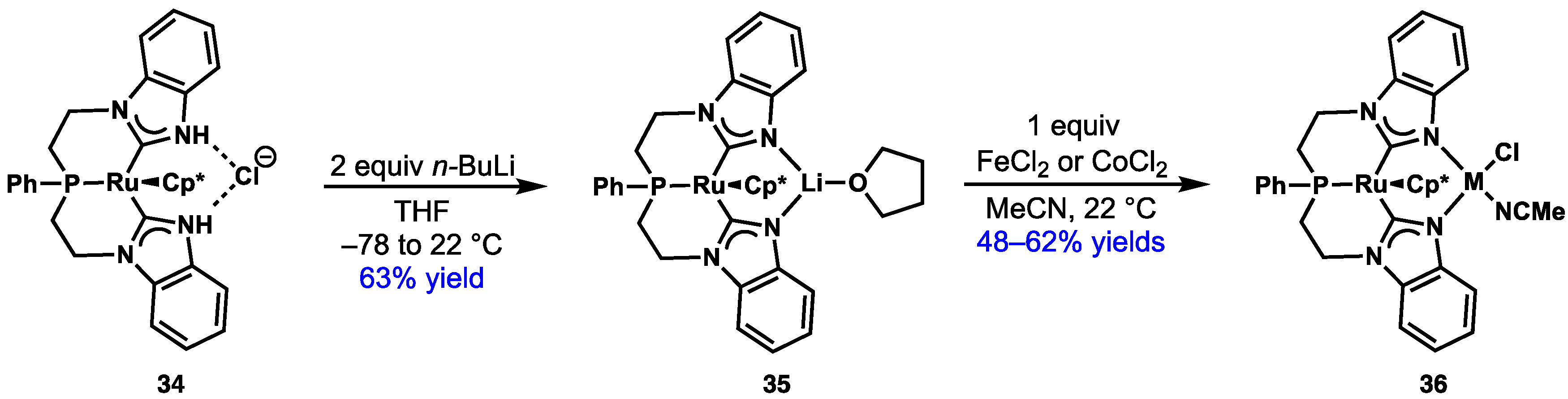
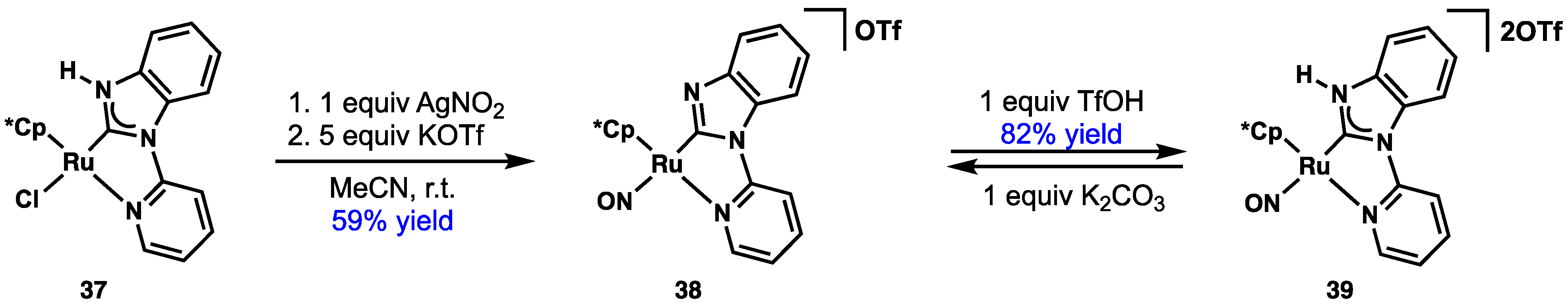
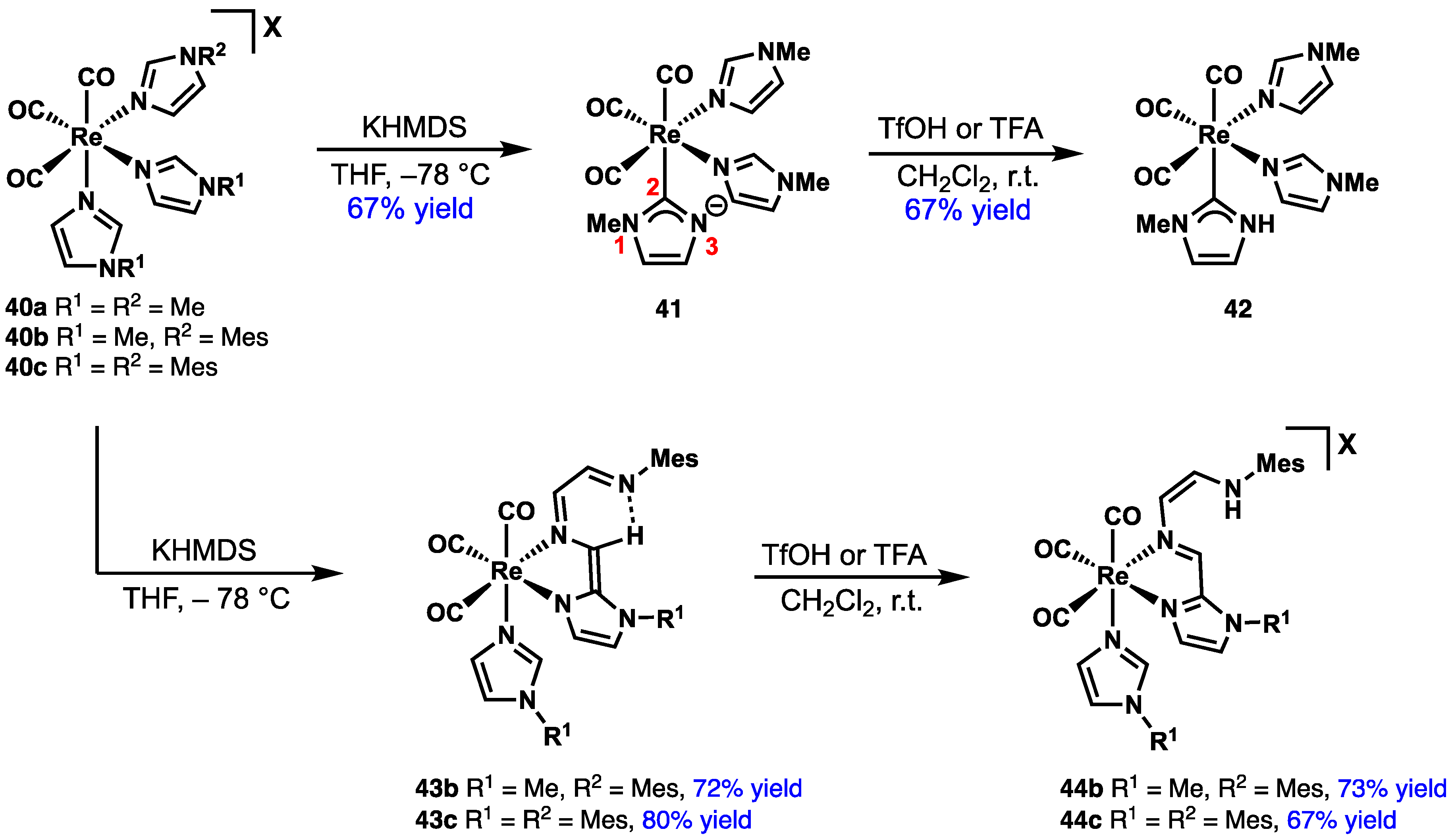
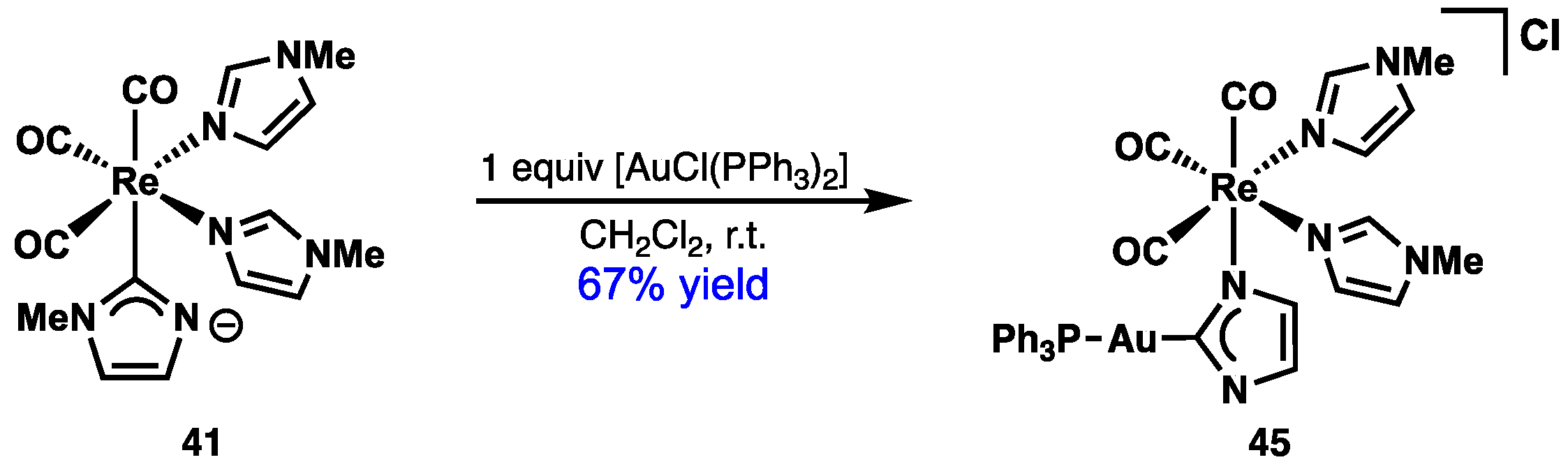
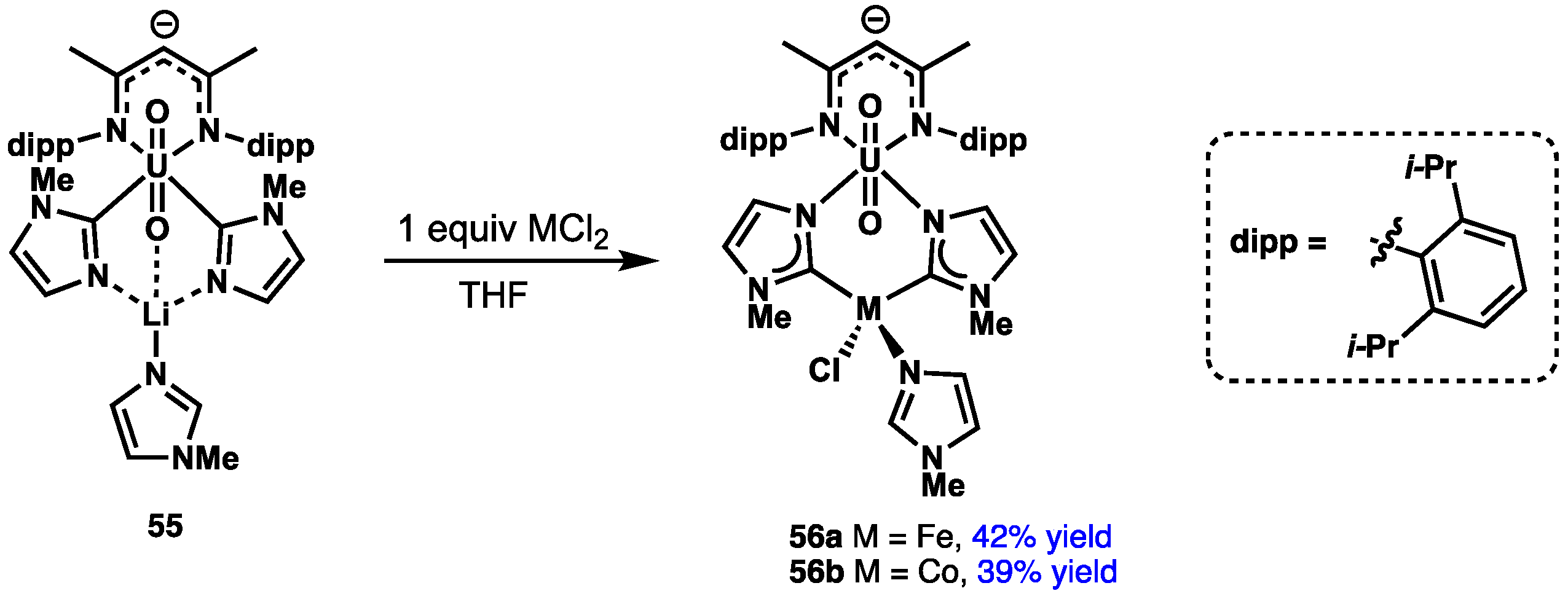
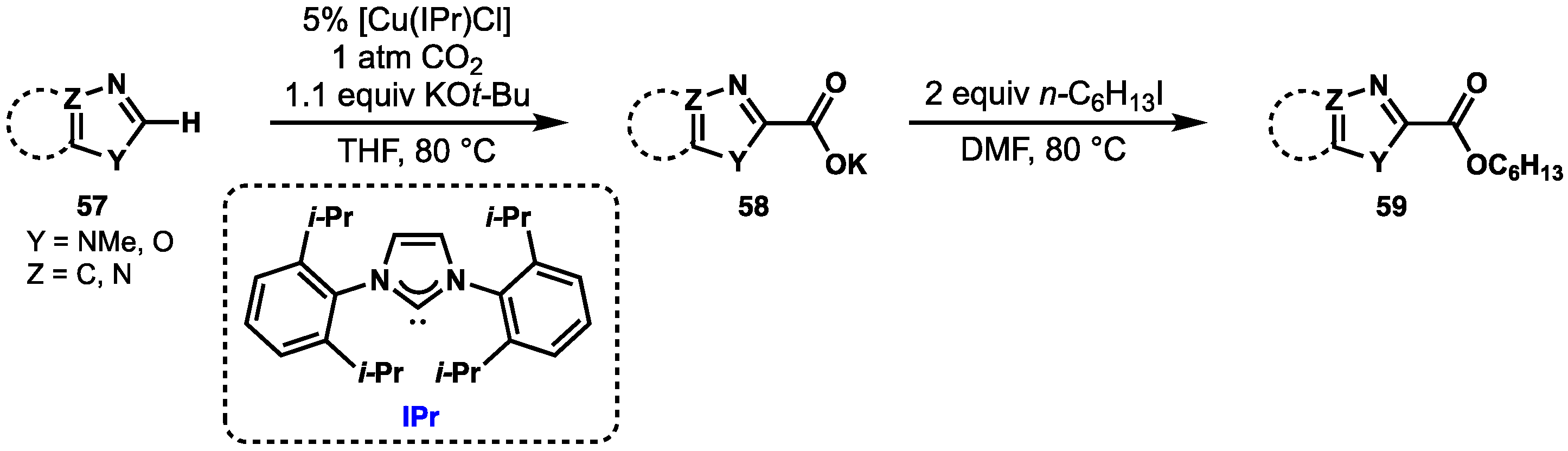
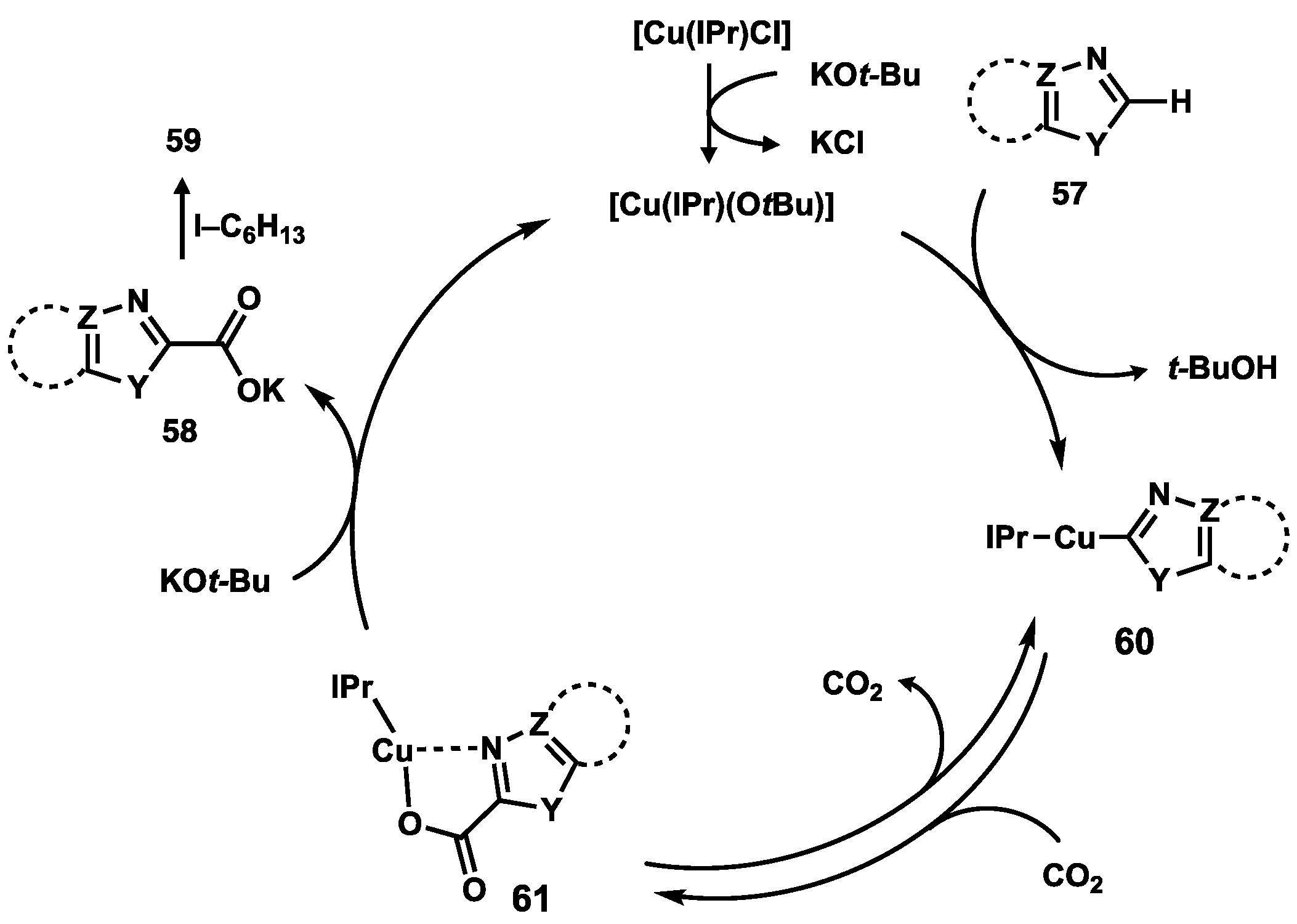
2.4. Preparation of N(M),NR Complexes and Applications to Synthesis and Catalysis
3. Summary and Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Igau, A.; Grutzmacher, H.; Baceiredo, A.; Bertrand, G. Analogous α,α′-Bis-Carbenoid Triply Bonded Species: Synthesis of a Stable λ3-Phosphinocarbene-λ5-Phosphaacetylene. J. Am. Chem. Soc. 1988, 110, 6463–6466. [Google Scholar] [CrossRef]
- Arduengo, A.J., III; Harlow, R.L.; Kline, M. A Stable Crystalline Carbene. J. Am. Chem. Soc. 1991, 113, 361–363. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Elison, M.; Fischer, J.; Kocher, C.; Artus, G.R.J. Metal Complexes of N-Heterocyclic Carbenes—A New Structural Principle for Catalysts in Homogeneous Catalysis. Angew. Chem. Int. Ed. 1995, 34, 2371–2374. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Kocher, C. N-Heterocyclic Carbenes. Angew. Chem. Int. Ed. 1997, 2162–2187. [Google Scholar] [CrossRef]
- Herrmann, W.A. N-Heterocyclic Carbenes: A New Concept in Organometallic Catalysis. Angew. Chem. Int. Ed. 2002, 41, 1290–1309. [Google Scholar] [CrossRef]
- Crabtree, R.H. Carbenes in Synthesis and Homogeneous Catalysis. Oil Gas Sci. Technol. 2007, 62, 739–744. [Google Scholar] [CrossRef] [Green Version]
- Hahn, F.E.; Jahnke, M.C. Heterocyclic Carbenes: Synthesis and Coordination Chemistry. Angew. Chem. Int. Ed. 2008, 47, 3122–3172. [Google Scholar] [CrossRef] [PubMed]
- Öfele, K.; Tosh, E.; Taubmann, C.; Herrmann, W.A. Carbocyclic Carbene Metal Complexes. Chem. Rev. 2009, 109, 3408–3444. [Google Scholar] [CrossRef] [PubMed]
- Díez-González, S.; Marion, N.; Nolan, S.P. N-Heterocyclic Carbenes in Late Transition Metal Catalysis. Chem. Rev. 2009, 109, 3612–3676. [Google Scholar] [CrossRef] [PubMed]
- Bugaut, X.; Glorius, F. Organocatalytic umpolung: N-heterocyclic carbenes and beyond. Chem. Soc. Rev. 2012, 41, 3511–3522. [Google Scholar] [CrossRef]
- Flanigan, D.M.; Romanov-Michailidis, F.; White, N.A.; Rovis, T. Organocatalytic Reactions Enabled by N-Heterocyclic Carbenes. Chem. Rev. 2015, 115, 9307–9387. [Google Scholar] [CrossRef] [PubMed]
- Schuster, O.; Yang, L.; Raubenheimer, H.G.; Albrecht, M. Beyond Conventional N-Heterocyclic Carbenes: Abnormal, Remote, and Other Classes of NHC Ligands with Reduced Heteroatom Stabilization. Chem. Rev. 2009, 109, 3445–3478. [Google Scholar] [CrossRef] [PubMed]
- Arduengo, A.J., III; Krafczyk, R.; Schmutzler, R.; Craig, H.A.; Goerlich, J.R.; Marshall, W.J.; Unverzagt, M. Imidazolylidenes, imidazolinylidenes and imidazolidines. Tetrahedron 1999, 55, 14523–14534. [Google Scholar] [CrossRef]
- Davies, C.J.E.; Page, M.J.; Ellul, C.E.; Mahon, M.F.; Whittlesey, M.K. Ni(I) and Ni(II) ring-expanded N-heterocyclic carbene complexes: C–H activation, indole elimination and catalytic hydrodehalogenation. Chem. Commun. 2010, 46, 5151–5153. [Google Scholar] [CrossRef] [PubMed]
- Hauwert, P.; Dunsford, J.J.; Tromp, D.S.; Weigand, J.J.; Lutz, M.; Cavell, K.J.; Elsevier, C.J. Zerovalent [Pd(NHC)(Alkene)1,2] Complexes Bearing Expanded-Ring N-Heterocyclic Carbene Ligands in Transfer Hydrogenation of Alkynes. Organometallics 2013, 32, 131–140. [Google Scholar] [CrossRef]
- Dunsford, J.J.; Cavell, K.J. Pd–PEPPSI-Type Expanded Ring N-Heterocyclic Carbene Complexes: Synthesis, Characterization, and Catalytic Activity in Suzuki–Miyaura Cross Coupling. Organometallics 2014, 33, 2902–2905. [Google Scholar] [CrossRef]
- Türkmen, H.; Pape, T.; Hahn, F.E.; Çetinkaya, B. Annulated N-Heterocyclic Carbene Ligands Derived from 2-Methylaminopiperidine: Their Complexes and Catalytic Applications. Organometallics 2008, 27, 571–575. [Google Scholar] [CrossRef]
- Martin, D.; Lassauque, N.; Donnadieu, B.; Bertrand, G. A Cyclic Diaminocarbene with Pyramidalized Nitrogen Atom: A Stable N-Heterocyclic Carbene with Enhanced Nucleophilicity. Angew. Chem. Int. Ed. 2012, 51, 6172–6175. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.-M.; Xiang, K.; Tu, Y.-Q.; Zhang, S.-H.; Yang, D.-T.; Wang, S.-H.; Zhang, F.-M. Spiro-fused six-membered N-heterocyclic carbene: A new scaffold toward unique properties and activities. Chem. Commun. 2014, 50, 7163–7165. [Google Scholar] [CrossRef] [PubMed]
- Rao, B.; Tang, H.; Zeng, X.; Liu, L.; Melaimi, M.; Bertrand, G. Cyclic (Amino)(aryl)carbenes (CAArCs) as Strong σ-Donating and π-Accepting Ligands for Transition Metals. Angew. Chem. Int. Ed. 2015, 54, 14915–14919. [Google Scholar] [CrossRef] [PubMed]
- Peris, E.; Crabtree, R.H. Recent homogeneous catalytic applications of chelate and pincer N-heterocyclic carbenes. Coord. Chem. Rev. 2004, 248, 2239–2246. [Google Scholar] [CrossRef]
- Vignolle, J.; Cattoën, X.; Bourissou, D. Stable Noncyclic Singlet Carbenes. Chem. Rev. 2009, 109, 3333–3384. [Google Scholar] [CrossRef] [PubMed]
- Mata, J.A.; Poyatos, M.; Peris, E. Structural and catalytic properties of chelating bis- and tris-N-heterocyclic carbenes. Coord. Chem. Rev. 2007, 251, 841–859. [Google Scholar] [CrossRef]
- Pugh, D.; Danopoulos, A.A. Metal complexes with ‘pincer’-type ligands incorporating N-heterocyclic carbene functionalities. Coord. Chem. Rev. 2007, 251, 610–641. [Google Scholar] [CrossRef]
- Poyatos, M.; Mata, J.A.; Peris, E. Complexes with Poly(N-heterocyclic carbene) Ligands: Structural Features and Catalytic Applications. Chem. Rev. 2009, 109, 3677–3707. [Google Scholar] [CrossRef] [PubMed]
- Hopkinson, M.N.; Ritcher, C.; Schedler, M.; Glorius, F. An overview of N-heterocylic carbenes. Nature 2014, 510, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zi, G. N-heterocyclic carbene (NHC) complexes of group 4 transition metals. Chem. Soc. Rev. 2015, 44, 1898–1921. [Google Scholar] [CrossRef]
- Peris, E. Smart N-Heterocyclic Carbene Ligands in Catalysis. Chem. Rev. 2018, 118, 9988–10031. [Google Scholar] [CrossRef]
- Charra, V.; de Fremont, P.; Braunstein, P. Multidentate N-heterocyclic carbene complexes of the 3d metals: Synthesis, structure, reactivity and catalysis. Coord. Chem. Rev. 2017, 341, 53–176. [Google Scholar] [CrossRef]
- Gardiner, M.G.; Ho, C.C. Recent advances in bidentate bis(N-heterocyclic carbene) transition metal complexes and their applications in metal-mediated reactions. Coord. Chem. Rev. 2018, 375, 373–388. [Google Scholar] [CrossRef]
- Kuwata, S.; Hahn, F.E. Complexes Bearing Protic N-Heterocyclic Carbene Ligands. Chem. Rev. 2018, 118, 9642–9677. [Google Scholar] [CrossRef]
- Liu, M.; Namyslo, J.C.; Nieger, M.; Polamo, M.; Schmidt, A. From betaines to anionic N-heterocyclic carbenes. Borane, gold, rhodium, and nickel complexes starting from an imidazoliumphenolate and its carbene tautomer. Beilstein J. Org. Chem. 2016, 12, 2673–2681. [Google Scholar] [CrossRef] [PubMed]
- César, V.; Lugan, N.; Lavigne, G. A Stable Anionic N-Heterocyclic Carbene and Its Zwitterionic Complexes. J. Am. Chem. Soc. 2008, 130, 11286–11287. [Google Scholar] [CrossRef] [PubMed]
- Danopoulos, A.A.; Monakhov, K.Y.; Braunstein, P. Anionic N-Heterocyclic Carbene Ligands from Mesoionic Imidazolium Precursors: Remote Backbone Arylimino Substitution Directs Carbene Coordination. Chem. Eur. J. 2013, 19, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Liddle, S.T.; Edworthy, I.S.; Arnold, P.L. Anionic tethered N-heterocyclic carbene chemistry. Chem. Soc. Rev. 2007, 36, 1732–1744. [Google Scholar] [CrossRef] [PubMed]
- Benhamou, L.; César, V.; Gornitzka, H.; Lugan, N.; Lavigne, G. Imidazol-2-ylidene-4-olate: An anionic N-heterocyclic carbene pre-programmed for further derivatization. Chem. Commun. 2009, 4720–4722. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, H.; Li, X. Ir(III)-induced C-bound to N-bound tautomerization of a N-heterocyclic carbene. Organometallics 2007, 26, 4684–4687. [Google Scholar] [CrossRef]
- Heinemann, C.; Thiel, W. Ab initio study on the stability of diaminocarbenes. Chem. Phys. Lett. 1994, 217, 11–16. [Google Scholar] [CrossRef]
- McGibbon, G.A.; Heinemann, C.; Lavorato, D.J.; Schwarz, H. Imidazol-2-ylidene: Generation of a Missing Carbene and Its Dication by Neutralization- Reionization and Charge-Stripping Mass Spectrometry. Angew. Chem. Int. Ed. 1997, 36, 1478–1481. [Google Scholar] [CrossRef]
- Häller, L.J.L.; Macgregor, S.A. Computational Study of the C- and N-Bound Tautomers of [Ru(Cl)(H)(CO)-(PPh3)2 (IiPrMe2)] (IiPrMe2 = 3-Isopropyl-4,5-dimethylimidazol-2-ylidene). Eur. J. Inorg. Chem. 2009, 2009, 2000–2006. [Google Scholar] [CrossRef]
- Sini, G.; Eisenstein, O.; Crabtree, R.H. Preferential C-binding versus N-binding in imidazole depends on the metal fragment involved. Inorg. Chem. 2002, 41, 602–604. [Google Scholar] [CrossRef] [PubMed]
- Fehlhammer, W.P.; Völkl, A.; Plaia, U.; Beck, G. Metallkomplexe funktioneller Isocyanide, XVI1) 1,3-Dipolare Cycloadditionen von Heteroallenen an die metallorganischen Nitrilylide [(OC)5M–C≡N–CHR]− (M = Cr, W; R = CO2Et). Chem. Ber. 1987, 120, 2031–2040. [Google Scholar] [CrossRef]
- Michelin, R.A.; Zanotto, L.; Braga, D.; Sabatino, P.; Angelici, R.J. Transition-Metal-Promoted Cyclization Reactions of Isocyanide Ligands. Synthesis of Cyclic Diaminocarbenes from Isocyanide Complexes of Palladium(II) and Platinum(II) and X-ray Structure of cis-Br2Pt[CN(C6H4-p-Me)CH2CH2N(H)](PPh3). Inorg. Chem. 1988, 27, 93–99. [Google Scholar] [CrossRef]
- Michelin, R.A.; Mozzon, M.; Zecca, M.; Corain, B.; Piazzi, O.; Zanotti, G. Palladium(II) cyclic carbene complexes from 3-isocyanopropylacrylate. X-ray structure of cis-{PdCl2(PPh3)[C–N(H)–(CH2)3–O–C(O)–CH–CH2]}. Inorg. Chim. Acta 1990, 174, 3–7. [Google Scholar] [CrossRef]
- Hahn, F.E.; Langenhahn, V.; Pape, T. Template synthesis of tungsten complexes with saturated N-heterocyclic carbene ligands. Chem. Commun. 2005, 5, 5390–5392. [Google Scholar] [CrossRef] [PubMed]
- Kaufhold, O.; Flores-Figueroa, A.; Pape, T.; Hahn, F.E. Template Synthesis of Ruthenium Complexes with Saturated and Benzannulated NH,NH-Stabilized N-Heterocyclic Carbene Ligands. Organometallics 2009, 28, 896–901. [Google Scholar] [CrossRef]
- Hahn, F.E.; Langenhahn, V.; Meier, N.; Lügger, T.; Fehlhammer, W.P. Template synthesis of benzannulated N-heterocyclic carbene ligands. Chem. Eur. J. 2003, 9, 704–712. [Google Scholar] [CrossRef]
- Liu, C.Y.; Chen, D.Y.; Lee, G.H.; Peng, S.M.; Liu, S.T. Synthesis of cyclic diamino-substituted metal carbene complexes. Organometallics 1996, 15, 1055–1061. [Google Scholar] [CrossRef]
- Brendler, E.; Hill, A.F.; Wagler, J. A donor-stabilized silanethione or a Si-substituted N-heterocyclic platinum carbene? Chem. A Eur. J. 2008, 14, 11300–11304. [Google Scholar] [CrossRef]
- Dobereiner, G.E.; Chamberlin, C.A.; Schley, N.D.; Crabtree, R.H. Acyl protection strategy for synthesis of a protic NHC complex via N-acyl methanolysis. Organometallics 2010, 29, 5728–5731. [Google Scholar] [CrossRef]
- Fehlhammer, W.P.; Bliß, T.; Fuchs, J.; Holzmann, G. Tetrakis (N-alkylimidazolin-2-yliden) palladium and -platin. Zeitschrift für Naturforsch. B 1992, 59, 79–89. [Google Scholar] [CrossRef]
- Meier, N.; Hahn, F.E.; Pape, T.; Siering, C.; Waldvogel, S.R. Molecular recognition utilizing complexes with NH,NR-stabilized carbene ligands. Eur. J. Inorg. Chem. 2007, 1210–1214. [Google Scholar] [CrossRef]
- Bonati, F.; Burini, A.; Pietroni, B.R.; Bovio, B. Reactions of C-imidazolyllithium derivatives with Group Ib compounds: Tris[μ-(1-alkylimidazolato-N3,C2)]tri-gold(I) and -silver(I). Crystal structure of bis(1-benzylimidazolin-2-yliden)gold(I) chloride. J. Organomet. Chem. 1989, 375, 147–160. [Google Scholar] [CrossRef]
- Raubenheimer, H.G.; Lindeque, L.; Cronje, S. Synthesis and characterization of neutral and cationic diamino carbene complexes of gold(I). J. Organomet. Chem. 1996, 511, 177–184. [Google Scholar] [CrossRef]
- Huertos, M.A.; Pérez, J.; Riera, L.; Menéndez-Velázquez, A. From N-alkylimidazole ligands at a rhenium center: Ring opening or formation of NHC complexes. J. Am. Chem. Soc. 2008, 130, 13530–13531. [Google Scholar] [CrossRef] [PubMed]
- Sundberg, R.J.; Bryan, R.F.; Taylor, I.F.; Taube, H. Nitrogen-Bound and Carbon-Bound Imidazole Complexes of Ruthenium Ammines. J. Am. Chem. Soc. 1974, 96, 381–392. [Google Scholar] [CrossRef]
- Hahn, F.E.; Naziruddin, A.R.; Hepp, A.; Pape, T. Synthesis, characterization, and H-bonding abilities of ruthenium(II) complexes bearing bidentate NR,NH-carbene/phosphine ligands. Organometallics 2010, 29, 5283–5288. [Google Scholar] [CrossRef]
- Eguillor, B.; Esteruelas, M.A.; García-Raboso, J.; Oliván, M.; Oñate, E.; Pastor, I.M.; Peñafiel, I.; Yus, M. Osmium NHC complexes from alcohol-functionalized imidazoles and imidazolium salts. Organometallics 2011, 30, 1658–1667. [Google Scholar] [CrossRef]
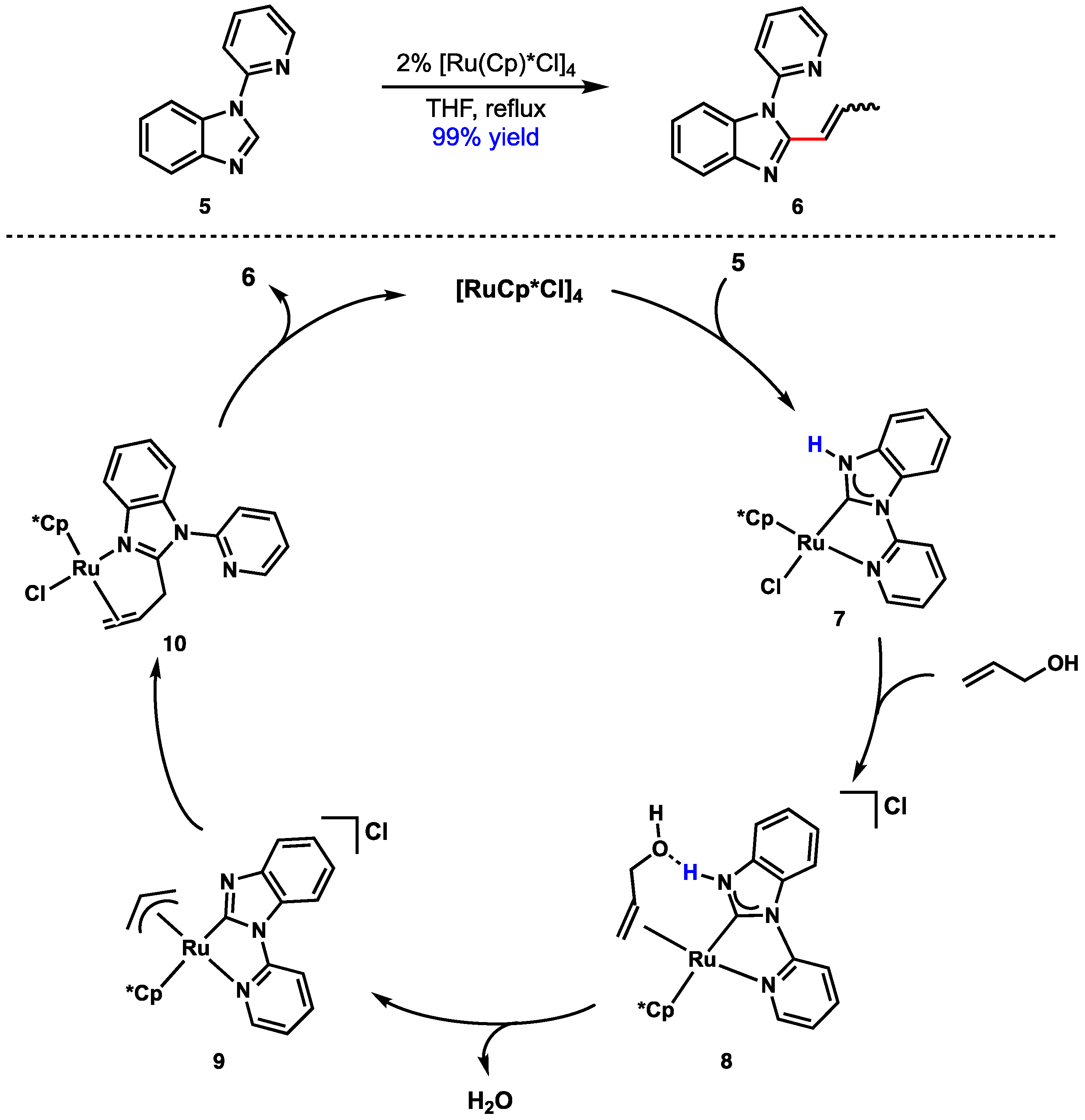
- Araki, K.; Kuwata, S.; Ikariya, T. Isolation and interconversion of protic N-heterocyclic carbene and imidazolyl complexes: Application to catalytic dehydrative condensation of N-(2-Pyridyl)benzimidazole and allyl alcohol. Organometallics 2008, 27, 2176–2178. [Google Scholar] [CrossRef]
- Miranda-Soto, V.; Grotjahn, D.B.; Cooksy, A.L.; Golen, J.A.; Moore, C.E.; Rheingold, A.L. A Labile and Catalytically Active Imidazol-2-yl Fragment System. Angew. Chem. Int. Ed. 2011, 50, 631–635. [Google Scholar] [CrossRef]
- Miranda-Soto, V.; Grotjahn, D.B.; DiPasquale, A.G.; Rheingold, A.L. Imidazol-2-yl Complexes of Cp*Ir as Bifunctional Ambident Reactants. J. Am. Chem. Soc. 2008, 130, 13200–13201. [Google Scholar] [CrossRef] [PubMed]
- Naziruddin, A.R.; Hepp, A.; Pape, T.; Hahn, F.E. Synthesis of Rhodium(I) Complexes Bearing Bidentate NH,NR-NHC/Phosphine Ligands. Organometallics 2011, 30, 5859–5866. [Google Scholar] [CrossRef]
- Flowers, S.E.; Cossairt, B.M. Mono- and Dimetalation of a Tridentate Bisimidazole-Phosphine Ligand. Organometallics 2014, 33, 4341–4344. [Google Scholar] [CrossRef]
- He, F.; Braunstein, P.; Wesolek, M.; Danopoulos, A.A. Imine-functionalised protic NHC complexes of Ir: Direct formation by C–H activation. Chem. Commun. 2015, 51, 2814–2817. [Google Scholar] [CrossRef] [PubMed]
- Cepa, S.; Schulte To Brinke, C.; Roelfes, F.; Hahn, F.E. Hydrogen Activation by an Iridium(III) Complex Bearing a Bidentate Protic NH,NR-NHC^Phosphine Ligand. Organometallics 2015, 34, 5454–5460. [Google Scholar] [CrossRef]
- Wiedemann, S.H.; Lewis, J.C.; Ellman, J.A.; Bergman, R.G. Experimental and Computational Studies on the Mechanism of N-Heterocycle C−H Activation by Rh(I). J. Am. Chem. Soc. 2006, 128, 2452–2462. [Google Scholar] [CrossRef]
- Hawkes, K.J.; Cavell, K.J.; Yates, B.F. Rhodium-Catalyzed C−C Coupling Reactions: Mechanistic Considerations. Organometallics 2008, 27, 4758–4771. [Google Scholar] [CrossRef]
- Lewis, J.C.; Wu, J.Y.; Bergman, R.G.; Ellman, J.A. Microwave-Promoted Rhodium-Catalyzed Arylation of Heterocycles through C−H Bond Activation. Angew. Chem. Int. Ed. 2006, 45, 1589–1591. [Google Scholar] [CrossRef]
- Johnson, L.K.; Angelici, R.J. Synthesis of Aminooxycarbene Complexes of Iron with N-Alkyl, -Allyl, and -Carbamoyl Groups. Inorg. Chem. 1987, 26, 973–976. [Google Scholar] [CrossRef]
- Hahn, F.E.; Langenhahn, V.; Lügger, T.; Pape, T.; Le Van, D. Template Synthesis of a Coordinated Tetracarbene Ligand with Crown Ether Topology. Angew. Chem. Int. Ed. 2005, 44, 3759–3763. [Google Scholar] [CrossRef]
- Kaufhold, O.; Stasch, A.; Edwards, P.G.; Hahn, F.E. Template Controlled Synthesis of a Coordinated [11]ane- P2CNHC Macrocycle. Chem. Commun 2007, 1822–1824. [Google Scholar] [CrossRef] [PubMed]
- Kaufhold, O.; Stasch, A.; Pape, T.; Hepp, A.; Edwards, P.G.; Newman, P.D.; Hahn, F.E. Metal Template Controlled Formation of [11]ane- P2CNHC Macrocycles. J. Am. Chem. Soc. 2009, 131, 306–317. [Google Scholar] [CrossRef]
- Blase, V.; Pape, T.; Hahn, F.E. Template Synthesis of a Macrocycle with a Mixed NHC/phosphine Donor Set. J. Organomet. Chem. 2011, 696, 3337–3342. [Google Scholar] [CrossRef]
- Flores-Figueroa, A.; Kaufhold, O.; Hepp, A.; Fröhlich, R.; Hahn, F.E. Synthesis of a Ruthenium(II) Complex Containing an [11]ane-P2CNHC (NHC = Imidazolidin-2-ylidene) Macrocycle. Organometallics 2009, 28, 6362–6369. [Google Scholar] [CrossRef]
- Flores-Figueroa, A.; Pape, T.; Weigand, J.J.; Hahn, F.E. Template-Controlled Formation of an [11]ane- P2CNHC Macrocyclic Ligand at an Iron(II) Template. Eur. J. Inorg. Chem. 2010, 2907–2910. [Google Scholar] [CrossRef]
- Boche, G.; Hilf, C.; Harms, K.; Marsch, M.; Lohrenz, J.C.W. Crystal Structure of the Dimeric(4-tert-Butylthiazolato)(glyme)lithium: Carbene Character of a Formyl Anion Equivalent. Angew. Chem. Int. Ed. 1995, 34, 487–489. [Google Scholar] [CrossRef]
- Kösterke, T.; Kösters, J.; Würthwein, E.U.; Mück-Lichtenfeld, C.; Schulte To Brinke, C.; Lahoz, F.; Hahn, F.E. Synthesis of complexes containing an anionic NHC ligand with an unsubstituted ring-nitrogen atom. Chem. Eur. J. 2012, 18, 14594–14598. [Google Scholar] [CrossRef]
- Arduengo, A.J.; Dixon, D.A.; Harlow, R.L.; Dias, H.V.R.; Booster, W.T.; Koetzle, T.F. Electron Distribution in a Stable Carbene. J. Am. Chem. Soc. 1994, 116, 6812–6822. [Google Scholar] [CrossRef]
- Cernochvá, J.; Necas, M.; Kuritka, I.; Vicha, R. 1-(1-Adamantylmethyl)-1H-benzimidazole. Acta Crystallogr. Sect. E 2011, E67, 2906. [Google Scholar] [CrossRef] [PubMed]
- Hille, U.E.; Zimmer, C.; Vock, C.A.; Hartmann, R.W. First Selective CPB11B1 Inhibitors for the Treatment of Cortisol-Dependent Diseases. ACS Med. Chem. Lett. 2011, 2, 2–6. [Google Scholar] [CrossRef]
- Huertos, M.A.; Pérez, J.; Riera, L.; Díaz, J.; López, R. Effect of the nature of the substituent in N-alkylimidazole ligands on the outcome of deprotonation: Ring opening versus the formation of N-heterocyclic carbene complexes. Chem. Eur. J. 2010, 16, 8495–8507. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, J.; Perandones, B.F. Base-Promoted Tautomerization of Imidazole Ligand to NHC & Subsequent Transmetalation Reaction. J. Am. Chem. Soc. 2007, 129, 9298–9299. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, J.; Berros, Á.; Perandones, B.F.; Vivanco, M. NHC–manganese(I) complexes as carbene transfer agents. Dalton Trans. 2009, 6999. [Google Scholar] [CrossRef] [PubMed]
- Hering, F.; Radius, U. From NHC to Imidazolyl Ligand: Synthesis of Platinum and Palladium Complexes d10-[M(NHC)2] (M = Pd, Pt) of the NHC 1,3-Diisopropylimidazolin-2-ylidene. Organometallics 2015, 34, 3236–3245. [Google Scholar] [CrossRef]
- Fortman, G.C.; Scott, N.M.; Linden, A.; Stevens, E.D.; Dorta, R.; Nolan, S.P. Unusual reactivities of N-heterocyclic carbenes upon coordination to the platinum(II)–dimethyl moiety. Chem. Commun. 2010, 46, 1050–1052. [Google Scholar] [CrossRef] [Green Version]
- Schettini, M.F.; Wu, G.; Hayton, T.W. Synthesis and reactivity of a uranyl-imidazolyl complex. Chem. Commun. 2012, 48, 1484–1486. [Google Scholar] [CrossRef]
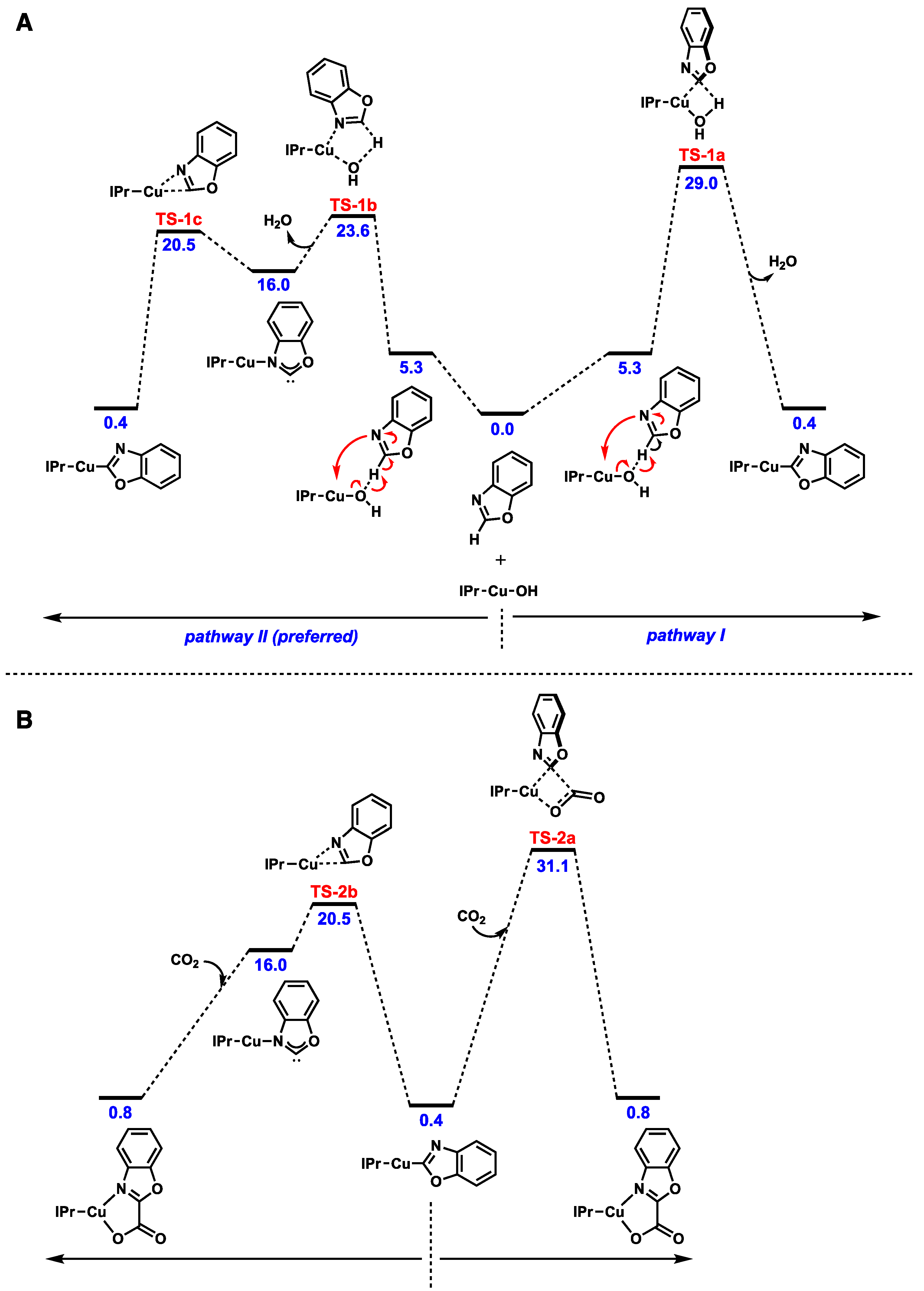
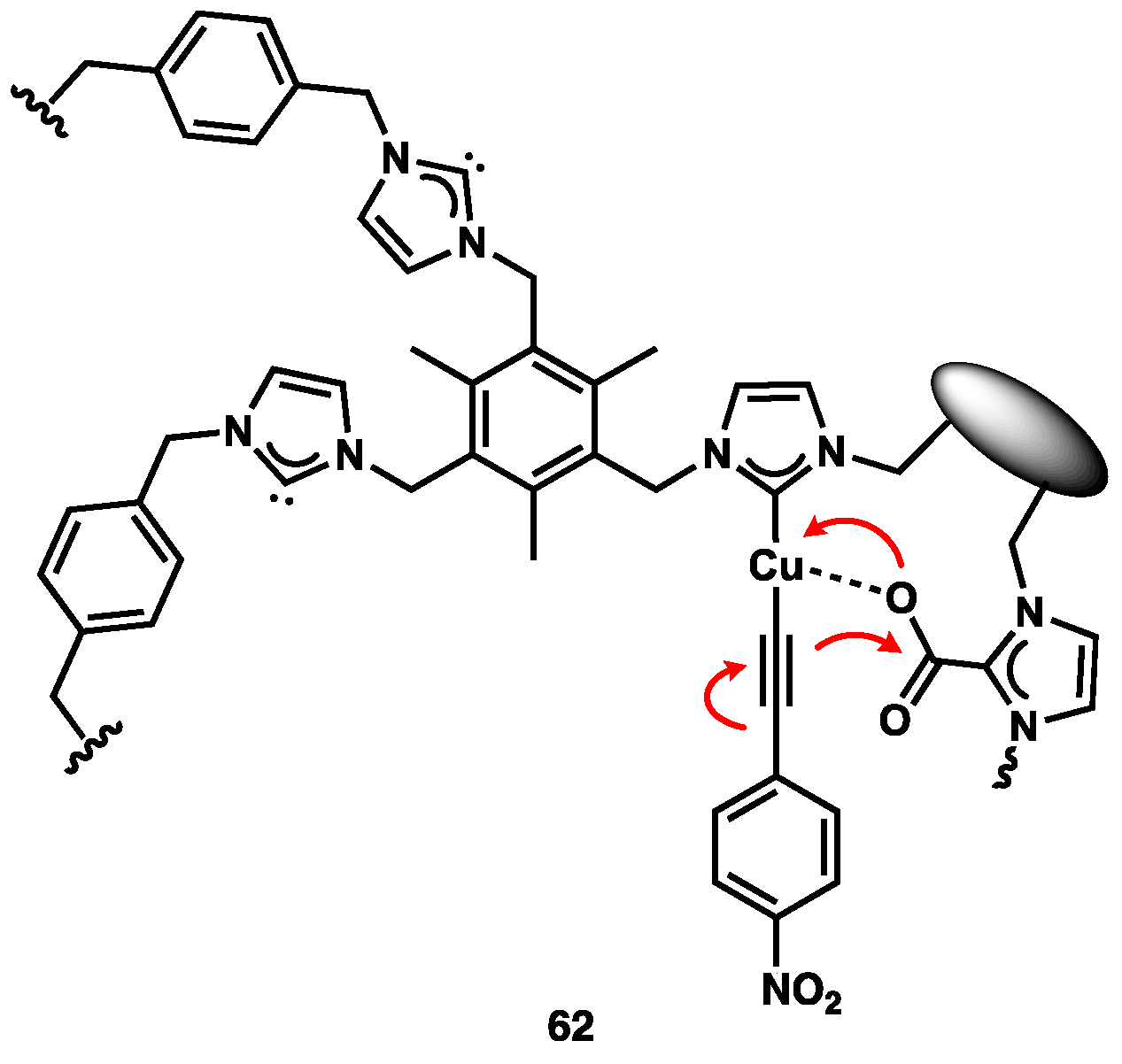
- Ariafard, A.; Zarkoob, F.; Batebi, H.; Stranger, R.; Yates, B.F. DFT studies on the carboxylation of the C-H bond of heteroarenes by copper(I) complexes. Organometallics 2011, 30, 6218–6224. [Google Scholar] [CrossRef]
- Zhang, L.; Cheng, J.; Ohishi, T.; Hou, Z. Copper-catalyzed direct carboxylation of C-H bonds with carbon dioxide. Angew. Chem. Int. Ed. 2010, 49, 8670–8673. [Google Scholar] [CrossRef]
- Boogaerts, I.I.F.; Fortman, G.C.; Furst, M.R.L.; Cazin, C.S.J.; Nolan, S.P. Carboxylation of N–H/C–H bonds using N-heterocyclic carbene copper(I) complexes. Angew. Chem. Int. Ed. 2010, 49, 8674–8677. [Google Scholar] [CrossRef]
- Boogaerts, I.I.F.; Nolan, S.P. Carboxylation of C–H bonds using N-heterocyclic carbene gold(I) complexes. J. Am. Chem. Soc. 2010, 132, 8858–8859. [Google Scholar] [CrossRef]
- Zhang, X.; Geng, Z.; Wang, Y.; Hou, X.; Wang, D. Theoretical studies on the mechanism of oxazole with CO2 catalyzed by gold(I) complexes. J. Mol. Catal. A Chem. 2012, 363–364, 31–40. [Google Scholar] [CrossRef]
- Yu, D.; Zhang, Y. Copper- and copper–N-heterocyclic carbene-catalyzed C–H activating carboxylation of terminal alkynes with CO2 at ambient conditions. Proc. Natl. Acad. Sci. USA 2010, 107, 20184–20189. [Google Scholar] [CrossRef] [PubMed]
- Stephan, D.W.; Erker, G. Frustrated Lewis Pair Chemistry: Development and Perspectives. Angew. Chem. Int. Ed. 2015, 54, 6400–6441. [Google Scholar] [CrossRef] [PubMed]





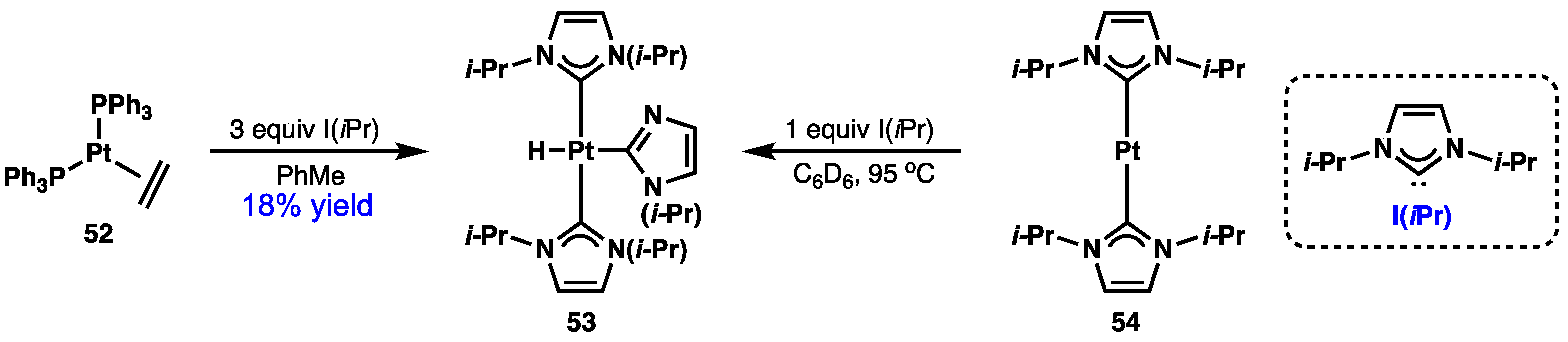

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | 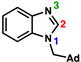 | 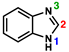 | 20; N3 = Protonated [77] | 17; N3 = Naked | 18; N3 = Alkylated |
|---|---|---|---|---|---|
| N1–C2–N3 (°) | 114.83(11) [79] | 114.41(17) | 107.1(4) | 111.37(17) | 106.7(2) |
| C2–N1 (Å); “A” | 1.3629(14) [79] | 1.345(2) | 1.350(6) | 1.389(2) | 1.351(3) |
| C2–N3 (Å); “B” | 1.3096(14) [79] | 1.312(2) | 1.349(6) | 1.319(3) | 1.364(3) |
| ΔA–B (Å) | 0.053 | 0.033 | 0.001 | 0.070 | 0.013 |
| C2–Pt (Å) | - | - | 1.972(4) | 1.987(2) | 1.973(2) |
| 13C NMR δC2 (ppm) | 144.3 (CDCl3) [80] | 141.9 (d6-DMSO) | 157.8 (CD2Cl2) | 149.4 (CD2Cl2) | 163.3 (CD2Cl2) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leow, M.Y.; Ho, C.C.; Gardiner, M.G.; Bissember, A.C. Non-Classical Anionic Naked N-Heterocyclic Carbenes: Fundamental Properties and Emerging Applications in Synthesis and Catalysis. Catalysts 2018, 8, 620. https://doi.org/10.3390/catal8120620
Leow MY, Ho CC, Gardiner MG, Bissember AC. Non-Classical Anionic Naked N-Heterocyclic Carbenes: Fundamental Properties and Emerging Applications in Synthesis and Catalysis. Catalysts. 2018; 8(12):620. https://doi.org/10.3390/catal8120620
Chicago/Turabian StyleLeow, Mei Yi, Curtis C. Ho, Michael G. Gardiner, and Alex C. Bissember. 2018. "Non-Classical Anionic Naked N-Heterocyclic Carbenes: Fundamental Properties and Emerging Applications in Synthesis and Catalysis" Catalysts 8, no. 12: 620. https://doi.org/10.3390/catal8120620