Highly Selective Continuous Flow Hydrogenation of Cinnamaldehyde to Cinnamyl Alcohol in a Pt/SiO2 Coated Tube Reactor

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
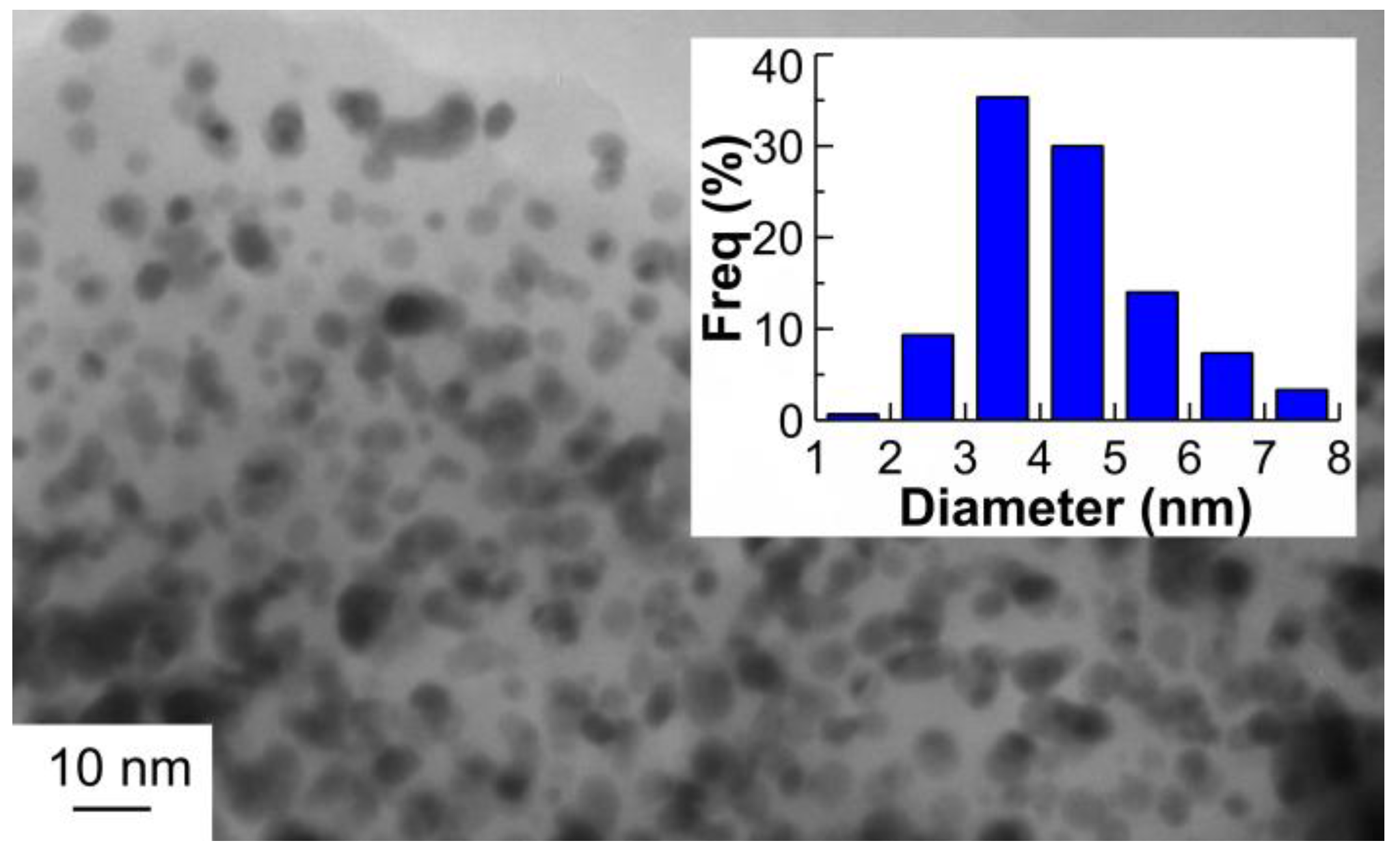
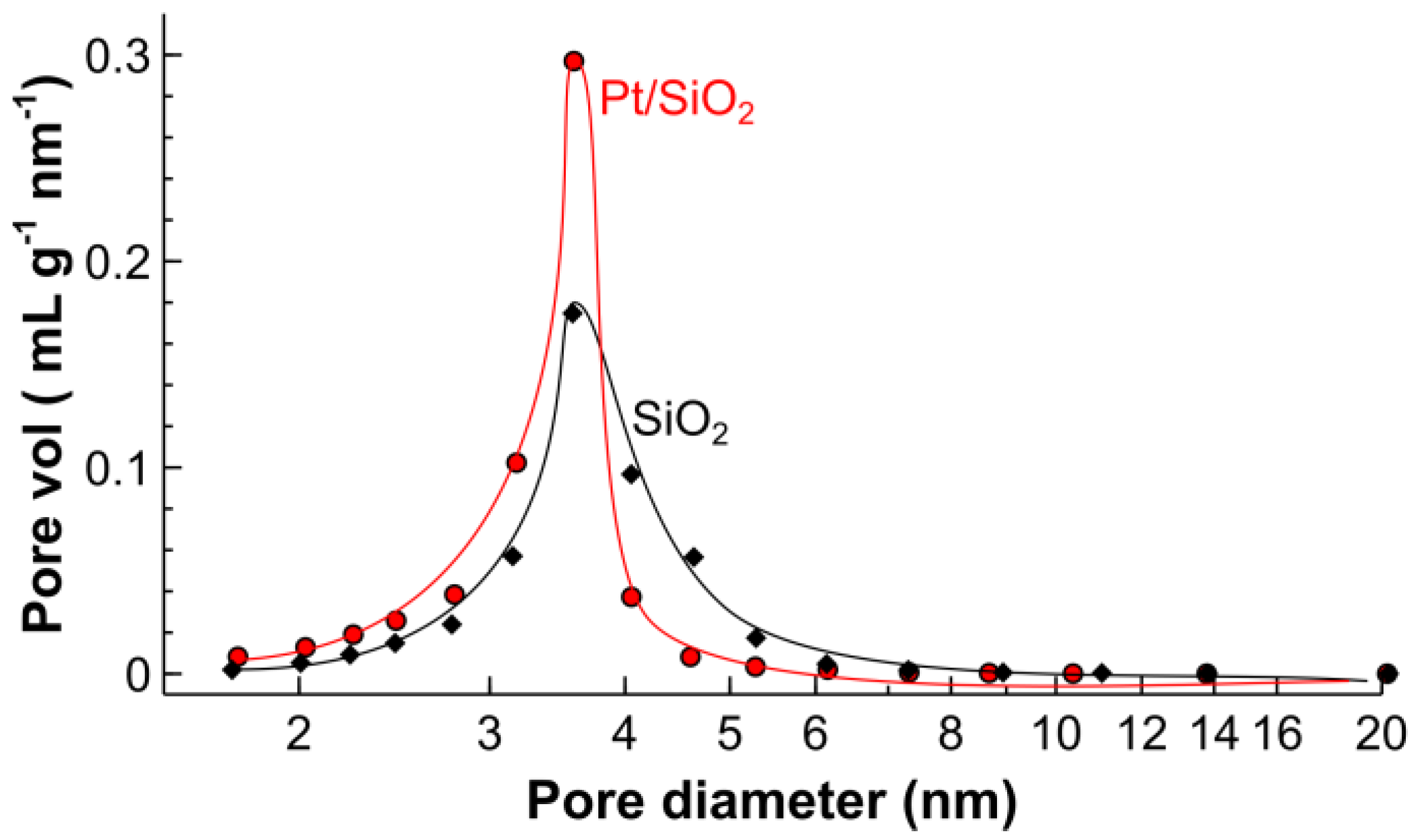
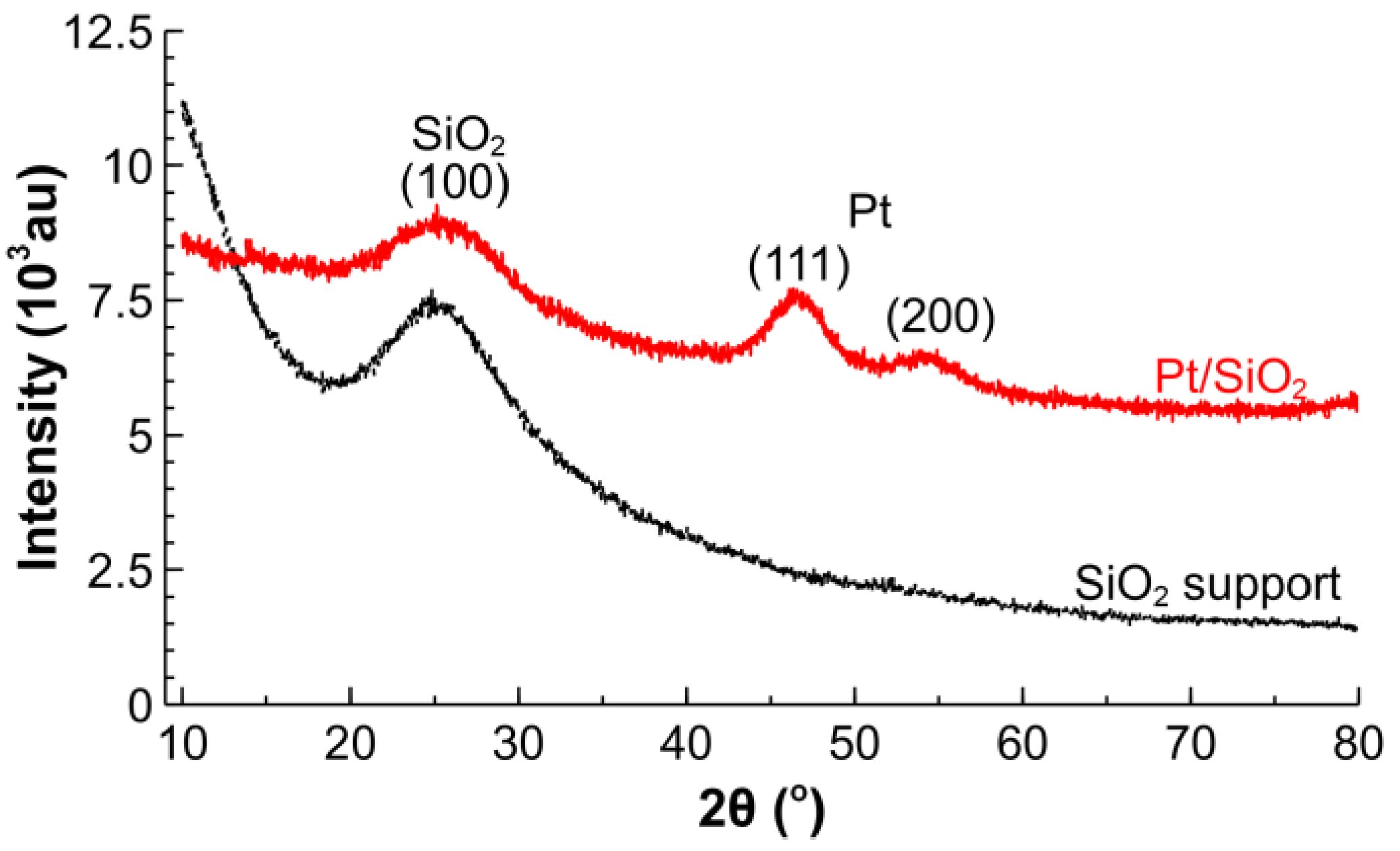
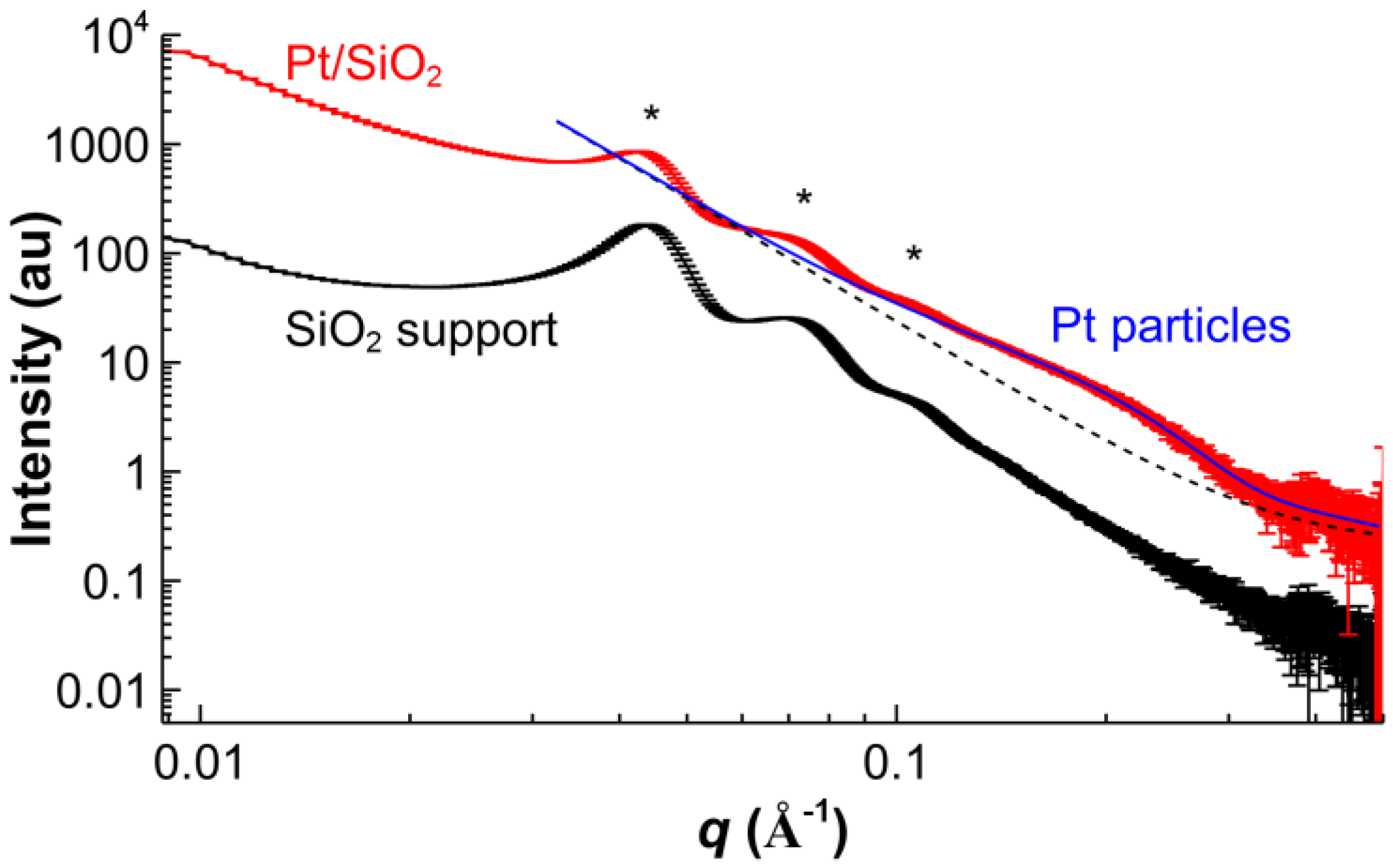
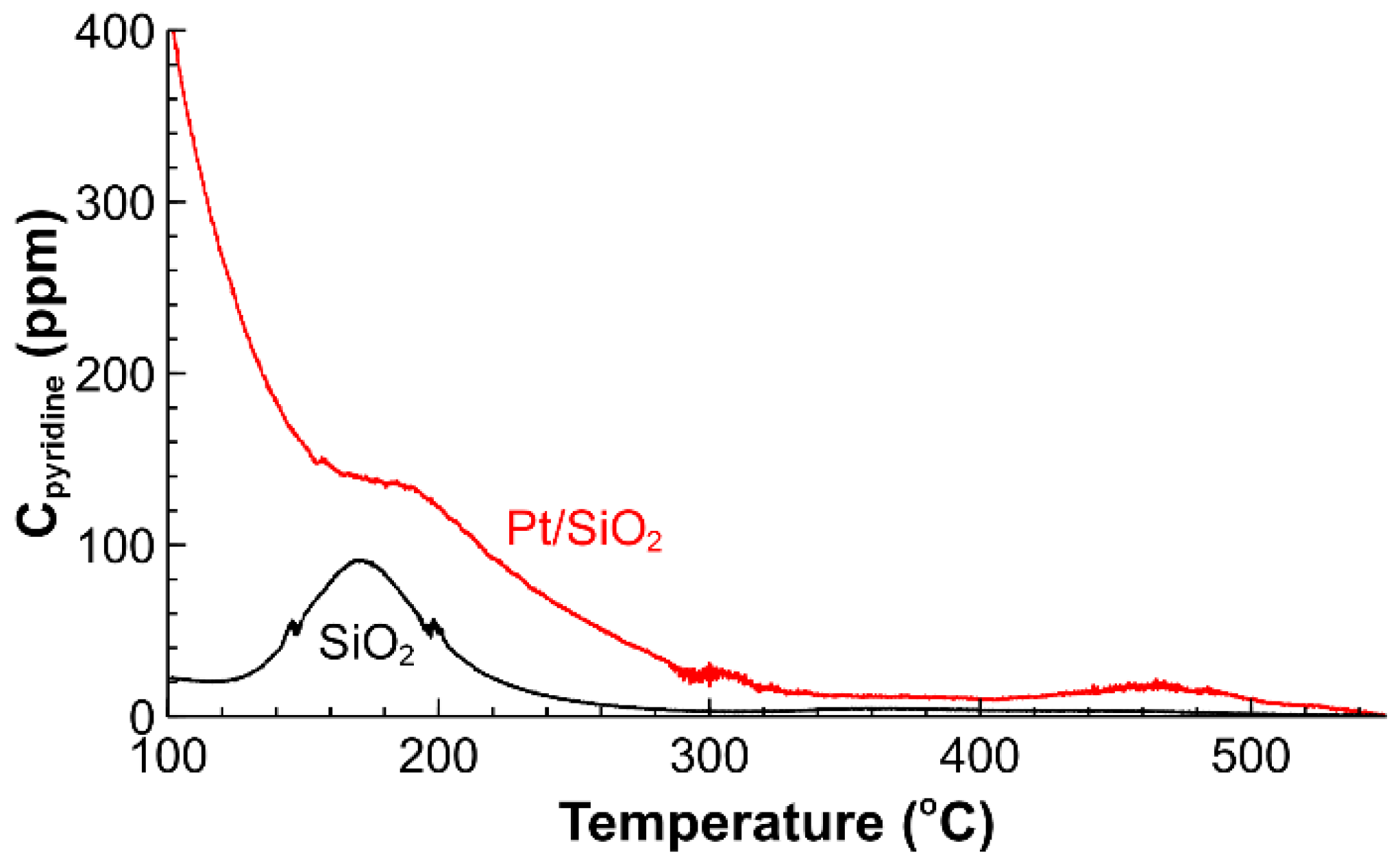
2.1. Catalyst Characterization
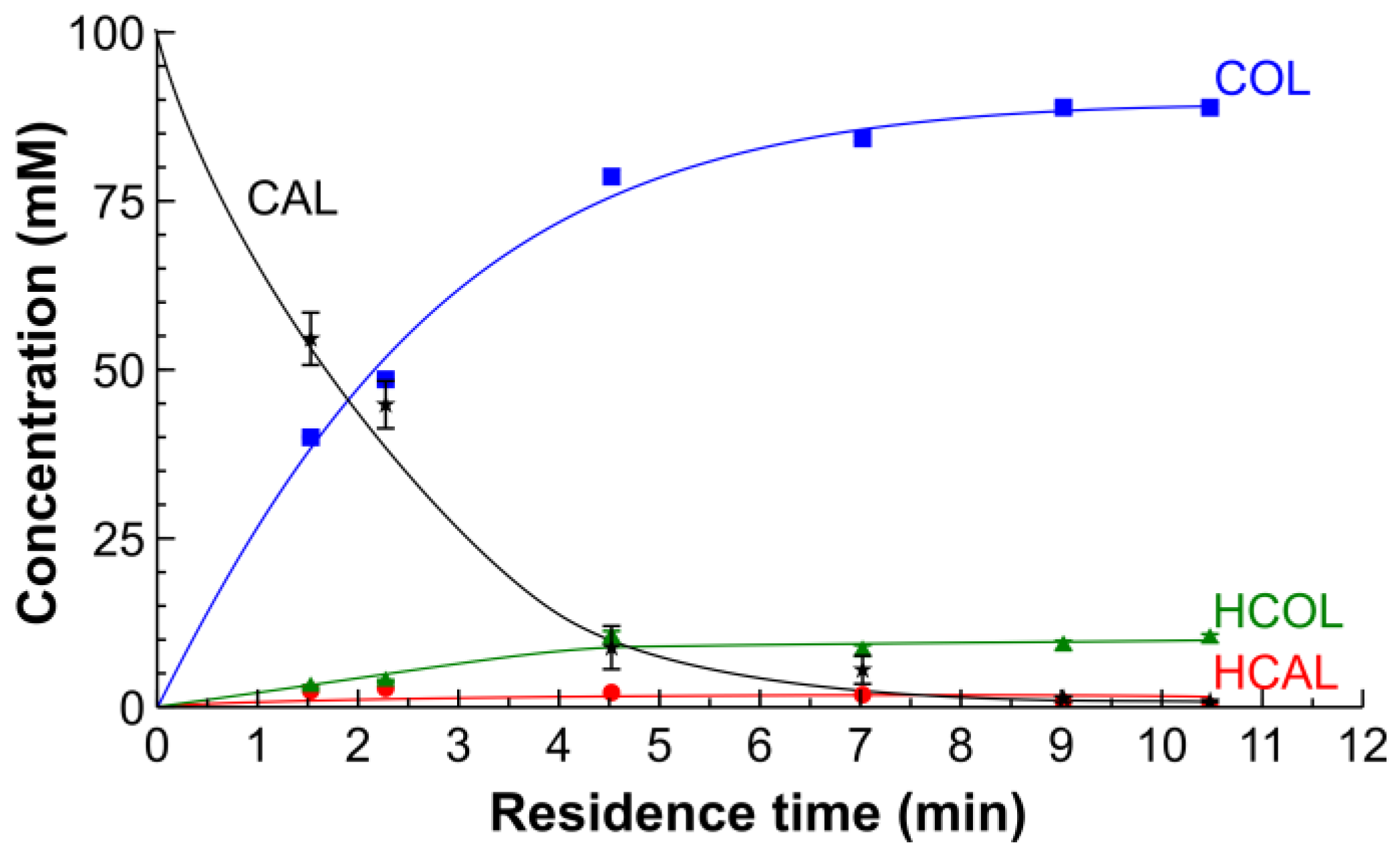
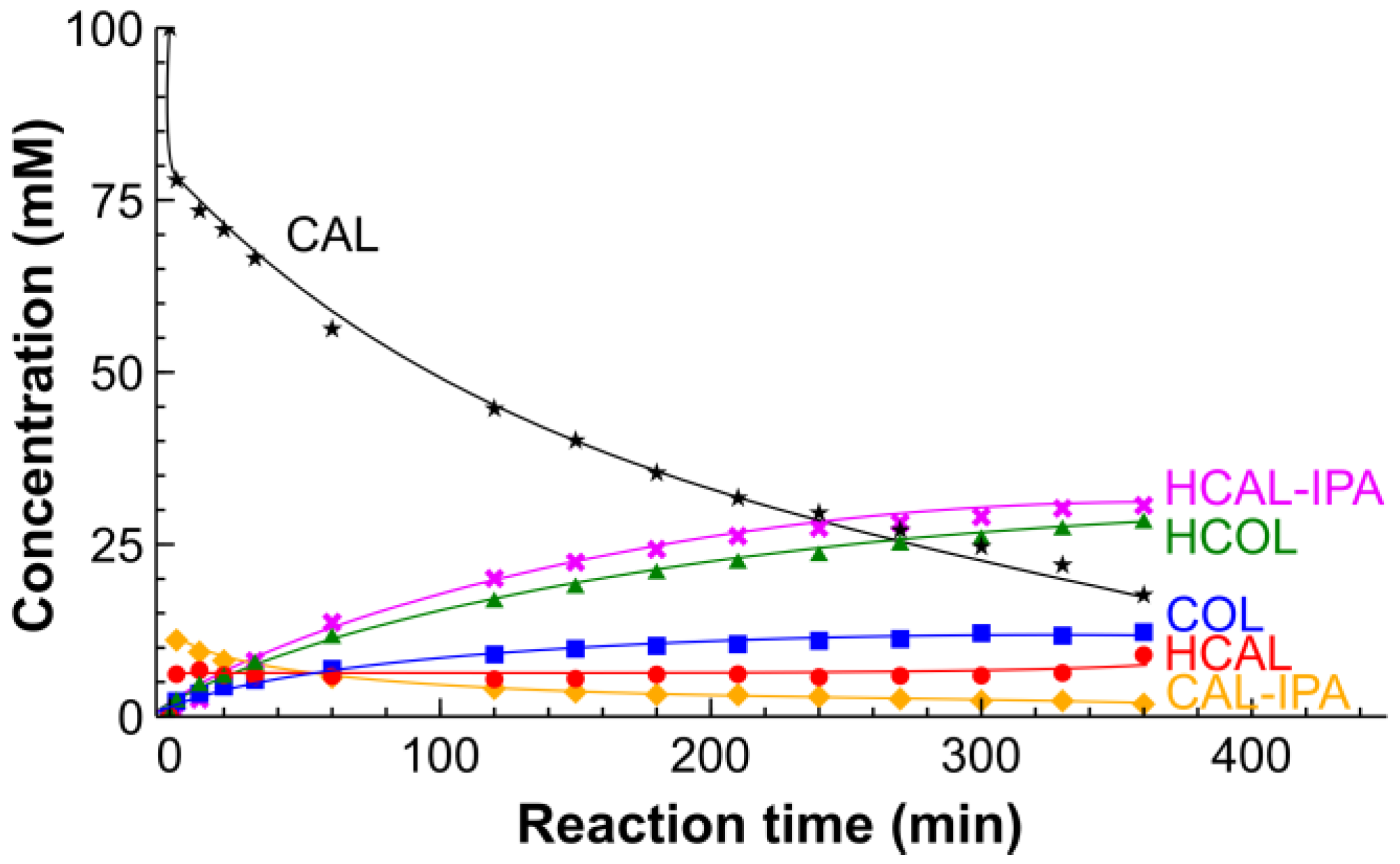
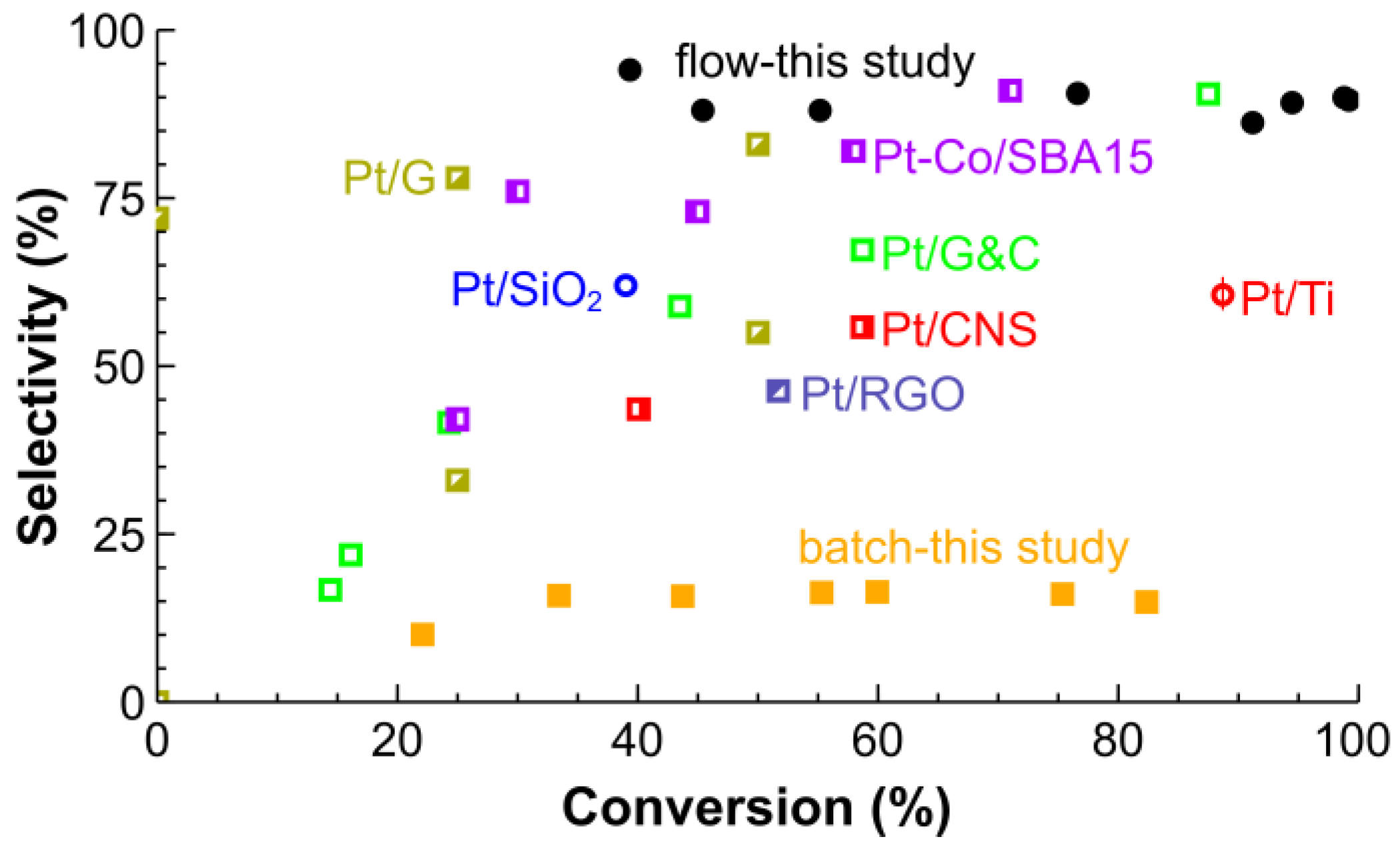
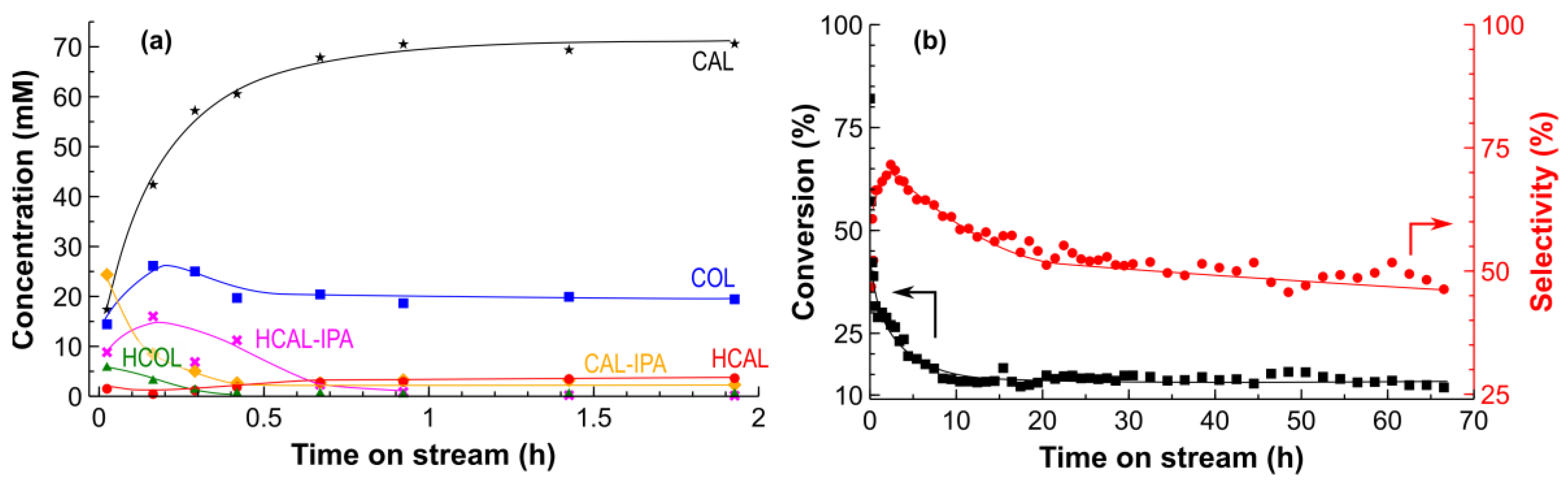
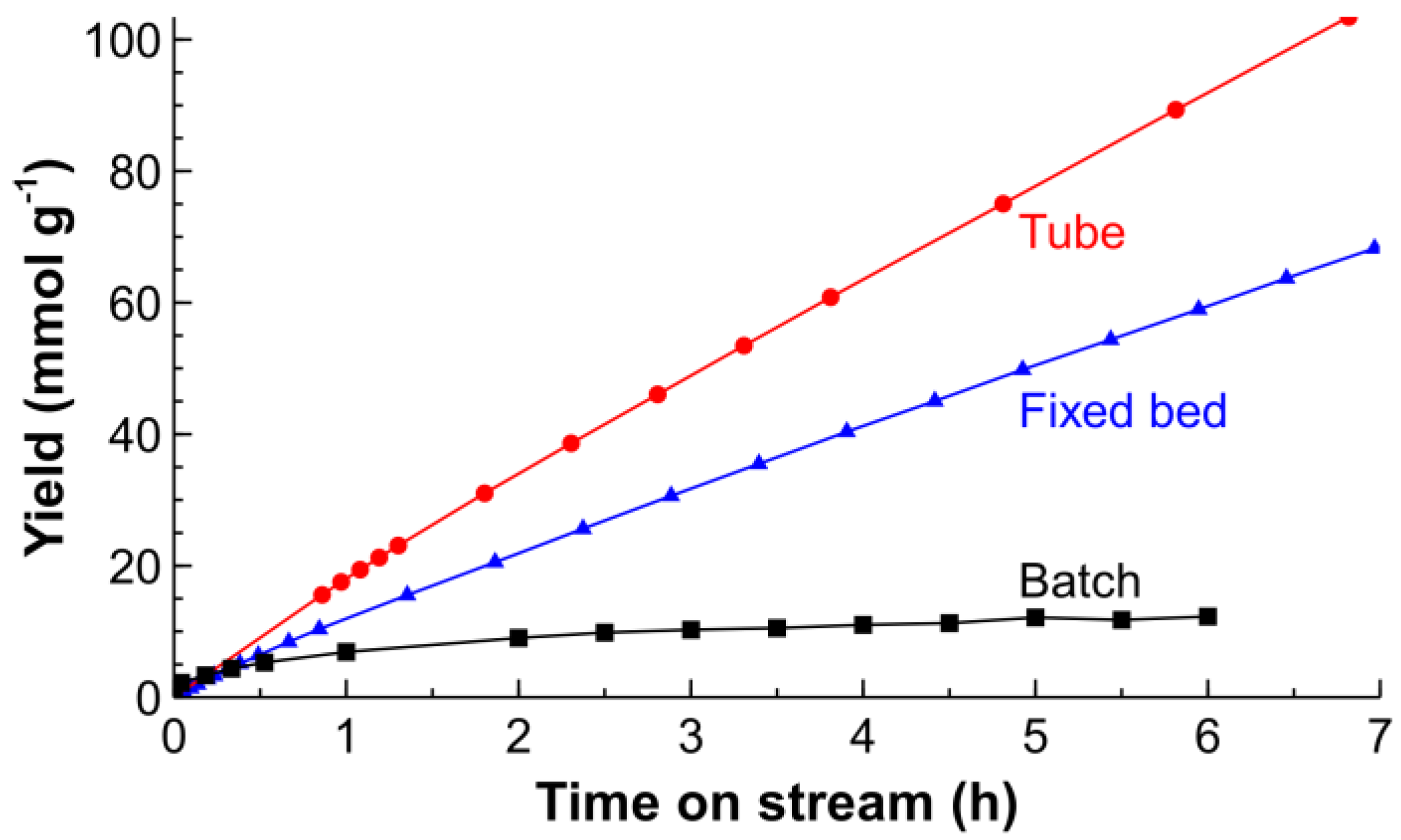
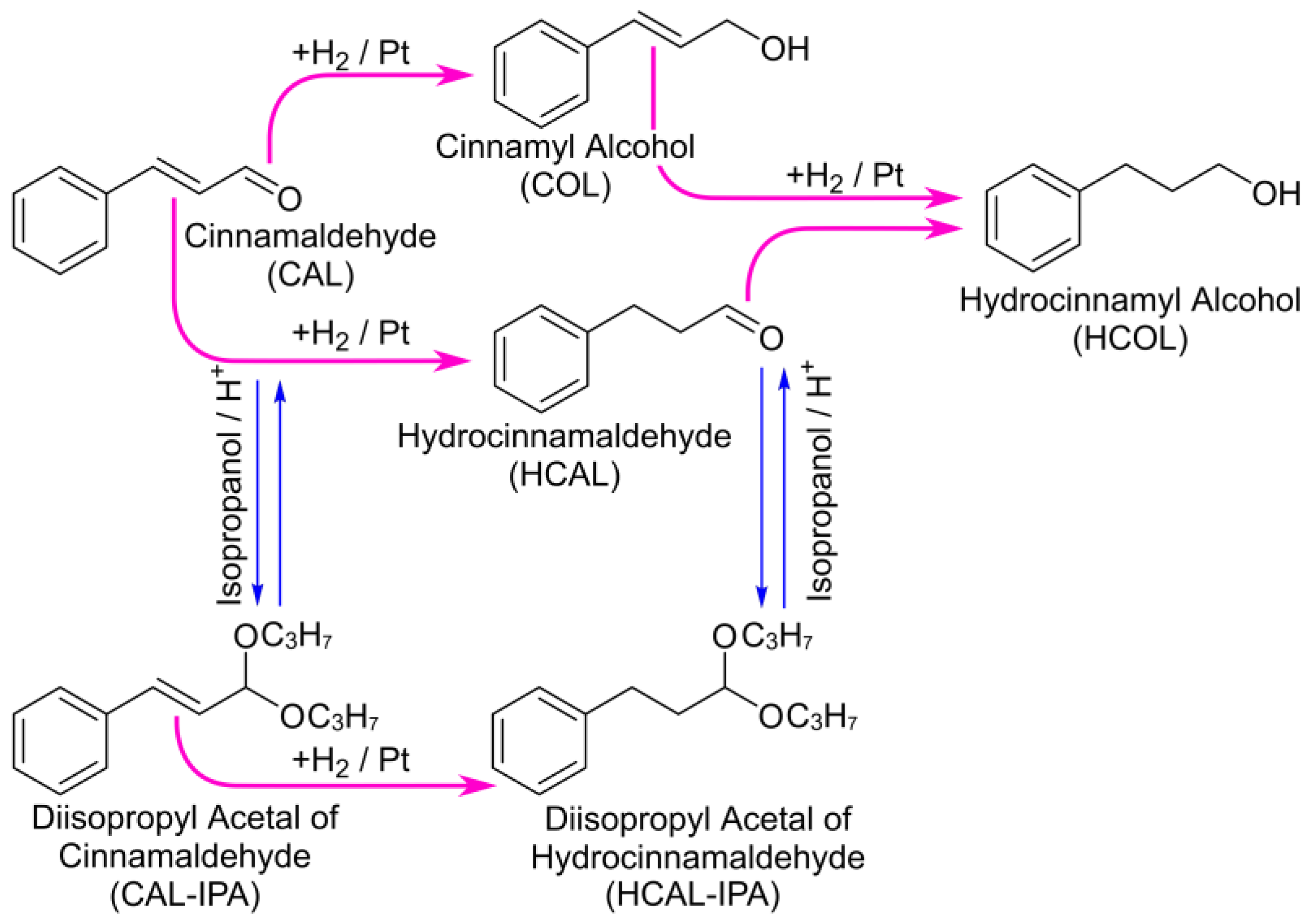
2.2. Hydrogenation of CAL over the Pt/SiO2 Catalysts in Batch and Tube Reactors
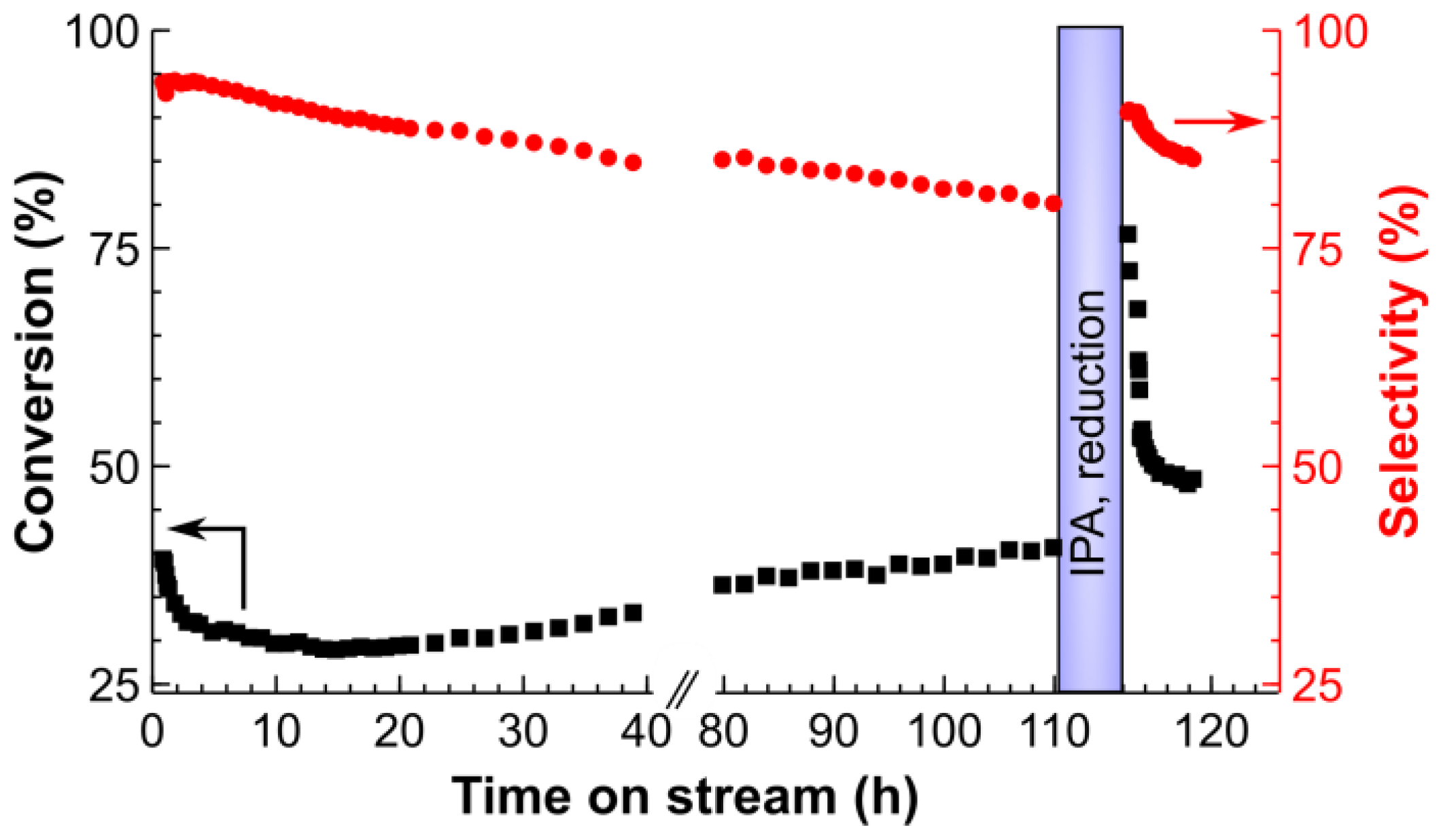
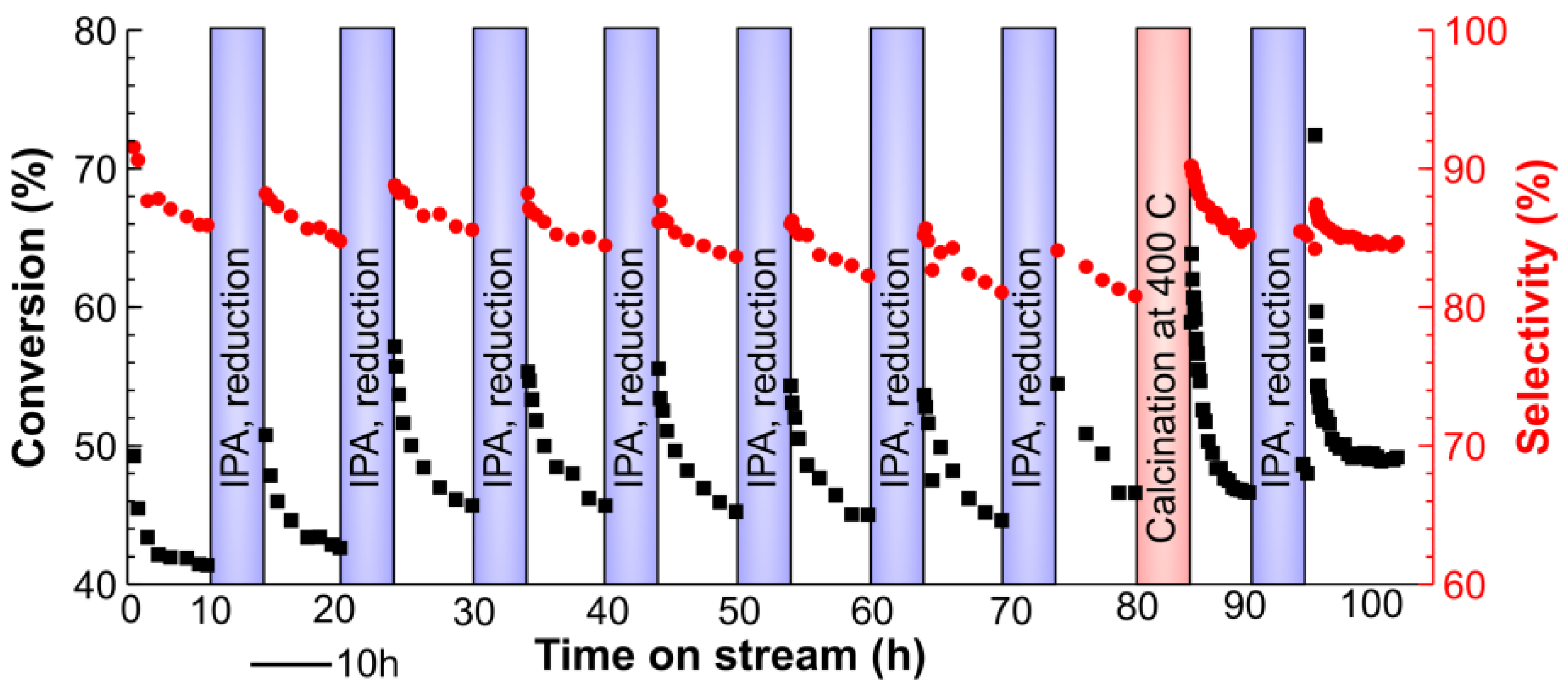
2.3. Long-Term Stability
3. Materials and Methods
3.1. Materials
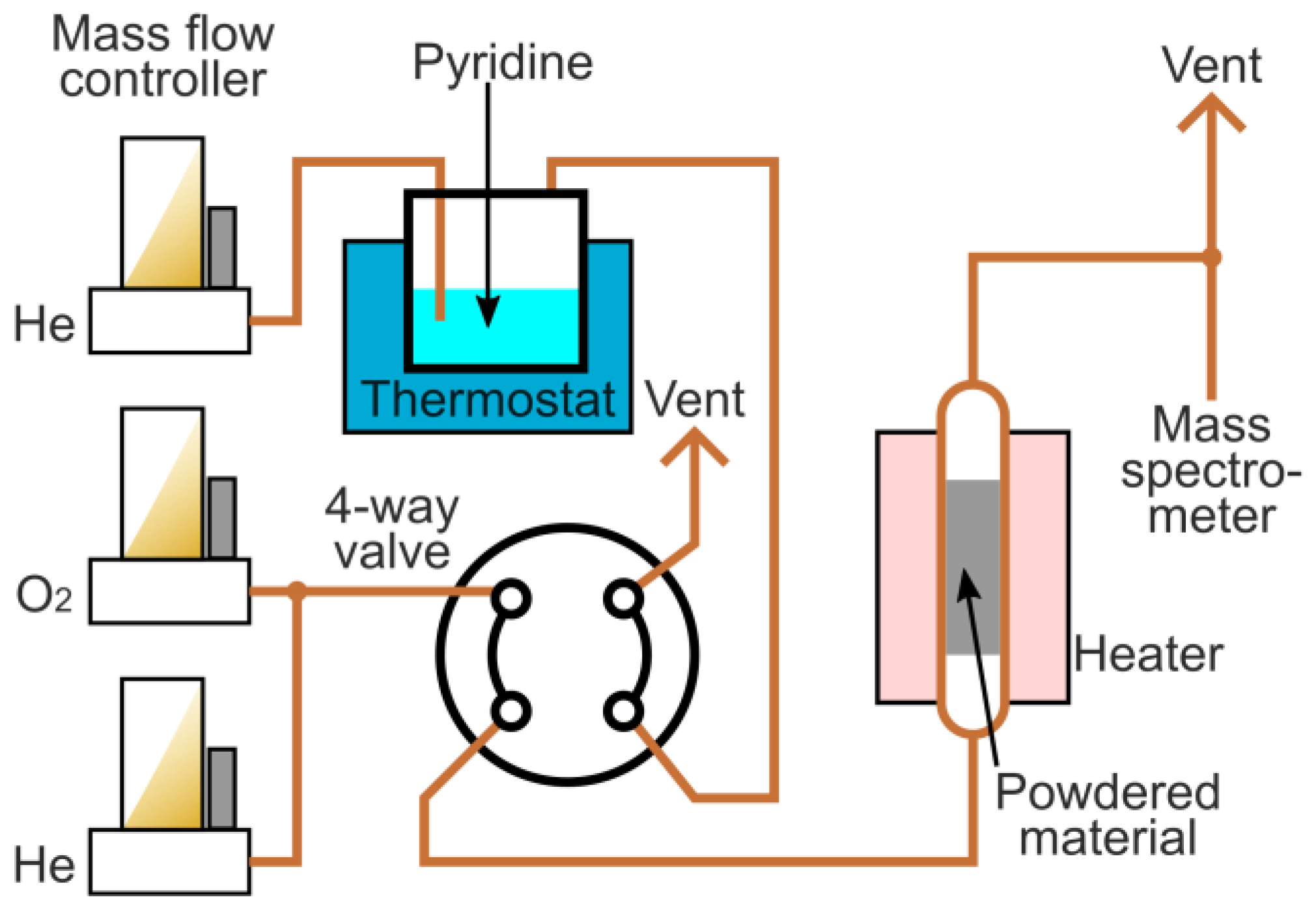
3.2. Materials Characterization
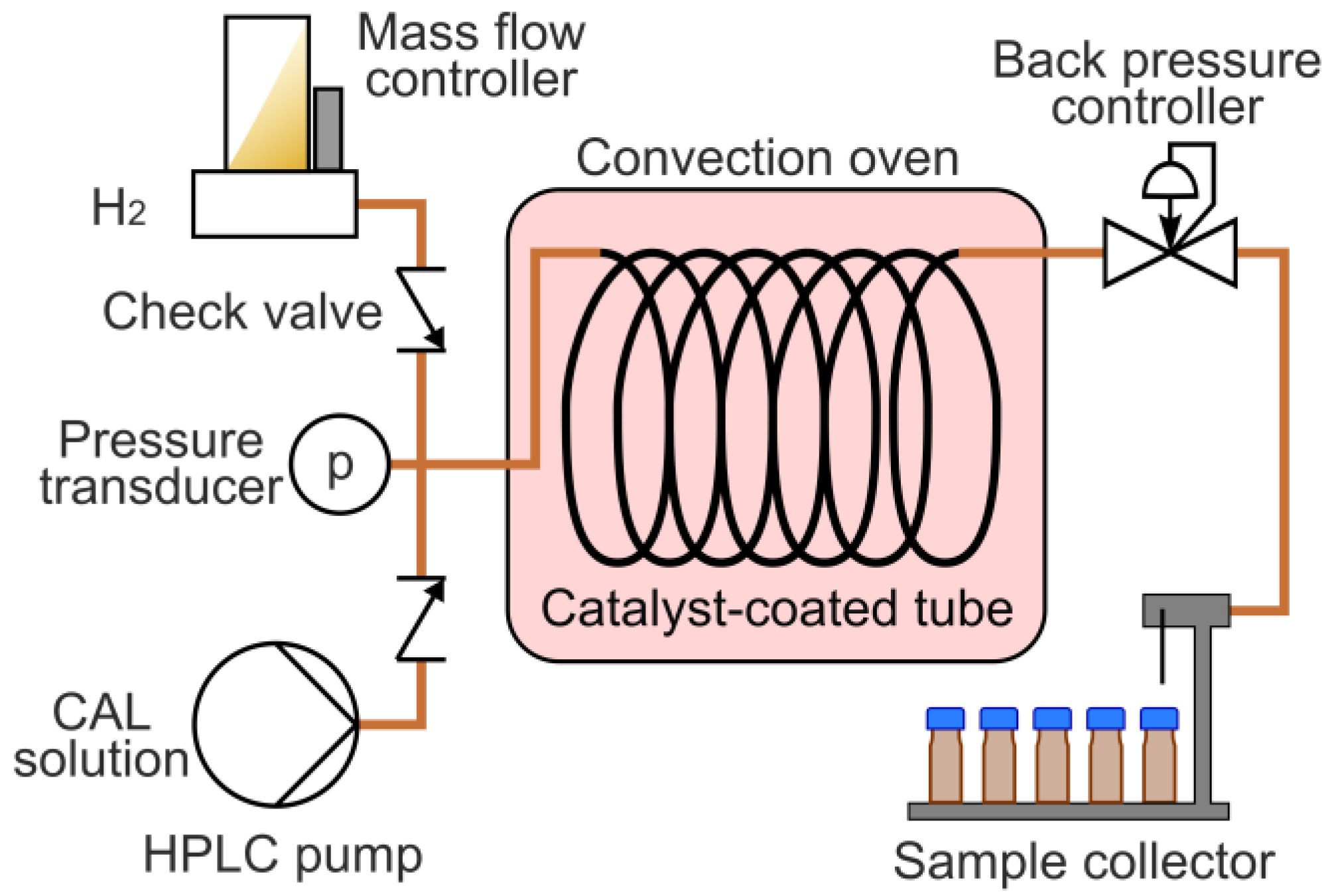
3.3. Hydrogenation in a Continuous Flow System
3.4. Hydrogenation in a Batch Reactor
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhang, Y.; Zhang, S.; Pan, X.; Bao, M.; Huang, J.; Shen, W. Selective Hydrogenation of Cinnamaldehyde to Cinnamyl Alcohol over Au Catalysts: Influence of the Oxide-Supports. Catal. Lett. 2017, 147, 102–109. [Google Scholar] [CrossRef]
- Yang, X.; Chen, D.; Liao, S.; Song, H.; Li, Y.; Fu, Z.; Su, Y. High-performance Pd-Au bimetallic catalyst with mesoporous silica nanoparticles as support and its catalysis of cinnamaldehyde hydrogenation. J. Catal. 2012, 291, 36–43. [Google Scholar] [CrossRef]
- Chen, S.; Meng, L.; Chen, B.; Chen, W.; Duan, X.; Huang, X.; Zhang, B.; Fu, H.; Wan, Y. Poison Tolerance to the Selective Hydrogenation of Cinnamaldehyde in Water over an Ordered Mesoporous Carbonaceous Composite Supported Pd Catalyst. ACS Catal. 2017. [Google Scholar] [CrossRef]
- Song, S.; Liu, X.; Li, J.; Pan, J.; Wang, F.; Xing, Y.; Wang, X.; Liu, X.; Zhang, H. Confining the Nucleation of Pt to In Situ Form (Pt-Enriched Cage)@CeO2 Core@Shell Nanostructure as Excellent Catalysts for Hydrogenation Reactions. Adv. Mater. 2017, 29, 1700495. [Google Scholar] [CrossRef] [PubMed]
- Plessers, E.; De Vos, D.E.; Roeffaers, M.B.J. Chemoselective reduction of α, β-unsaturated carbonyl compounds with UiO-66 materials. J. Catal. 2016, 340, 136–143. [Google Scholar] [CrossRef]
- Tian, Z.; Li, Q.; Hou, J.; Li, Y.; Ai, S. Highly selective hydrogenation of α,β-unsaturated aldehydes by Pt catalysts supported on Fe-based layered double hydroxides and derived mixed metal oxides. Catal. Sci. Technol. 2016, 6, 703–707. [Google Scholar] [CrossRef]
- Gallezot, P.; Richard, D. Selective Hydrogenation of α, β-Unsaturated Aldehydes. Catal. Rev. 1998, 40, 81–126. [Google Scholar] [CrossRef]
- Mäki-Arvela, P.; Hájek, J.; Salmi, T.; Murzin, D.Y. Chemoselective hydrogenation of carbonyl compounds over heterogeneous catalysts. Appl. Catal. A Gen. 2005, 292, 1–49. [Google Scholar] [CrossRef]
- Kumar, N.; Salmi, T.; Murzin, D.Y. Impact of Catalyst Reduction Mode on Selective Hydrogenation of Cinnamaldehyde over Ru-Sn Sol-Gel Catalysts. Ind. Eng. Chem. Res. 2003, 42, 295–305. [Google Scholar]
- Hájek, J.; Kumar, N.; Mäki-arvela, P.; Salmi, T.; Murzin, D.Y.; Paseka, I.; Heikkilä, T.; Laine, E.; Laukkanen, P.; Väyrynen, J. Ruthenium-modified MCM-41 mesoporous molecular sieve and Y zeolite catalysts for selective hydrogenation of cinnamaldehyde. Appl. Catal. A Gen. 2003, 251, 385–396. [Google Scholar] [CrossRef]
- Rebrov, E.V. Heterogeneous Catalysis in Microreactors. In Microreactors in Preparative Chemistry: Practical Aspects in Bioprocessing, Nanotechnology, Catalysis and More; John Wiley & Sons: Hoboken, NJ, USA, 2013; pp. 243–271. ISBN 9783527652891. [Google Scholar]
- Giroir-Fendler, A.; Richard, D.; Gallezot, P. Selectivity in cinnamaldehyde hydrogenation of group-VIII metals supported on graphite and carbon. Stud. Surf. Sci. Catal. 1988, 41, 171–178. [Google Scholar] [CrossRef]
- Claus, P. Selective hydrogenation of alpha,beta-unsaturated aldehydes and other C=O and C=C bonds containing compounds. Top. Catal. 1998, 5, 51–62. [Google Scholar] [CrossRef]
- Reyes, P.; Rodr, C. Promoting effect of Mo on the selective hydrogenation of cinnamaldehyde on Rh/SiO2 catalysts. Catal. Lett. 2000, 69, 27–32. [Google Scholar] [CrossRef]
- Liu, X.; Cheng, S.; Long, J.; Zhang, W.; Liu, X.; Wei, D. MOFs-Derived Co@CN bi-functional catalysts for selective transfer hydrogenation of α, β-unsaturated aldehydes without use of base additives. Mater. Chem. Front. 2017. [Google Scholar] [CrossRef]
- Coq, B.; Kumbhar, P.S.; Moreau, C.; Moreau, P.; Warawdekar, M.G. Liquid-Phase Hydrogenation of Cinnamaldehyde Over Supported Ruthenium Catalysts—Influence of Particle-Size, Bimetallics and Nature of Support. J. Mol. Catal. 1993, 85, 215–228. [Google Scholar] [CrossRef]
- Galvagno, S.; Donato, A.; Neri, G.; Pietropaolo, R.; Pietropaolo, D. Hydrogenation of cinnamaldehyde over platinum catalysts: Influence of addition of metal chlorides. J. Mol. Catal. 1989, 49, 223–232. [Google Scholar] [CrossRef]
- GaIvagno, S.; Poltarzewski, Z.; Donato, A.; Neri, G.; Pietropaolo, R. Selective hydrogenation of α,β-unsaturated aldehydes to give unsaturated alcohols over platinum–germanium catalysts. J. Chem. Soc. Commun. 1998, 5, 51–62. [Google Scholar]
- Zheng, Q.; Wang, D.; Yuan, F.; Han, Q.; Dong, Y.; Liu, Y.; Niu, X.; Zhu, Y. An Effective Co-promoted Platinum of Co–Pt/SBA-15 Catalyst for Selective Hydrogenation of Cinnamaldehyde to Cinnamyl Alcohol. Catal. Lett. 2016, 146, 1535–1543. [Google Scholar] [CrossRef]
- Shi, Y.-S.; Yuan, Z.-F.; Wei, Q.; Sun, K.-Q.; Xu, B.-Q. Pt–FeOx/SiO2 catalysts prepared by galvanic displacement show high selectivity for cinnamyl alcohol production in the chemoselective hydrogenation of cinnamaldehyde. Catal. Sci. Technol. 2016, 6, 7033. [Google Scholar] [CrossRef]
- Toebes, M.L.; Zhang, Y.; Hájek, J.; Alexander Nijhuis, T.; Bitter, J.H.; Jos Van Dillen, A.; Murzin, D.Y.; Koningsberger, D.C.; De Jong, K.P. Support effects in the hydrogenation of cinnamaldehyde over carbon nanofiber-supported platinum catalysts: Characterization and catalysis. J. Catal. 2004, 226, 215–225. [Google Scholar] [CrossRef]
- He, S.; Xie, L.; Che, M.; Chan, H.C.; Yang, L.; Shi, Z.; Tang, Y.; Gao, Q. Chemoselective hydrogenation of α, β-unsaturated aldehydes on hydrogenated MoOx nanorods supported iridium nanoparticles. J. Mol. Catal. A Chem. 2016, 425, 248–254. [Google Scholar] [CrossRef]
- Han, Q.; Liu, Y.; Wang, D.; Yuan, F.; Niu, X.; Zhu, Y. Effect of carbon nanosheets with different graphitization degrees as a support of noble metals on selective hydrogenation of cinnamaldehyde. RSC Adv. 2016, 6, 98356–98364. [Google Scholar] [CrossRef]
- Guo, Z.; Xiao, C.; Maligal-Ganesh, R.V.; Zhou, L.; Goh, T.W.; Li, X.; Tesfagaber, D.; Thiel, A.; Huang, W. Pt nanoclusters confined within metal-organic framework cavities for chemoselective cinnamaldehyde hydrogenation. ACS Catal. 2014, 4, 1340–1348. [Google Scholar] [CrossRef]
- van de Moesdijk, Cornelis G.M.; Bosma, Marcel A.R. Process for the Preparation of α-β Unsaturated Alcohols. U.S. Patent 4,745,234, 17 May 1988. [Google Scholar]
- Shirai, M.; Tanaka, T.; Arai, M. Selective hydrogenation of α, β-unsaturated aldehyde to unsaturated alcohol with supported platinum catalysts at high pressures of hydrogen. J. Mol. Catal. A Chem. 2001, 168, 99–103. [Google Scholar] [CrossRef]
- Roberge, D.M.; Ducry, L.; Bieler, N.; Cretton, P.; Zimmermann, B. Microreactor Technology: A Revolution for the Fine Chemical and Pharmaceutical Industries? Chem. Eng. Technol. 2005, 28, 318–323. [Google Scholar] [CrossRef]
- Wiles, C.; Watts, P. Continuous flow reactors: A perspective. Green Chem. 2012, 14, 38–54. [Google Scholar] [CrossRef]
- Hessel, V.; Kralisch, D.; Kockmann, N. Novel Process Windows: Innovative Gates to Intensified and Sustainable Chemical Processes; John Wiley & Sons: Hoboken, NJ, USA, 2014; ISBN 3527328580. [Google Scholar]
- Jähnisch, K.; Hessel, V.; Löwe, H.; Baerns, M. Chemistry in microstructured reactors. Angew. Chem. Int. Ed. 2004, 43, 406–446. [Google Scholar] [CrossRef] [PubMed]
- Durndell, L.J.; Wilson, K.; Lee, A.F. Platinum-catalysed cinnamaldehyde hydrogenation in continuous flow. RSC Adv. 2015, 5, 80022–80026. [Google Scholar] [CrossRef]
- Cherkasov, N.; Al-rawashdeh, M.; Ibhadon, A.O.; Rebrov, E.V. Scale up study of capillary microreactors in solvent-free semihydrogenation of 2-methyl-3-butyn-2-ol. Catal. Today 2016, 273, 205–212. [Google Scholar] [CrossRef]
- Vaccaro, L.; Lanari, D.; Marrocchi, A.; Strappaveccia, G. Flow approaches towards sustainability. Green Chem. 2014, 16, 3680. [Google Scholar] [CrossRef]
- Niesz, K.; Hornyak, I.; Borcsek, B.; Darvas, F. Nanoparticle synthesis completed with in situ catalyst preparation performed on a high-pressure high-temperature continuous flow reactor. Microfluid. Nanofluid. 2008, 5, 411–416. [Google Scholar] [CrossRef]
- Avril, A.; Hornung, C.H.; Urban, A.; Fraser, D.; Horne, M.; Veder, J.-P.; Tsanaktsidis, J.; Rodopoulos, T.; Henry, C.; Gunasegaram, D.R. Continuous flow hydrogenations using novel catalytic static mixers inside a tubular reactor. React. Chem. Eng. 2017, 2, 180–188. [Google Scholar] [CrossRef]
- Borodziński, A.; Bonarowska, M. Relation between Crystallite Size and Dispersion on Supported Metal Catalysts. Langmuir 1997, 13, 5613–5620. [Google Scholar] [CrossRef]
- Holbrook, B.P.M.; Rajagopalan, R.; Dronvajjala, K.; Choudhary, Y.K.; Foley, H.C. Molecular sieving carbon catalysts for liquid phase reactions: Study of alkene hydrogenation using platinum embedded nanoporous carbon. J. Mol. Catal. A Chem. 2013, 367, 61–68. [Google Scholar] [CrossRef]
- Su, J.; Chen, J.S. Synthetic porous materials applied in hydrogenation reactions. Microporous Mesoporous Mater. 2017, 237, 246–259. [Google Scholar] [CrossRef]
- Ma, Y.; Han, L.; Miyasaka, K.; Oleynikov, P.; Che, S.; Terasaki, O. Structural study of hexagonal close-packed silica mesoporous crystal. Chem. Mater. 2013, 25, 2184–2191. [Google Scholar] [CrossRef]
- Beaucage, G. Approximations Leading to a Unified Exponential/Power-Law Approach to Small-Angle Scattering. J. Appl. Crystallogr. 1995, 28, 717–728. [Google Scholar] [CrossRef]
- Ilavsky, J.; Jemian, P.R. Irena: Tool suite for modeling and analysis of small-angle scattering. J. Appl. Crystallogr. 2009, 42, 347–353. [Google Scholar] [CrossRef]
- Kataoka, T.; Dumesic, J.A. Acidity of unsupported and silica-supported vanadia, molybdena, and titania as studied by pyridine adsorption. J. Catal. 1988, 112, 66–79. [Google Scholar] [CrossRef]
- Cardona-Martínez, N.; Dumesic, J.A. Acid strength of silica-alumina and silica studied by microcalorimetric measurements of pyridine adsorption. J. Catal. 1990, 125, 427–444. [Google Scholar] [CrossRef]
- Dang, Z.; Anderson, B.G.; Amenomiya, Y.; Morrow, B.A. Silica-supported zirconia. 1. Characterization by infrared spectroscopy, temperature-programmed desorption, and X-ray diffraction. J. Phys. Chem. 1995, 99, 14437–14443. [Google Scholar] [CrossRef]
- Wallin, M.; Grönbeck, H.; Spetz, A.L.; Eriksson, M.; Skoglundh, M. Vibrational analysis of H2 and D2 adsorption on Pt/SiO2. J. Phys. Chem. B 2005, 109, 9581–9588. [Google Scholar] [CrossRef] [PubMed]
- Regenhardt, S.A.; Trasarti, A.F.; Meyer, C.I.; Garetto, T.F.; Marchi, A.J. Selective gas-phase conversion of maleic anhydride to propionic acid on Pt-based catalysts. Catal. Commun. 2013, 35, 59–63. [Google Scholar] [CrossRef]
- Santiago-Pedro, S.; Tamayo-Galván, V.; Viveros-García, T. Effect of the acid-base properties of the support on the performance of Pt catalysts in the partial hydrogenation of citral. Catal. Today 2013, 213, 101–108. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, C.; Gong, W.; Song, J.; Su, Y.; Zhang, H.; Wang, G.; Zhao, H. One-pot redox synthesis of Pt/Fe3O4 catalyst for efficiently chemoselective hydrogenation of cinnamaldehyde. RSC Adv. 2017, 7, 21107–21113. [Google Scholar] [CrossRef]
- Wang, D.; Zhu, Y.; Tian, C.; Wang, L.; Zhou, W.; Dong, Y.; Han, Q.; Liu, Y.; Yuan, F.; Fu, H. Synergistic effect of Mo2N and Pt for promoted selective hydrogenation of cinnamaldehyde over Pt–Mo2 N/SBA-15. Catal. Sci. Technol. 2016, 6, 2403–2412. [Google Scholar] [CrossRef]
- Hu, Q.; Wang, S.; Gao, Z.; Li, Y.; Zhang, Q.; Xiang, Q.; Qin, Y. The precise decoration of Pt nanoparticles with Fe oxide by atomic layer deposition for the selective hydrogenation of cinnamaldehyde. Appl. Catal. B Environ. 2017, 218, 591–599. [Google Scholar] [CrossRef]
- Zhu, Y.; Zaera, F. Selectivity in the catalytic hydrogenation of cinnamaldehyde promoted by Pt/SiO2 as a function of metal nanoparticle size. Catal. Sci. Technol. 2014, 4, 955–962. [Google Scholar] [CrossRef]
- Garkhedkar, A.M.; Shingote, S.K.; Rane, V.H.; Kelkar, A.A.; Ranade, V.V. Intensifying Hydrogenation of Cinnamaldehyde to Cinnamyl Alcohol: Catalyst, Solvent and Operating Conditions. Indian Chem. Eng. 2015, 57, 219–239. [Google Scholar] [CrossRef]
- Shi, J.; Zhang, M.; Du, W.; Ning, W.; Hou, Z. SnO2-isolated Pt3Sn alloy on reduced graphene oxide: An efficient catalyst for selective hydrogenation of C=O in unsaturated aldehydes†. Catal. Sci. Technol. 2015, 5, 3108–3112. [Google Scholar] [CrossRef]
- Koo-Amornpattana, W.; Winterbottom, J.M. Pt and Pt-alloy catalysts and their properties for the liquid-phase hydrogenation of cinnamaldehyde. Catal. Today 2001, 66, 277–287. [Google Scholar] [CrossRef]
- Chatterjee, M.; Ikushima, Y.; Zhao, F.Y. Highly efficient hydrogenation of cinnamaldehyde catalyzed by Pt-MCM-48 in supercritical carbon dioxide. Catal. Lett. 2002, 82, 141–144. [Google Scholar] [CrossRef]
- Chatterjee, M.; Ikushima, Y.; Zhao, F. Completely selective hydrogenation of trans-cinnamaldehyde to cinnamyl alcohol promoted by a Ru-Pt bimetallic catalyst supported on MCM-48 in supercritical carbon dioxide. New J. Chem. 2002, 27, 510–513. [Google Scholar] [CrossRef]
- Shokouhimehr, M. Magnetically Separable and Sustainable Nanostructured Catalysts for Heterogeneous Reduction of Nitroaromatics. Catalysts 2015, 5, 534–560. [Google Scholar] [CrossRef]
- Cherkasov, N.; Jadvani, V.; Mann, J.; Losovyj, Y.B.; Shifrina, Z.B.; Bronstein, L.M.; Rebrov, E.V. Hydrogenation of bio-oil into higher alcohols over Ru/Fe3O4-SiO2 catalysts. Fuel Process. Technol. 2017, 167, 738–746. [Google Scholar] [CrossRef]
- Mahmoud, S.; Hammoudeh, A.; Gharaibeh, S.; Melsheimer, J. Hydrogenation of cinnamaldehyde over sol-gel Pd/SiO2 catalysts: Kinetic aspects and modification of catalytic properties by Sn, Ir and Cu additives. J. Mol. Catal. A Chem. 2002, 178, 161–167. [Google Scholar] [CrossRef]
- Yue, J. Multiphase flow processing in microreactors combined with heterogeneous catalysis for efficient and sustainable chemical synthesis. Catal. Today 2017, 1–17. [Google Scholar] [CrossRef]
- Tanimu, A.; Jaenicke, S.; Alhooshani, K. Heterogeneous catalysis in continuous flow microreactors: A review of methods and applications. Chem. Eng. J. 2017, 327, 792–821. [Google Scholar] [CrossRef]
- Van Herk, D.; Castaño, P.; Makkee, M.; Moulijn, J.A.; Kreutzer, M.T. Catalyst testing in a multiple-parallel, gas–liquid, powder-packed bed microreactor. Appl. Catal. A Gen. 2009, 365, 199–206. [Google Scholar] [CrossRef]
- Al-Rawashdeh, M.; Yu, F.; Nijhuis, T.A.; Rebrov, E.V.; Hessel, V.; Schouten, J.C. Numbered-up gas-liquid micro/milli channels reactor with modular flow distributor. Chem. Eng. J. 2012, 207–208, 645–655. [Google Scholar] [CrossRef] [Green Version]
- Al-Rawashdeh, M.M.; Yue, F.; Patil, N.G.; Nijhuis, T.A.; Hessel, V.; Schouten, J.C.; Rebrov, E.V. Designing Flow and Temperature Uniformities in Parallel Microchannels Reactor Ma’moun. AIChE J. 2014, 60, 1941–1952. [Google Scholar] [CrossRef]
- Cherkasov, N.; Ibhadon, A.O.; Rebrov, E.V. Method of Forming a Coating. WO2017220590, 20 June 2017. [Google Scholar]
- Cherkasov, N.; Rebrov, E.V. Updated Catalyst Coating Method for Use in Flow Fine Chemical Synthesis. UK Patent Application No. 17 150 86.3, 19 September 2017. [Google Scholar]
- Cherkasov, N.; Ibhadon, A.O.; Rebrov, E.V. Novel synthesis of thick wall coatings of titania supported Bi poisoned Pd catalysts and application in selective hydrogenation of acetylene alcohols in capillary microreactors. Lab Chip 2015, 15, 1952–1960. [Google Scholar] [CrossRef] [PubMed]
- Cherkasov, N.; Ibhadon, A.O.; McCue, A.; Anderson, J.A.; Johnston, S.K. Palladium–bismuth intermetallic and surface-poisoned catalysts for the semi-hydrogenation of 2-methyl-3-butyn-2-ol. Appl. Catal. A Gen. 2015, 497, 22–30. [Google Scholar] [CrossRef]
- Bai, Y.; Li, W.; Liu, C.; Yang, Z.; Feng, X.; Lu, X.; Chan, K.-Y. Stability of Pt nanoparticles and enhanced photocatalytic performance in mesoporous Pt-(anatase/TiO2(B)) nanoarchitecture. J. Mater. Chem. 2009, 19, 7055. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Vaittinen, O.; Metsälä, M.; Persijn, S.; Vainio, M.; Halonen, L. Adsorption of ammonia on treated stainless steel and polymer surfaces. Appl. Phys. B 2014, 115, 185–196. [Google Scholar] [CrossRef]
- Cherkasov, N.; Bai, Y.; Rebrov, E. Process Intensification of Alkynol Semihydrogenation in a Tube Reactor Coated with a Pd/ZnO Catalyst. Catalysts 2017, 7, 358. [Google Scholar] [CrossRef]
- Cherkasov, N.; Ibhadon, A.O.; Rebrov, E.V. Solvent-free Semihydrogenation of Acetylene Alcohols in a Capillary Reactor coated with a Pd-Bi/TiO2 Catalyst. Appl. Catal. A Gen. 2016, 515, 108–115. [Google Scholar] [CrossRef]

















© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, Y.; Cherkasov, N.; Huband, S.; Walker, D.; Walton, R.I.; Rebrov, E. Highly Selective Continuous Flow Hydrogenation of Cinnamaldehyde to Cinnamyl Alcohol in a Pt/SiO2 Coated Tube Reactor. Catalysts 2018, 8, 58. https://doi.org/10.3390/catal8020058
Bai Y, Cherkasov N, Huband S, Walker D, Walton RI, Rebrov E. Highly Selective Continuous Flow Hydrogenation of Cinnamaldehyde to Cinnamyl Alcohol in a Pt/SiO2 Coated Tube Reactor. Catalysts. 2018; 8(2):58. https://doi.org/10.3390/catal8020058
Chicago/Turabian StyleBai, Yang, Nikolay Cherkasov, Steven Huband, David Walker, Richard I. Walton, and Evgeny Rebrov. 2018. "Highly Selective Continuous Flow Hydrogenation of Cinnamaldehyde to Cinnamyl Alcohol in a Pt/SiO2 Coated Tube Reactor" Catalysts 8, no. 2: 58. https://doi.org/10.3390/catal8020058
APA StyleBai, Y., Cherkasov, N., Huband, S., Walker, D., Walton, R. I., & Rebrov, E. (2018). Highly Selective Continuous Flow Hydrogenation of Cinnamaldehyde to Cinnamyl Alcohol in a Pt/SiO2 Coated Tube Reactor. Catalysts, 8(2), 58. https://doi.org/10.3390/catal8020058