Vine Shoots and Grape Stalks as Carbon Sources for Hydrogen Evolution Reaction Electrocatalyst Supports




Abstract
:1. Introduction
2. Results and Discussion
2.1. Characterisation of Bio-Based Activated Carbon Supports
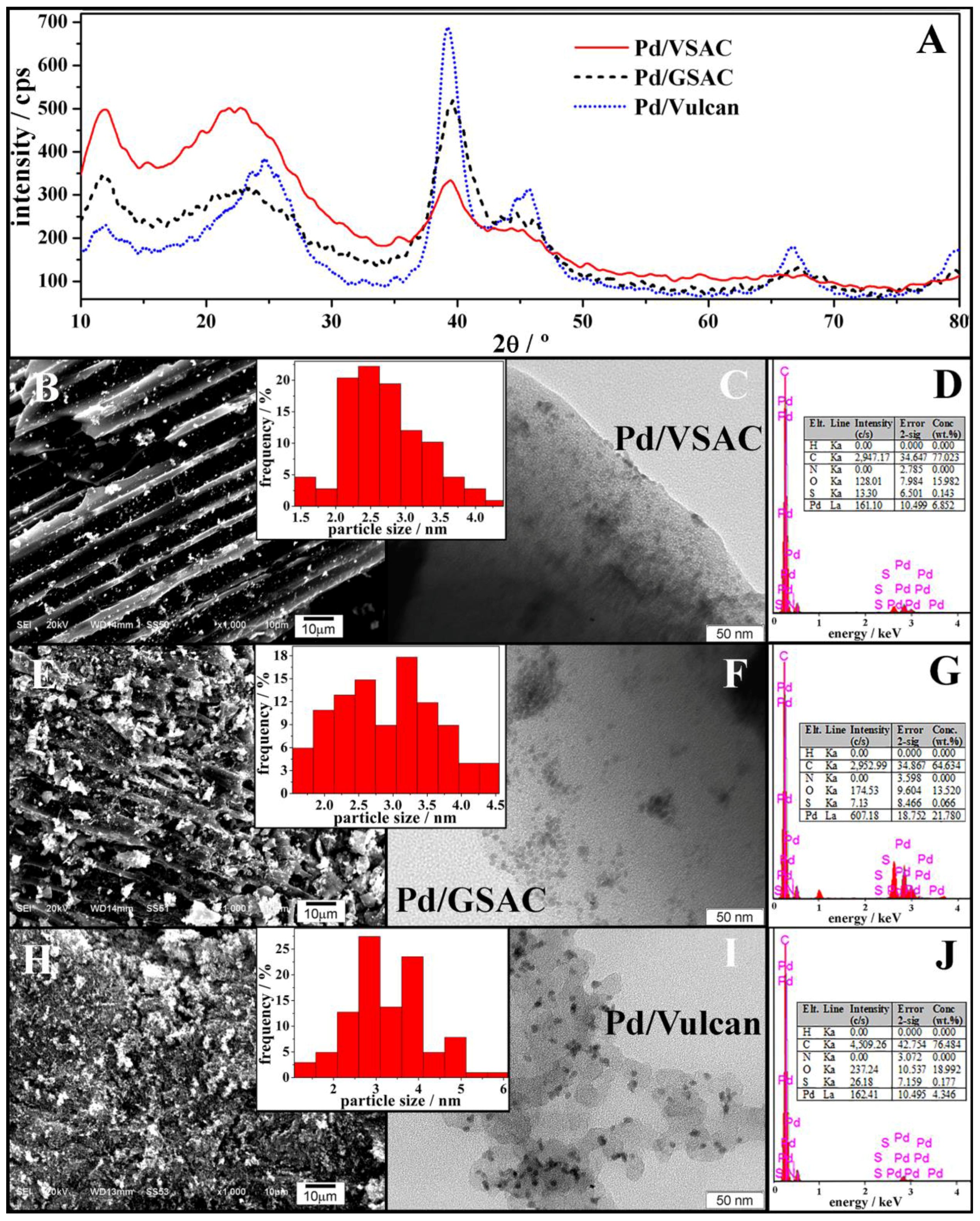
2.2. Characterisation of the Activated Carbon-Supported Pd Nanoparticles
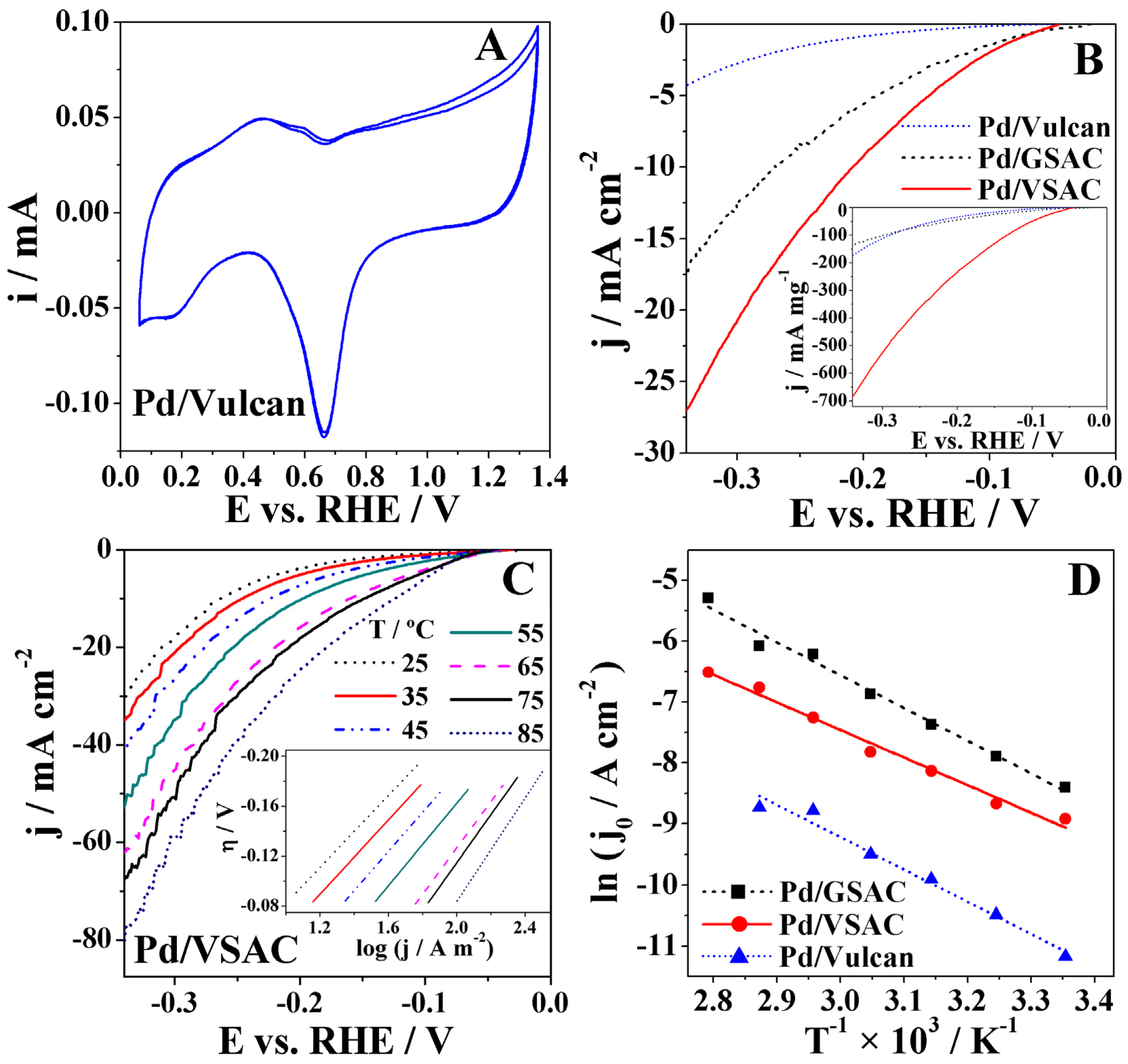
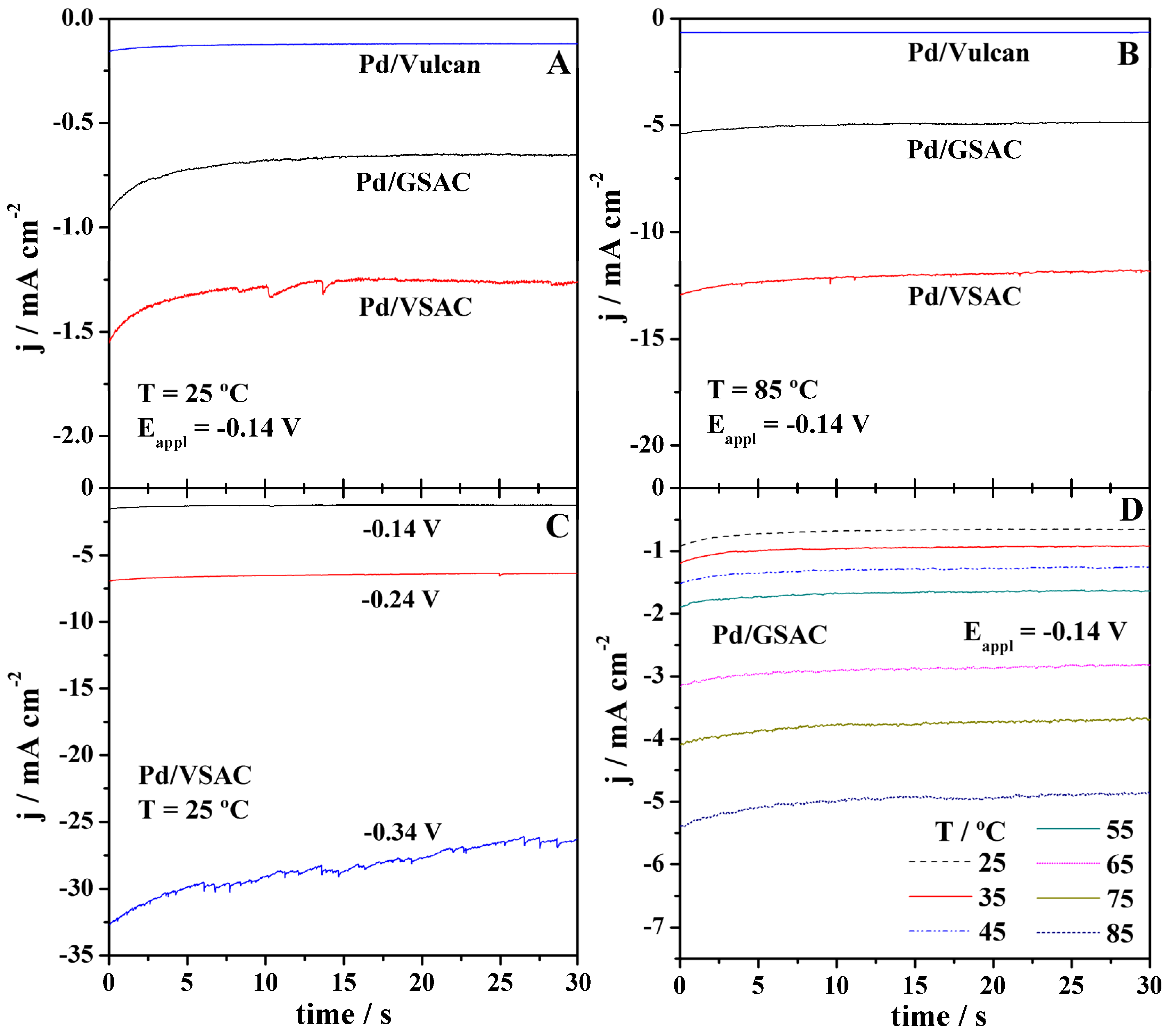
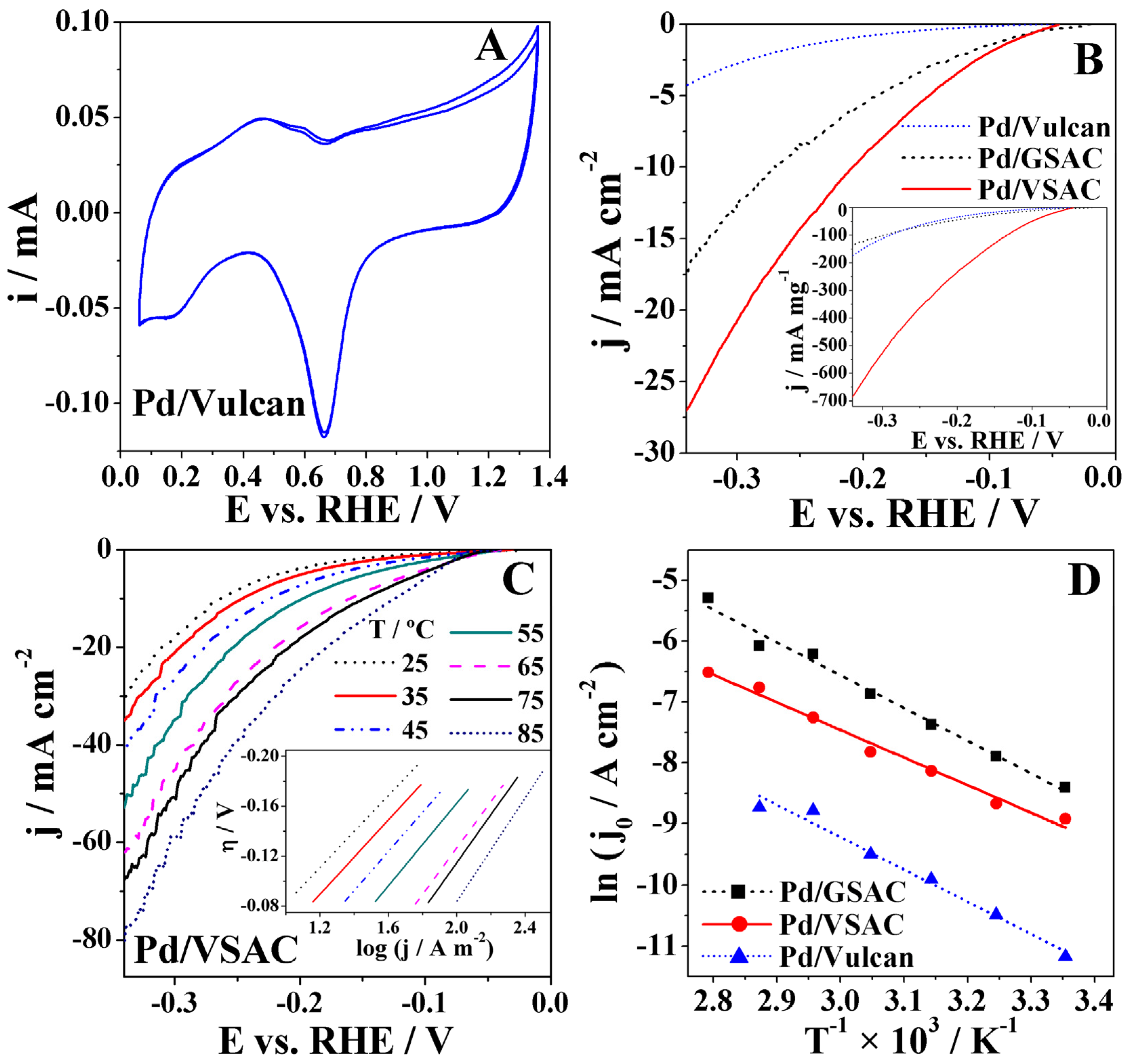
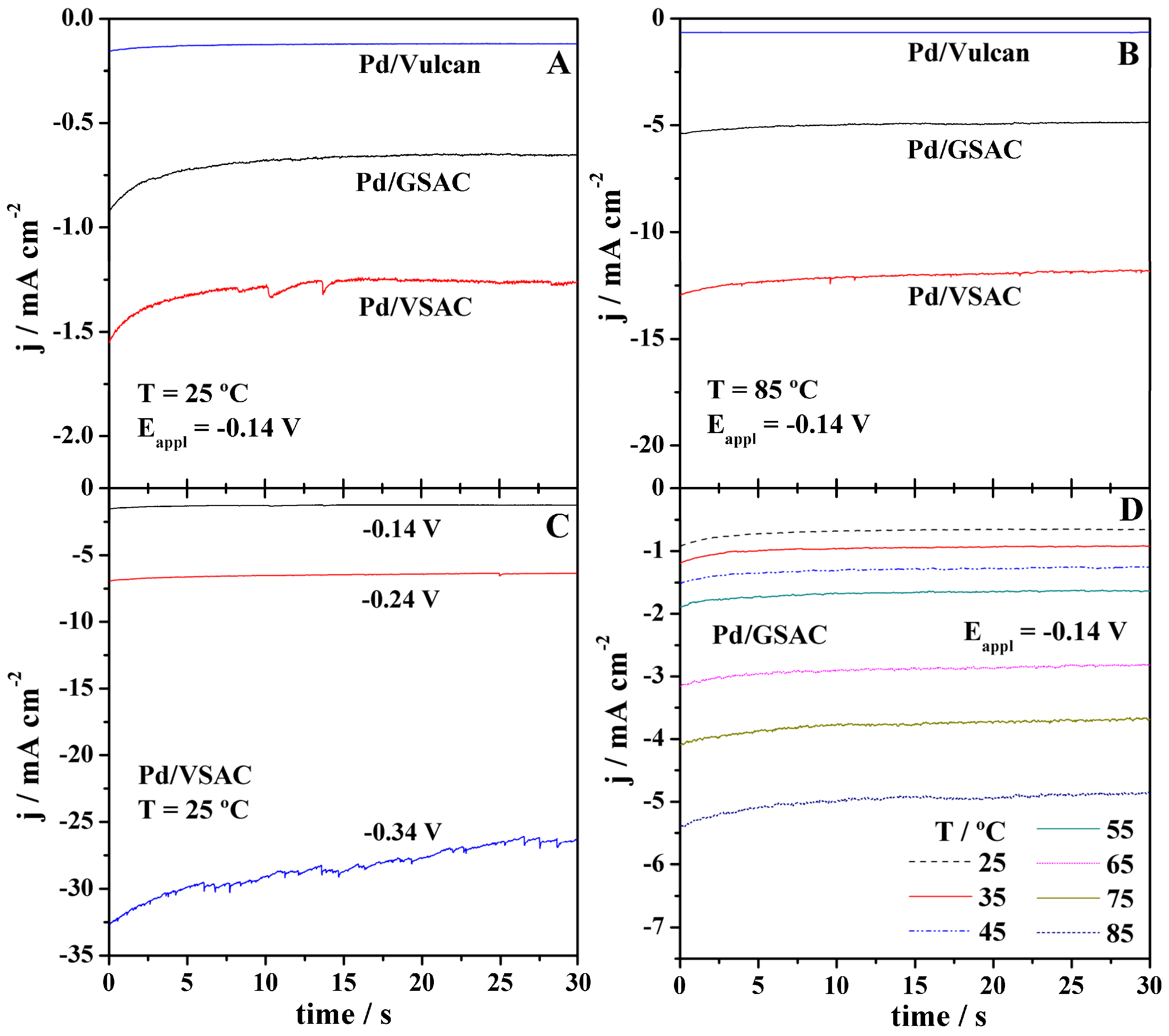
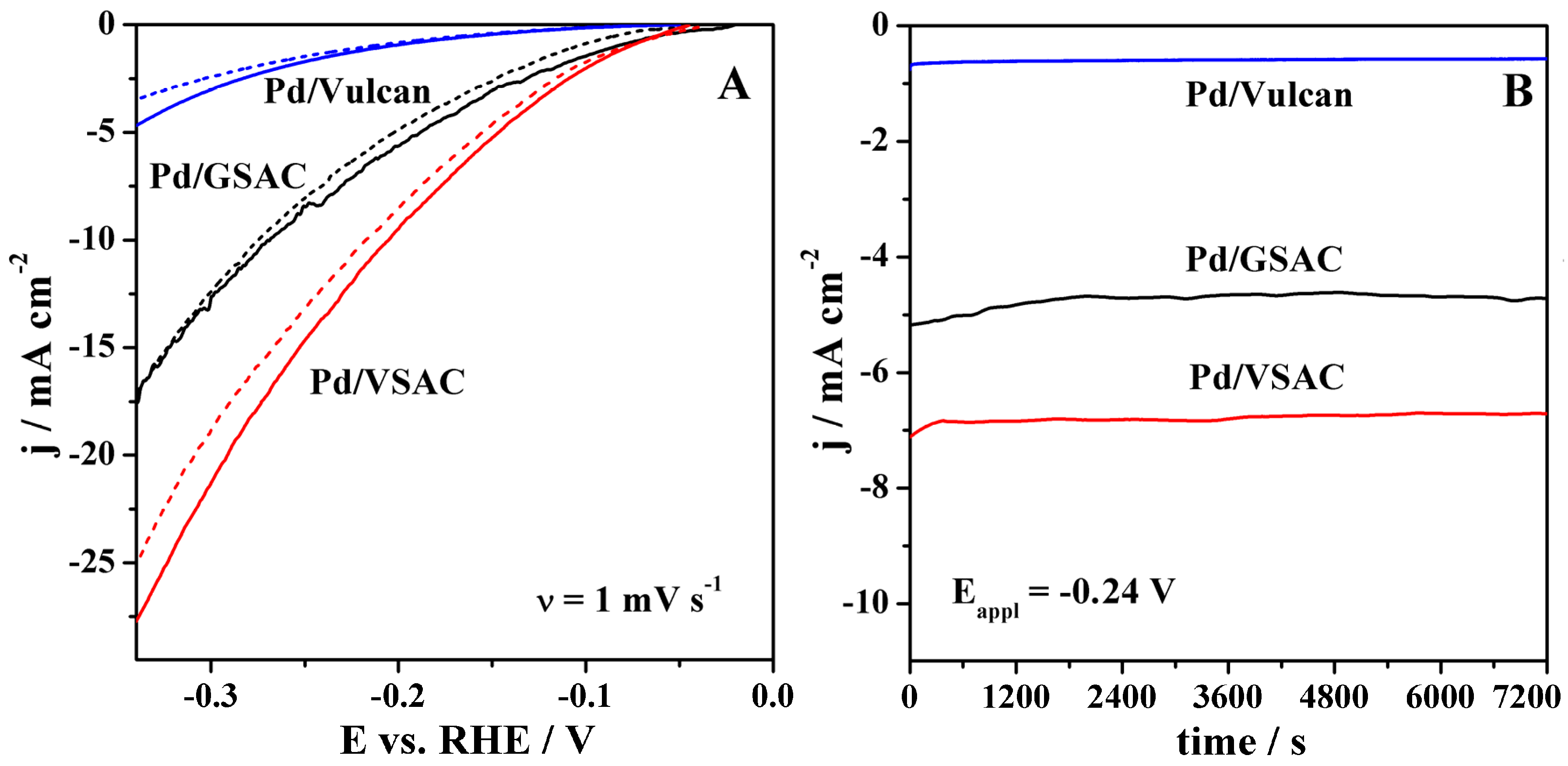
2.3. HER Studies
3. Experimental
3.1. Preparation and Characterisation of Bio-Based Activated Carbon Supports
3.2. Preparation and Characterisation of Activated Carbon-Supported Pd Nanoparticles
3.3. Electrode Preparation
3.4. Electrochemical Measurements
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Höök, M.; Tang, X. Depletion of fossil fuels and anthropogenic climate change—A review. Energy Policy 2003, 52, 797–809. [Google Scholar] [CrossRef]
- Safavi, A.; Kazemi, S.H.; Kazemi, H. Electrocatalytic behaviors of silver–palladium nanoalloys modified carbon ionic liquid electrode towards hydrogen evolution reaction. Fuel 2014, 118, 156–162. [Google Scholar] [CrossRef]
- Dixon, R.K. Advancing towards a hydrogen energy economy: Status, opportunities and barriers. Mitig. Adapt. Strateg. Glob. Chang. 2007, 12, 325–341. [Google Scholar] [CrossRef]
- Marini, S.; Salvi, P.; Nelli, P.; Pesenti, R.; Villa, M.; Berrettoni, M.; Zangari, G.; Kiros, Y. Advanced alkaline water electrolysis. Electrochim. Acta 2012, 82, 384–391. [Google Scholar] [CrossRef]
- Santos, D.M.F.; Sequeira, C.A.C.; Figueiredo, J.L. Hydrogen production by alkaline water electrolysis. Quim. Nova 2013, 36, 1176–1193. [Google Scholar] [CrossRef]
- Safizadeh, F.; Ghali, E.; Houlachi, G. Electrocatalysis developments for hydrogen evolution reaction in alkaline solutions—A review. Int. J. Hydrogen Energy 2015, 40, 256–274. [Google Scholar] [CrossRef]
- Sawyer, D.T.; Sobkowiak, A.; Roberts, J.L. Electrochemistry for Chemists, 2nd ed.; John Wiley & Sons: New York, NY, USA, 1995. [Google Scholar]
- Zinola, C.F.; Martins, M.E.; Tejera, E.P.; Neves, N.P., Jr. Electrocatalysis: Fundamentals and Applications. Int. J. Electrochem. 2012, 2012, 874687. [Google Scholar] [CrossRef]
- Santos, D.M.F.; Šljukić, B.; Sequeira, C.A.C.; Macciò, D.; Saccone, A.; Figueiredo, J.L. Electrocatalytic approach for the efficiency increase of electrolytic hydrogen production: Proof-of-concept using Pt-Dy. Energy 2013, 50, 486–492. [Google Scholar] [CrossRef]
- Yang, L.; Liu, P.; Li, J.; Xiang, B. Two-dimensional material molybdenum disulfides as electrocatalysts for hydrogen evolution. Catalysts 2017, 7, 285. [Google Scholar] [CrossRef]
- Eftekhari, A. Electrocatalysts for hydrogen evolution reaction. Int. J. Hydrogen Energy 2017, 42, 11053–11077. [Google Scholar] [CrossRef]
- Šljukić, B.; Santos, D.M.F.; Vujković, M.; Amaral, L.; Rocha, R.P.; Sequeira, C.A.C.; Figueiredo, J.L. Molybdenum Carbide Nanoparticles on Carbon Nanotubes and Carbon Xerogel: Low-Cost Cathodes for Hydrogen Production by Alkaline Water Electrolysis. ChemSusChem 2016, 9, 1200–1208. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, W.; Wang, X.; Wang, E. N-doped graphene supported WxC composite material as an efficient non-noble metal electrocatalyst for hydrogen evolution reaction. Electrochim. Acta 2017, 251, 660–671. [Google Scholar] [CrossRef]
- Li, C.; Bo, X.; Li, M.; Guo, L. Facile electrodeposition fabrication of molybdenum-tungsten sulfide on carbon cloth for electrocatalytic hydrogen evolution. Int. J. Hydrogen Energy 2017, 42, 15479–15488. [Google Scholar] [CrossRef]
- Gupta, U.; Rao, C.N.R. Hydrogen generation by water splitting using MoS2 and other transition metal dichalcogenides. Nano Energy 2017, 41, 49–65. [Google Scholar] [CrossRef]
- Su, J.; Zhou, J.; Wang, L.; Liu, C.; Chen, Y. Synthesis and application of transition metal phosphides as electrocatalyst for water splitting. Sci. Bull. 2017, 62, 633–644. [Google Scholar] [CrossRef]
- Pi, M.; Wu, T.; Guo, W.; Wang, X.; Chen, S. Phase-controlled synthesis of polymorphic tungsten diphosphide with hybridization of monoclinic and orthorhombic phases as a novel electrocatalyst for efficient hydrogen evolution. J. Power Sources 2017, 349, 138–143. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, P.; Lei, J.; Dong, W.; Wang, J. Integrated 3D MoSe2@Ni0.85Se nanowire network with synergistic cooperation as highly efficient electrocatalysts for hydrogen evolution reaction in alkaline medium. Electrochim. Acta 2017, 246, 712–719. [Google Scholar] [CrossRef]
- Zeng, M.; Li, Y. Recent advances in heterogeneous electrocatalysts for hydrogen evolution reaction. J. Mater. Chem. 2015, 3, 14942–14962. [Google Scholar] [CrossRef]
- Bhowmik, T.; Kundu, M.K.; Barman, S. Palladium nanoparticle-graphitic carbon nitride porous synergistic catalyst for hydrogen evolution/oxidation reactions over a broad range of pH correlation of its catalytic activity with measured hydrogen binding energy. ACS Catal. 2016, 6, 1929–1941. [Google Scholar] [CrossRef]
- Limpattayanate, S.; Hunson, M. Electrocatalytic activity of Pt-Pd electrocatalysts for the oxygen reaction in proton exchange membrane fuel cells: Effect of supports. Renew. Energy 2014, 63, 205–211. [Google Scholar] [CrossRef]
- Banadaki, A.D.; Amir, K. Recent advances in facile synthesis of bimetallic nanostructures: An overview. J. Nanomater. 2014, 2014, 985948. [Google Scholar]
- Tong, S.-S.; Wang, X.-J.; Li, Q.-C.; Han, X.-J. Progress on electrocatalysts of hydrogen evolution reaction based on carbon fiber materials. Chin. J. Anal. Chem. 2016, 44, 1447–1457. [Google Scholar] [CrossRef]
- Zhou, W.; Jia, J.; Lu, J.; Yang, L.; Hou, D.; Li, G.; Chen, S. Recent developments of carbon-based electrocatalysts for hydrogen evolution reaction. Nano Energy 2016, 28, 29–43. [Google Scholar] [CrossRef]
- Martins, M.; Šljukić, B.; Sequeira, C.A.C.; Metin, Ö.; Erdem, M.; Şener, T.; Santos, D.M.F. Biobased carbon-supported palladium electrocatalysts for borohydride fuel cells. Int. J. Hydrogen Energy 2016, 41, 10914–10922. [Google Scholar] [CrossRef]
- Sanders, I.J.; Peeten, T.L. Carbon Black: Production, Properties and Uses; Nova Science Publisher Inc.: New York, NY, USA, 2011. [Google Scholar]
- Erdem, M.; Orhan, R.; Şahin, M.; Aydın, E. Preparation and characterization of a novel activated carbon from vine shoots by ZnCl2. Water Air Soil Pollut. 2016, 227, 1–14. [Google Scholar] [CrossRef]
- Ozdemir, I.; Sahin, M.; Orhan, R.; Erdem, M. Preparation and characterization of activated carbon from grape stalk by zinc chloride activation. Fuel Process. Technol. 2014, 125, 4588–4589. [Google Scholar] [CrossRef]
- Mall, I.; Srivastava, V.; Kumar, G.; Mishra, I. Characterization and utilization of mesoporous fertilizer plant waste carbon for adsorptive removal of dyes from aqueous solution. Colloid Surf. A 2006, 278, 175–187. [Google Scholar] [CrossRef]
- Green, T.; Britz, D. Kinetics of the deuterium and hydrogen evolution reactions at palladium in alkaline solution. J. Electroanal. Chem. 1996, 412, 59–66. [Google Scholar] [CrossRef]
- Cardoso, J.A.S.B.; Amaral, L.; Metin, O.; Cardoso, D.S.P.; Sevim, M.; Sener, T.; Sequeira, C.A.C.; Santos, D.M.F. Reduced graphene oxide assembled Pd-based nanoalloys for hydrogen evolution reaction. Int. J. Hydrogen Energy 2017, 42, 3916–3925. [Google Scholar] [CrossRef]
- Vasić, M.; Čebela, M.; Pašti, I.; Amaral, L.; Hercigonja, R.; Santos, D.M.F.; Šljukić, B. Efficient hydrogen evolution electrocatalysis in alkaline medium using Pd-modified zeolite X. Electrochim. Acta 2018, 259, 882–892. [Google Scholar] [CrossRef]
- Sequeira, C.A.C.; Santos, D.M.F.; Šljukić, B.; Amaral, L. Physics of electrolytic gas evolution. Braz. J. Phys. 2013, 43, 199–208. [Google Scholar] [CrossRef]
- Santos, D.M.F.; Amaral, L.; Šljukić, B.; Macciò, D.; Saccone, A.; Sequeira, C.A.C. Electrocatalytic activity of nickel-cerium alloys for hydrogen evolution in alkaline electrolysis. J. Electrochem. Soc. 2014, 161, F386–F390. [Google Scholar] [CrossRef]
- Pluntke, Y.; Kibler, L.A. Hydrogen evolution electrocatalysts on AgPd(111) alloys. Electrocatalysis 2011, 2, 192–199. [Google Scholar] [CrossRef]
- Pierozynski, B.; Mikolajczyk, T.; Turemko, M.; Czerwosz, E.; Kozlowski, M. Hydrogen evolution reaction at Pd-modified carbon fibre in 0.1 M NaOH. Int. J. Hydrogen Energy 2015, 40, 1795–1799. [Google Scholar] [CrossRef]
- Pierozynski, B. Hydrogen evolution reaction at Pd-modified carbon fibre and nickel-coated carbon fibre materials. Int. J. Hydrogen Energy 2013, 38, 7733–7740. [Google Scholar] [CrossRef]
- Chen, J.; Chen, J.; Yu, D.; Zhang, M.; Zhu, H.; Du, M. Carbon nanofiber-supported PdNi alloy nanoparticles as highly bifunctional catalysts for hydrogen and oxygen evolution reactions. Electrochim. Acta 2017, 246, 17–26. [Google Scholar] [CrossRef]
- Durst, J.; Simon, C.; Hasché, F.; Gasteiger, H.A. Hydrogen oxidation and evolution reaction kinetics on carbon supported Pt, Ir, Rh, and Pd electrocatalysts in acidic media. J. Electrochem. Soc. 2015, 162, F190–F203. [Google Scholar] [CrossRef]
- Boehm, H.P. Surface oxides on carbon and their analysis: A critical assessment. Carbon 2002, 40, 145–149. [Google Scholar] [CrossRef]
- Mazumder, V.; Sun, S. Oleylamine-mediated synthesis of Pd nanoparticles for catalytic formic acid oxidation. J. Am. Chem. Soc. 2009, 131, 4588–4589. [Google Scholar] [CrossRef] [PubMed]
- Sanetuntikul, J.; Hang, T.; Shanmugam, S. Hollow nitrogen-doped carbon spheres as efficient and durable electrocatalysts for oxygen reduction. Chem. Commun. 2014, 50, 9473–9476. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Support Material | BET Surface Area/m2 g−1 | Total Pore Volume/cm3 g−1 | Pore Size/nm |
|---|---|---|---|
| VSAC | 1689 | 0.84 | 1.9 |
| GSAC | 1411 | 0.72 | 2.1 |
| Vulcan | 224 | 0.47 | 11 |
| Electrode | T/°C | α | b/V dec−1 | j0/mA cm−2 | j0.1 */mA cm−2 | η10 **/V |
|---|---|---|---|---|---|---|
| Pd/GSAC | 25 | 0.36 | 0.163 | 0.13 | 0.76 | 0.299 |
| 35 | 0.36 | 0.168 | 0.17 | 0.96 | 0.281 | |
| 45 | 0.35 | 0.178 | 0.29 | 1.27 | 0.269 | |
| 55 | 0.34 | 0.190 | 0.40 | 1.70 | 0.256 | |
| 65 | 0.31 | 0.213 | 0.71 | 2.83 | 0.222 | |
| 75 | 0.30 | 0.230 | 1.16 | 3.54 | 0.203 | |
| 85 | 0.34 | 0.208 | 1.48 | 4.64 | 0.171 | |
| Pd/VSAC | 25 | 0.41 | 0.144 | 0.22 | 1.43 | 0.224 |
| 35 | 0.41 | 0.148 | 0.37 | 2.02 | 0.207 | |
| 45 | 0.40 | 0.156 | 0.63 | 3.04 | 0.185 | |
| 55 | 0.39 | 0.165 | 1.04 | 4.50 | 0.158 | |
| 65 | 0.36 | 0.185 | 1.99 | 7.69 | 0.111 | |
| 75 | 0.36 | 0.192 | 2.29 | 8.90 | 0.109 | |
| 85 | 0.34 | 0.207 | 5.02 | 12.6 | 0.085 | |
| Pd/Vulcan | 25 | 0.56 | 0.105 | 0.01 | 0.10 | >0.3 |
| 35 | 0.44 | 0.137 | 0.03 | 0.18 | >0.3 | |
| 45 | 0.39 | 0.162 | 0.05 | 0.28 | >0.3 | |
| 55 | 0.37 | 0.175 | 0.07 | 0.39 | >0.3 | |
| 65 | 0.38 | 0.189 | 0.11 | 0.62 | >0.3 | |
| 75 | 0.35 | 0.194 | 0.15 | 0.69 | >0.3 | |
| 85 | 0.34 | 0.202 | 0.16 | 0.55 | >0.3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cardoso, J.A.S.B.; Šljukić, B.; Erdem, M.; Sequeira, C.A.C.; Santos, D.M.F. Vine Shoots and Grape Stalks as Carbon Sources for Hydrogen Evolution Reaction Electrocatalyst Supports. Catalysts 2018, 8, 50. https://doi.org/10.3390/catal8020050
Cardoso JASB, Šljukić B, Erdem M, Sequeira CAC, Santos DMF. Vine Shoots and Grape Stalks as Carbon Sources for Hydrogen Evolution Reaction Electrocatalyst Supports. Catalysts. 2018; 8(2):50. https://doi.org/10.3390/catal8020050
Chicago/Turabian StyleCardoso, J.A.S.B., B. Šljukić, M. Erdem, C.A.C. Sequeira, and D.M.F. Santos. 2018. "Vine Shoots and Grape Stalks as Carbon Sources for Hydrogen Evolution Reaction Electrocatalyst Supports" Catalysts 8, no. 2: 50. https://doi.org/10.3390/catal8020050
APA StyleCardoso, J. A. S. B., Šljukić, B., Erdem, M., Sequeira, C. A. C., & Santos, D. M. F. (2018). Vine Shoots and Grape Stalks as Carbon Sources for Hydrogen Evolution Reaction Electrocatalyst Supports. Catalysts, 8(2), 50. https://doi.org/10.3390/catal8020050