In Situ UV-Visible Assessment of Iron-Based High-Temperature Water-Gas Shift Catalysts Promoted with Lanthana: An Extent of Reduction Study



Abstract
:1. Introduction
2. Results and Discussion
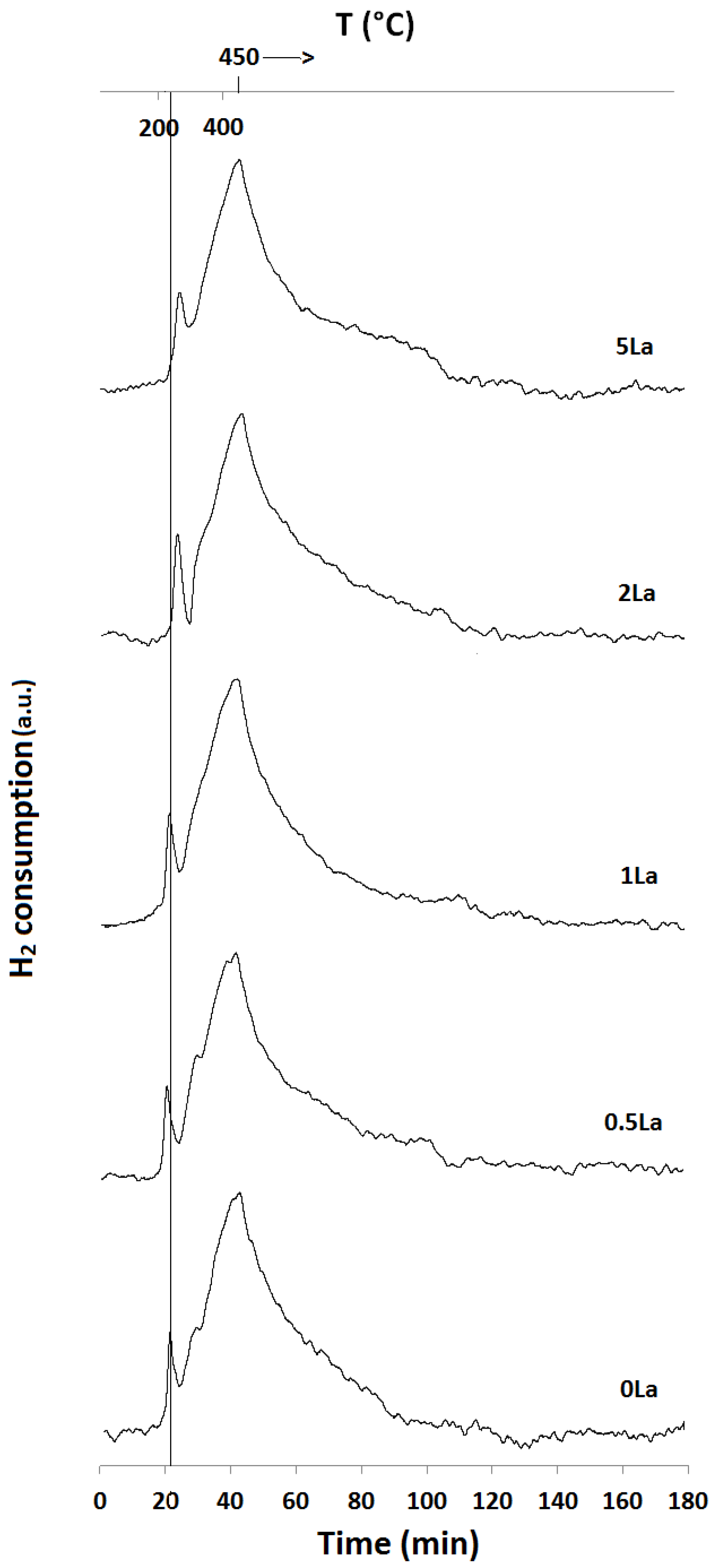
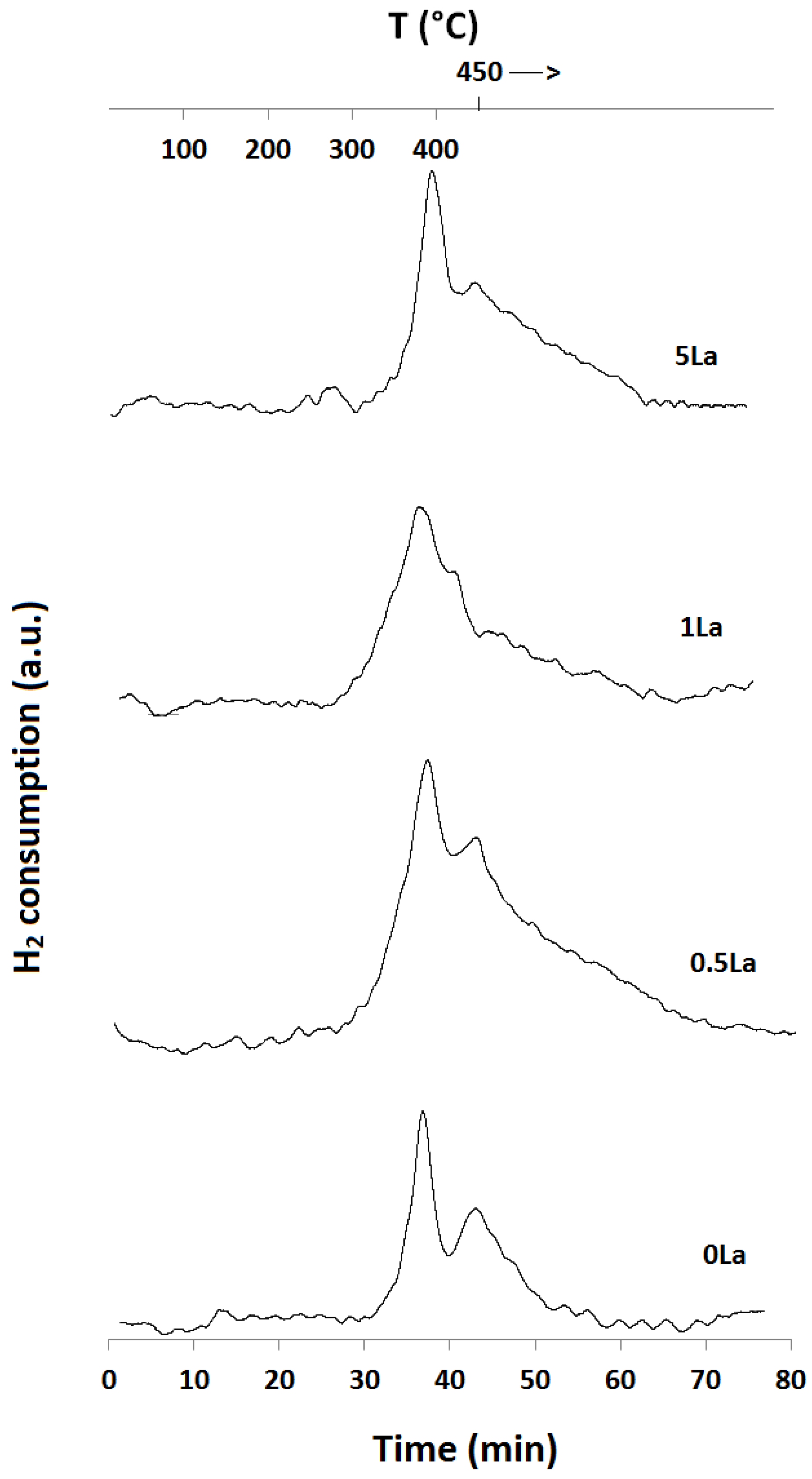
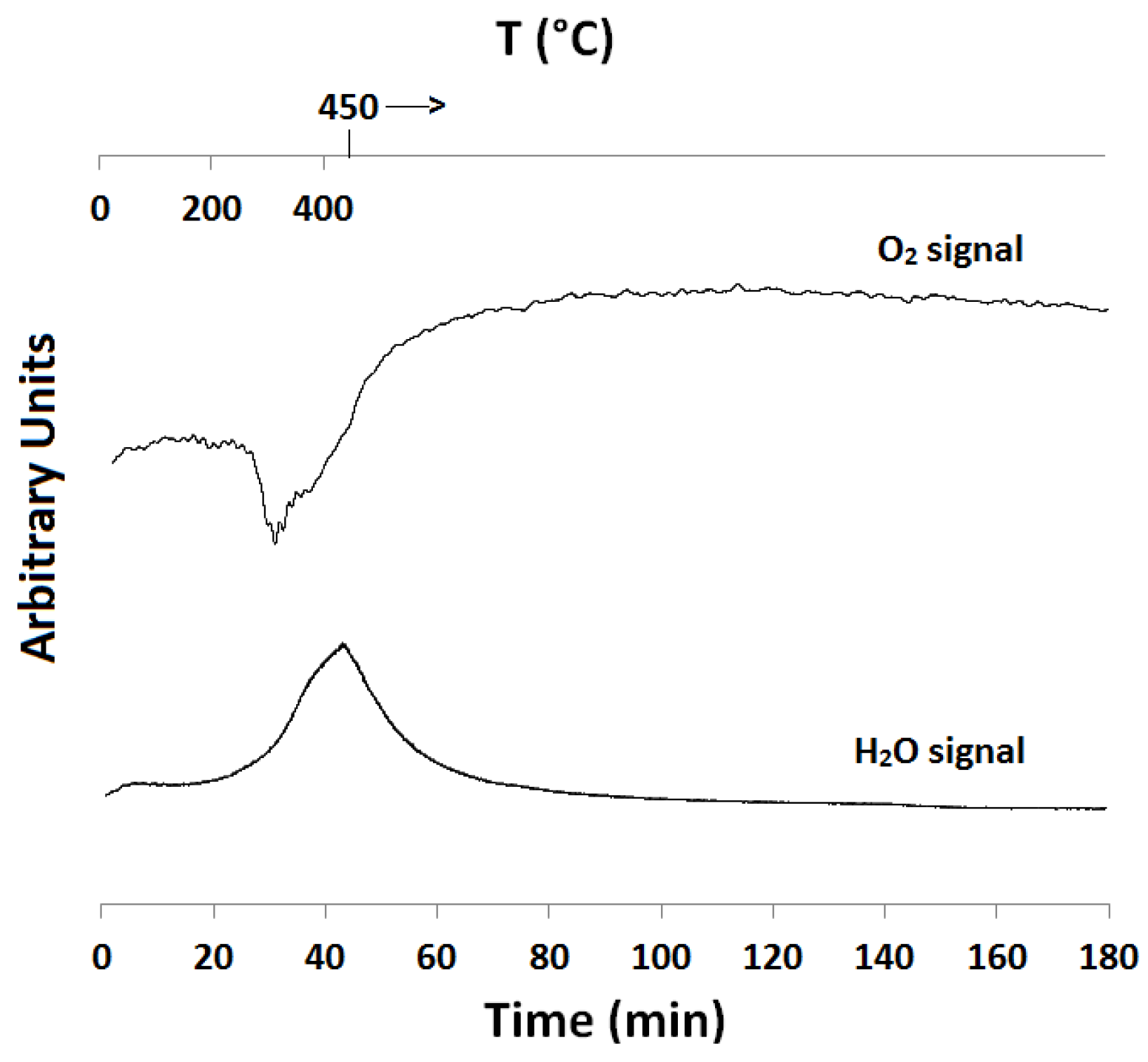
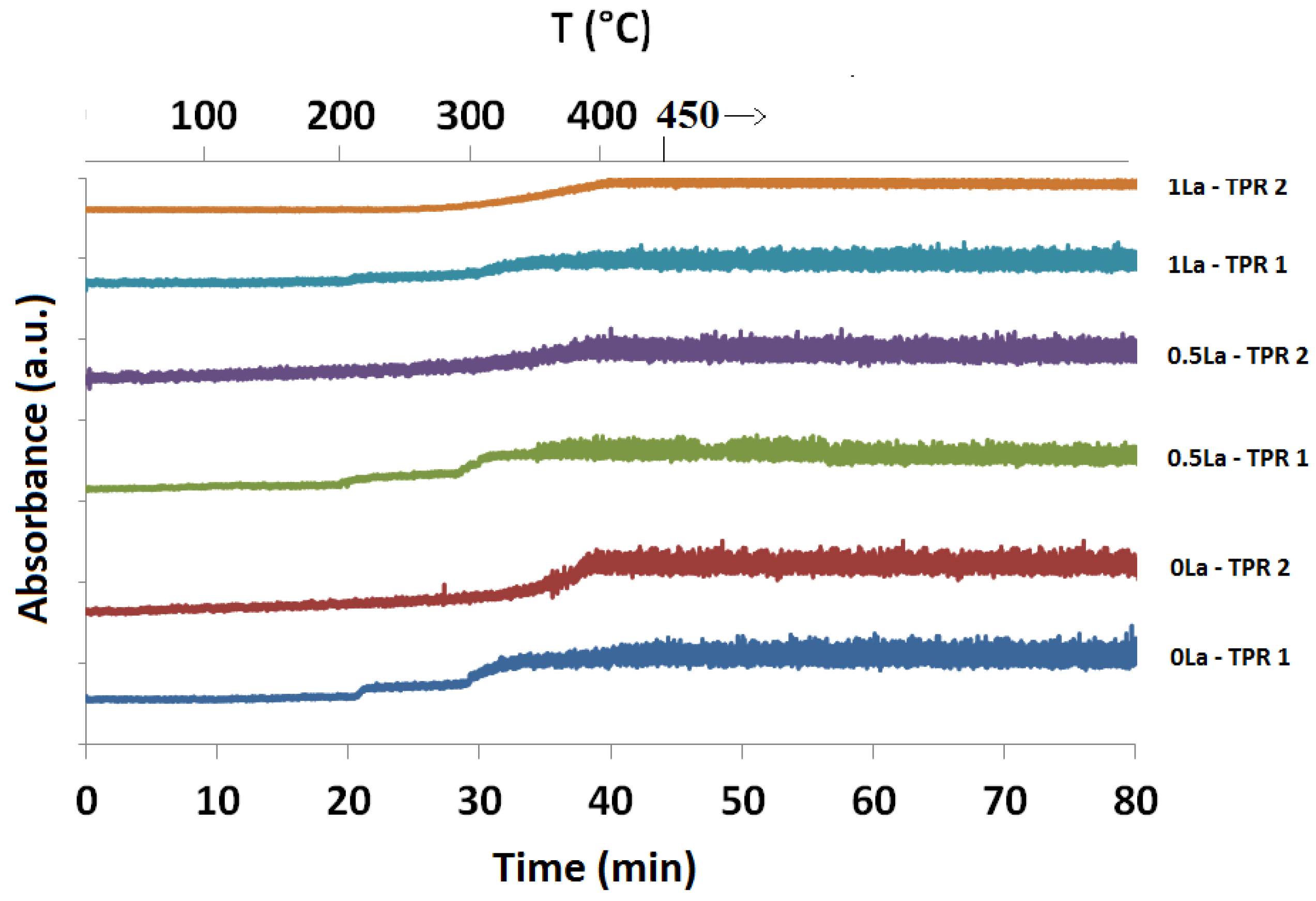
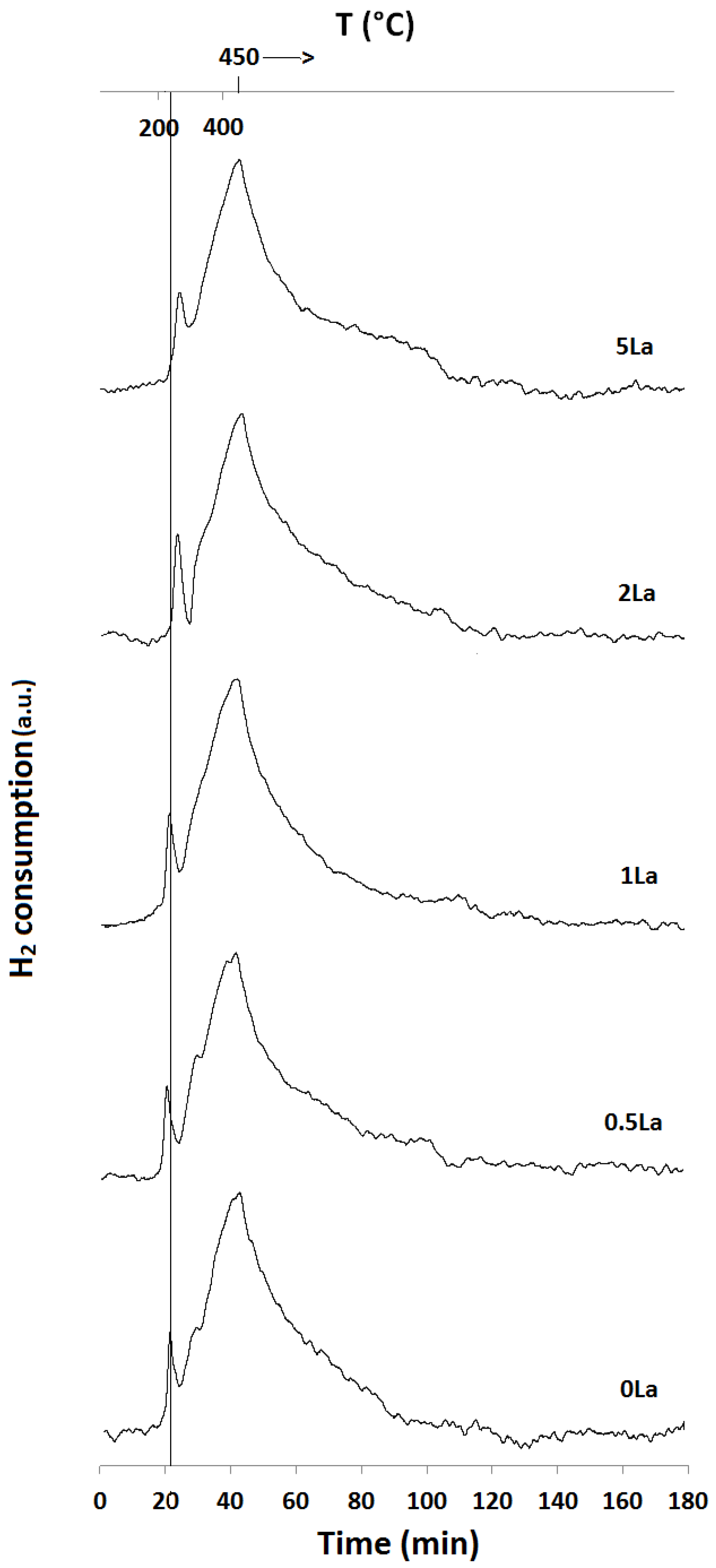
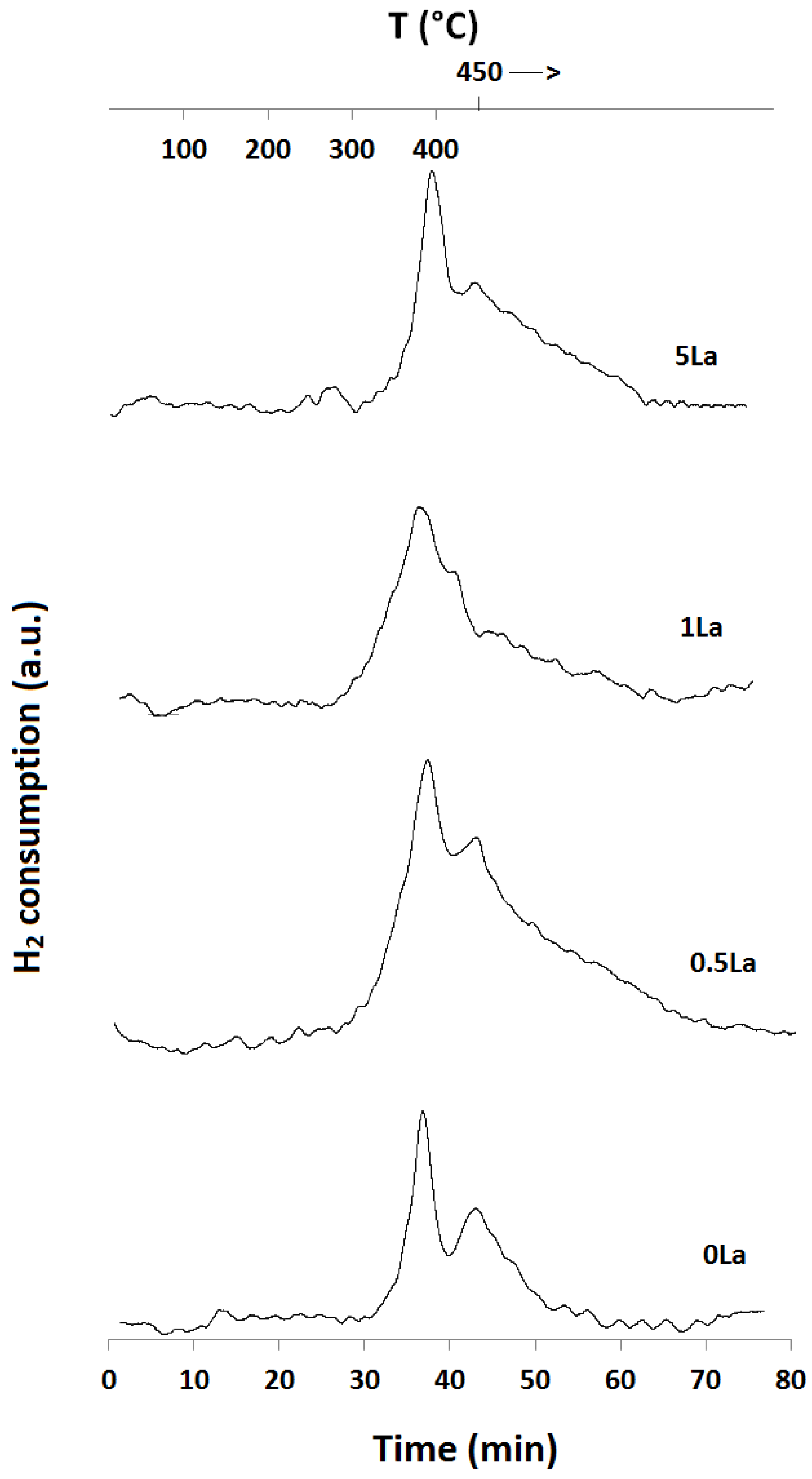
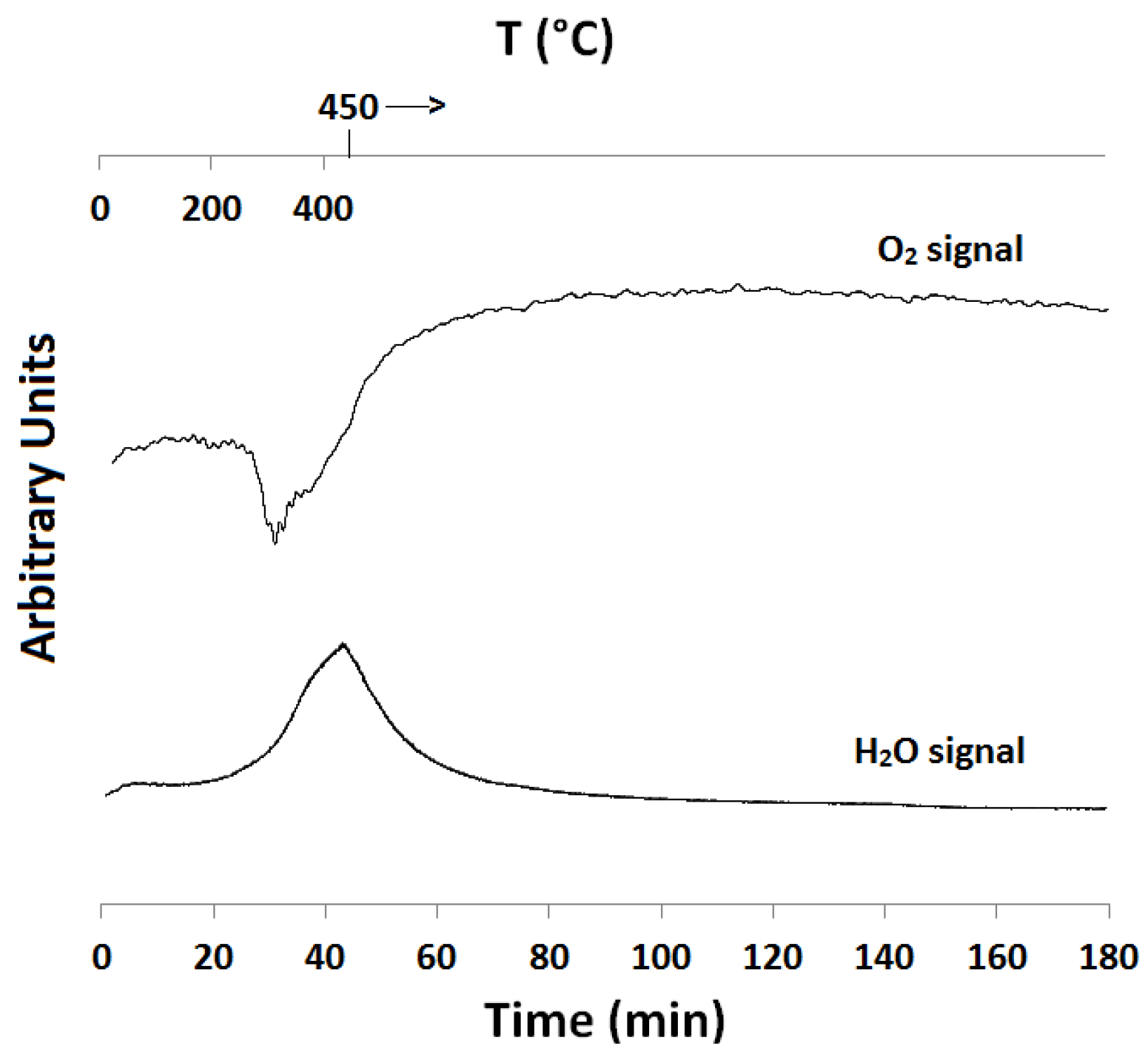
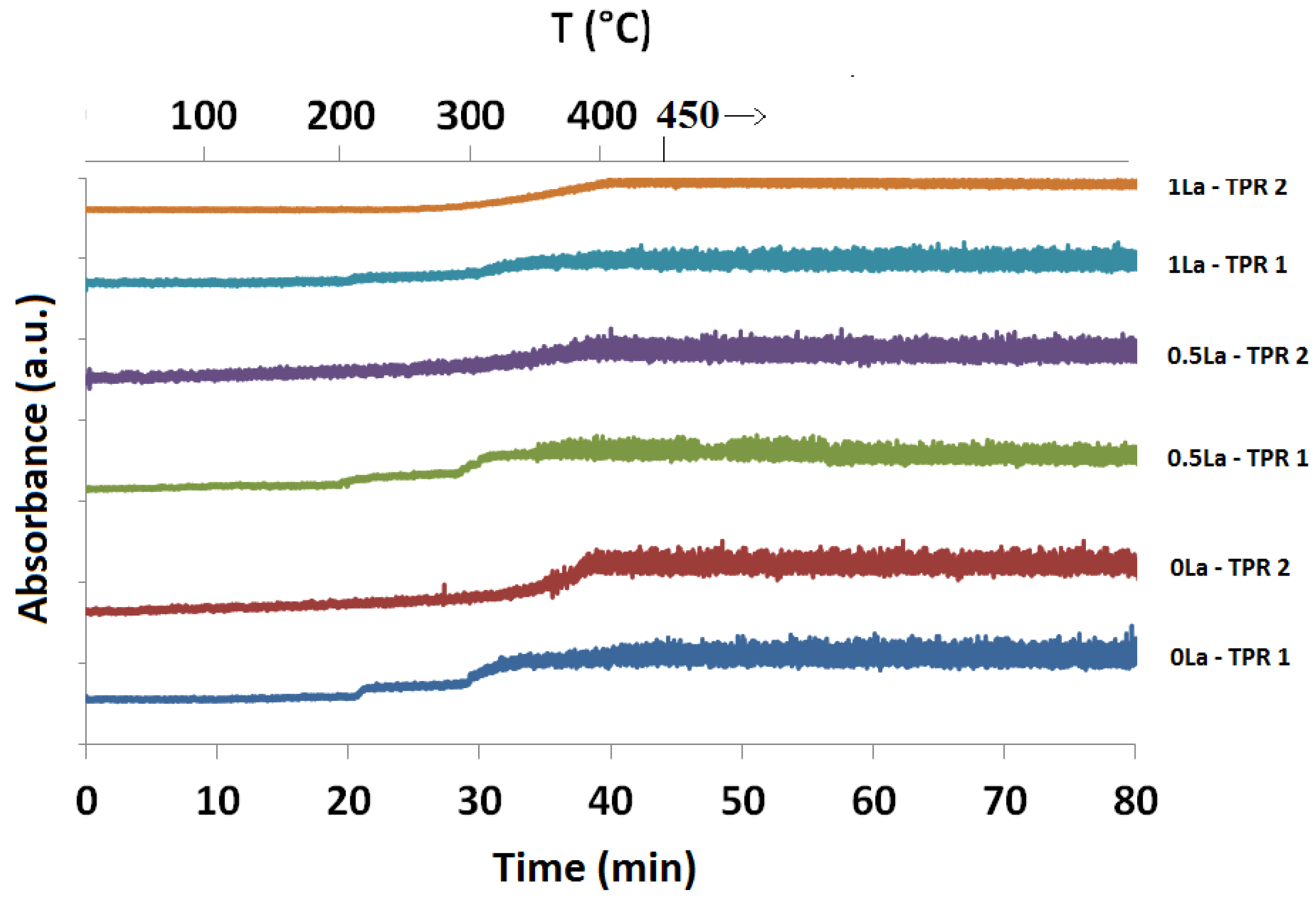
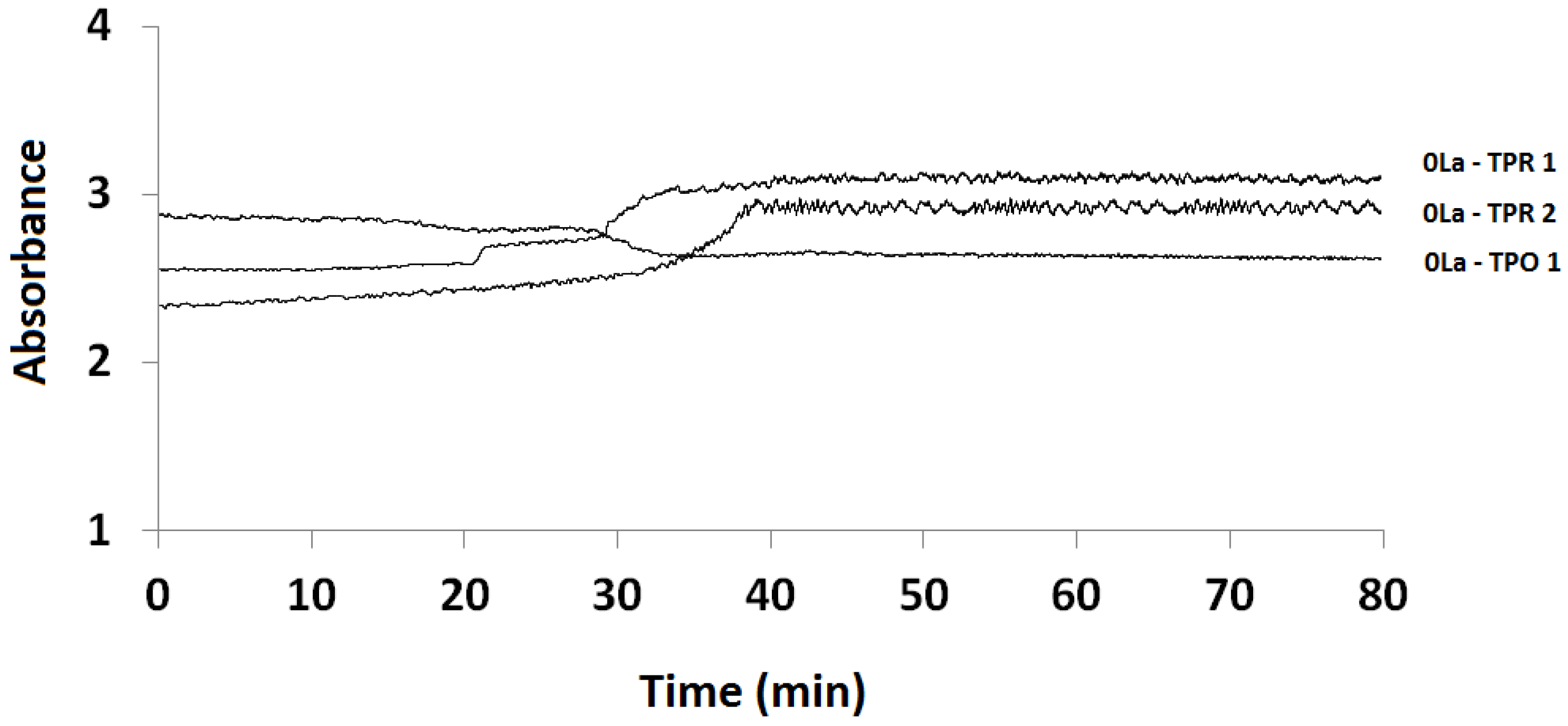
2.1. Temperature-Programmed Reduction and Oxidation (TPR and TPO)
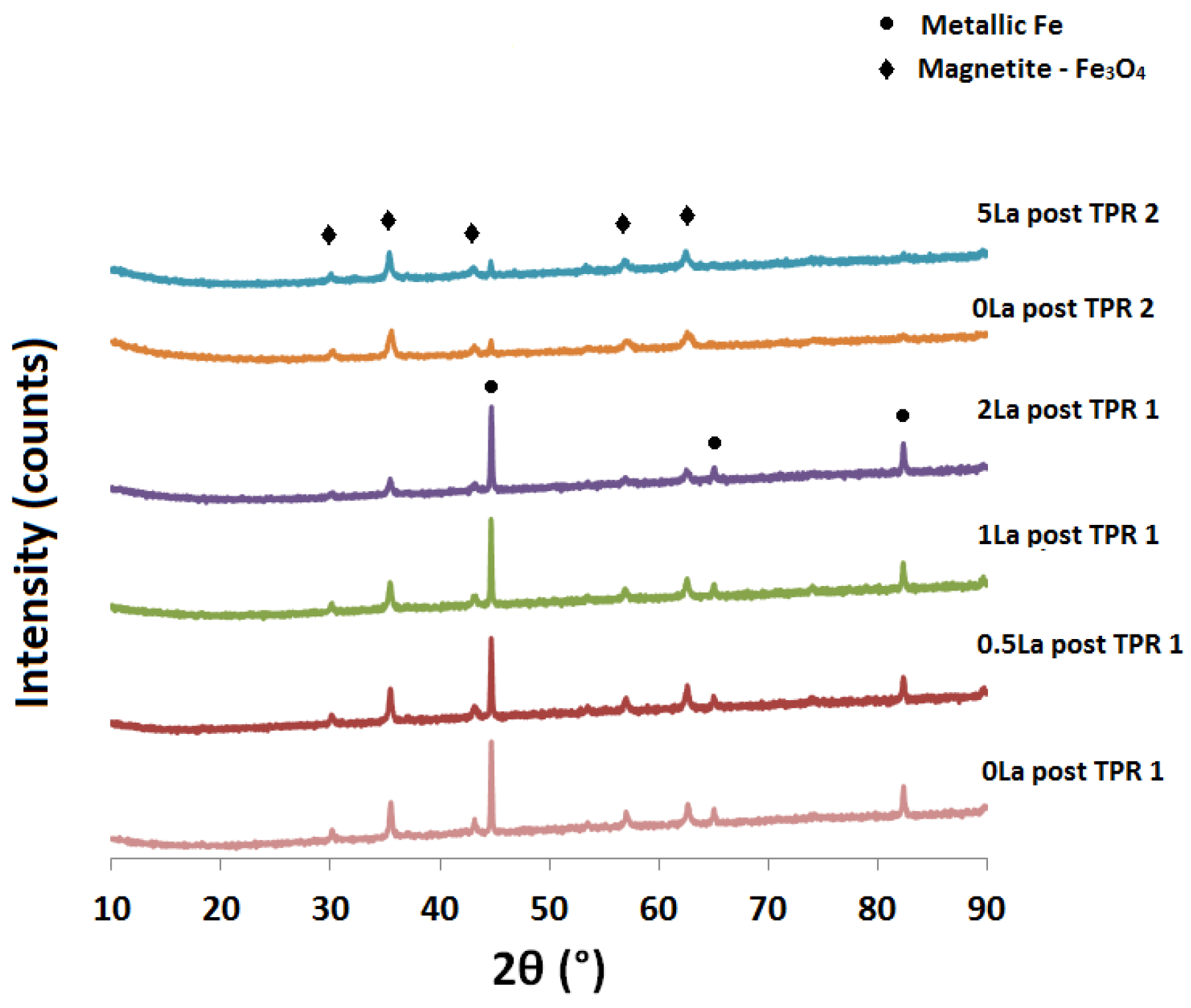
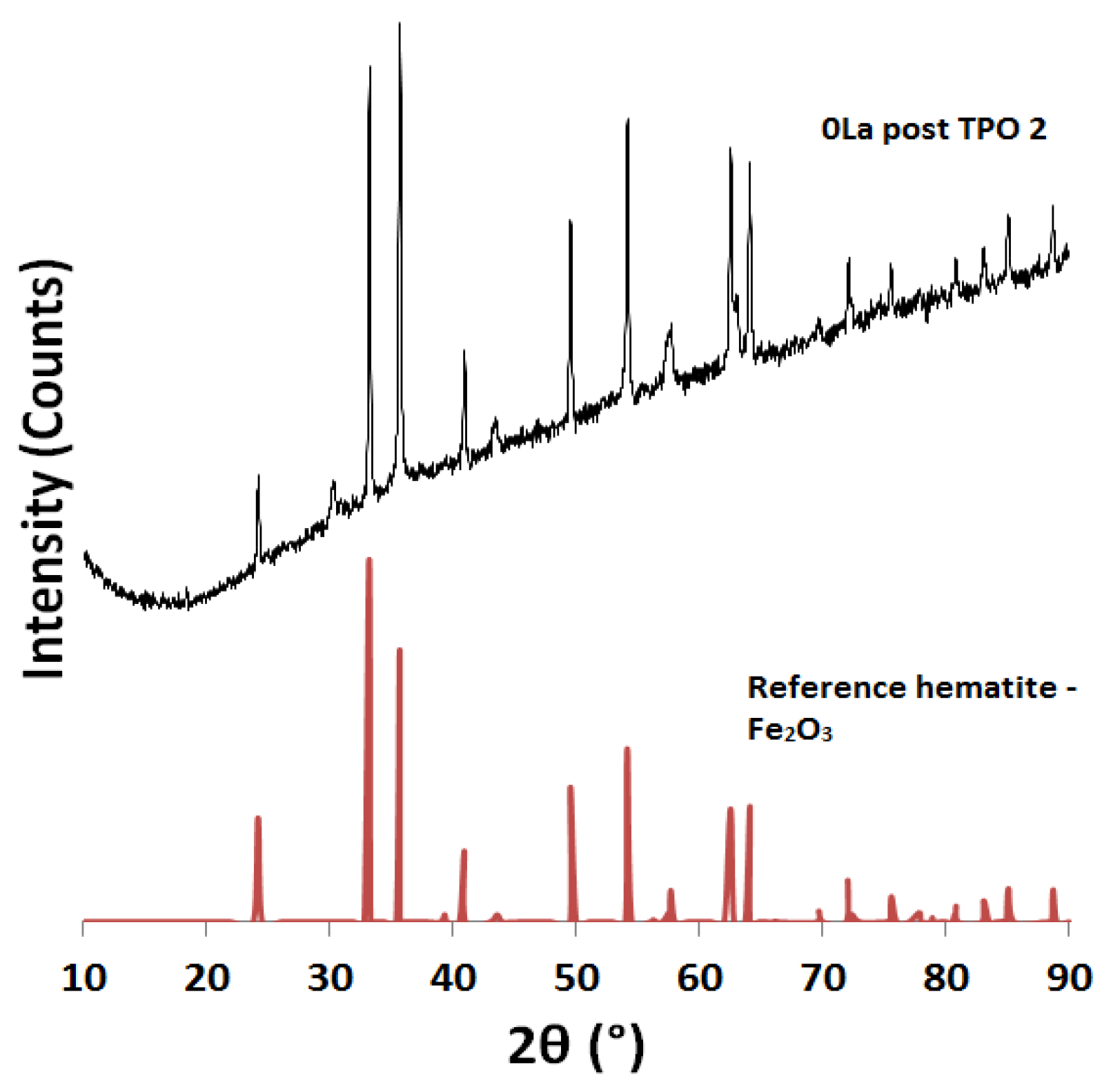
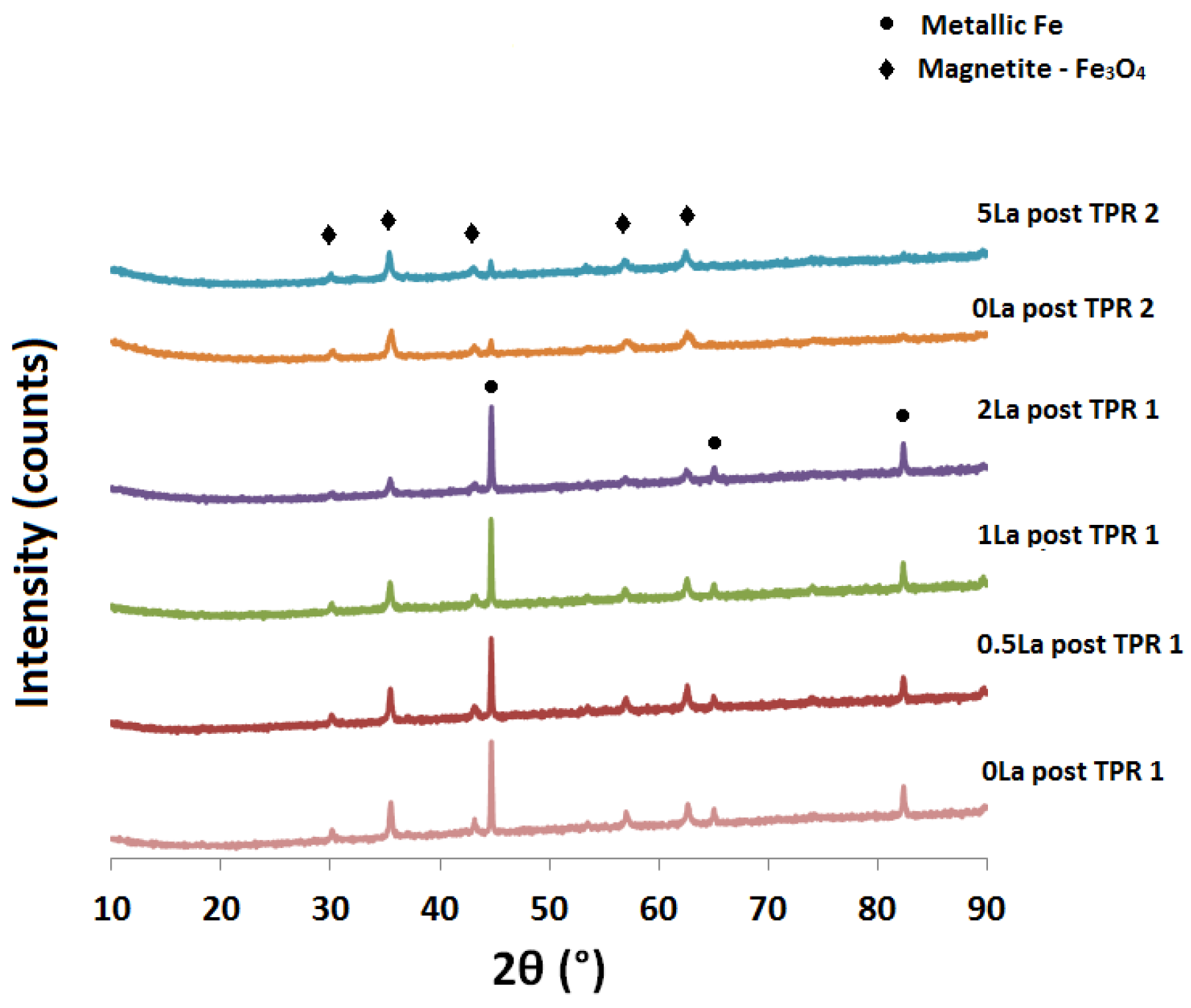
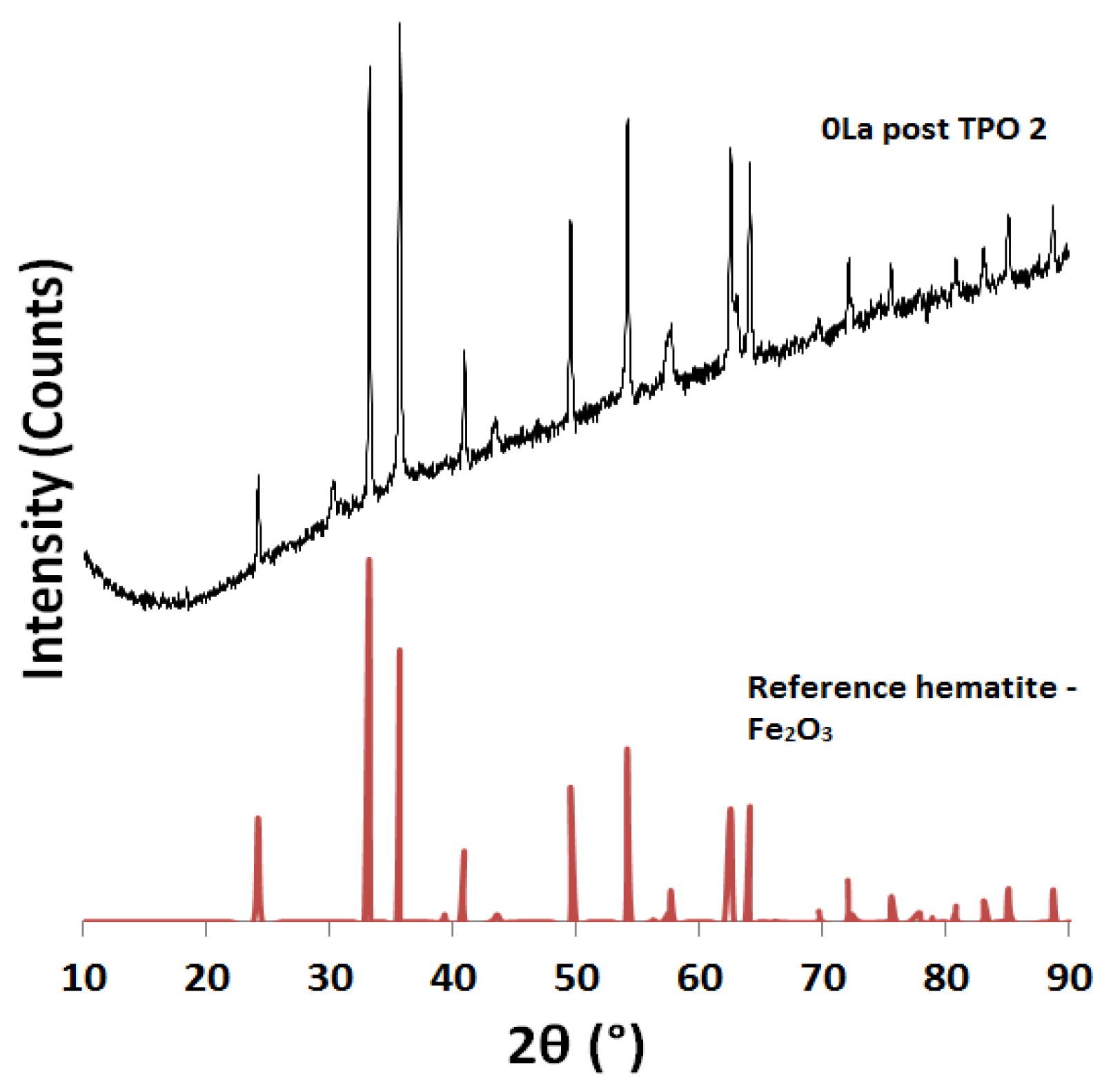
2.2. Powder X-ray Diffraction (XRD) Analysis
2.3. In Situ UV-Visible Spectroscopy
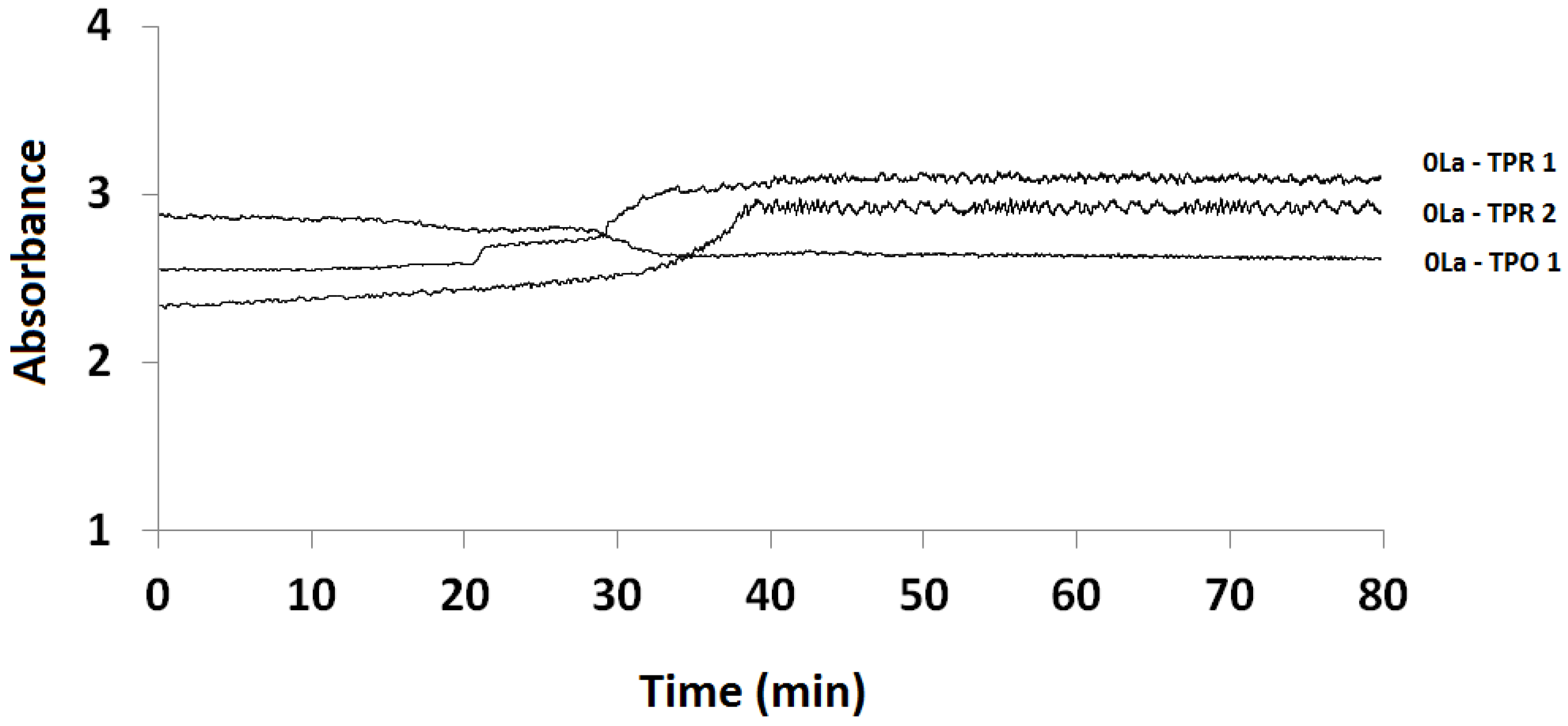
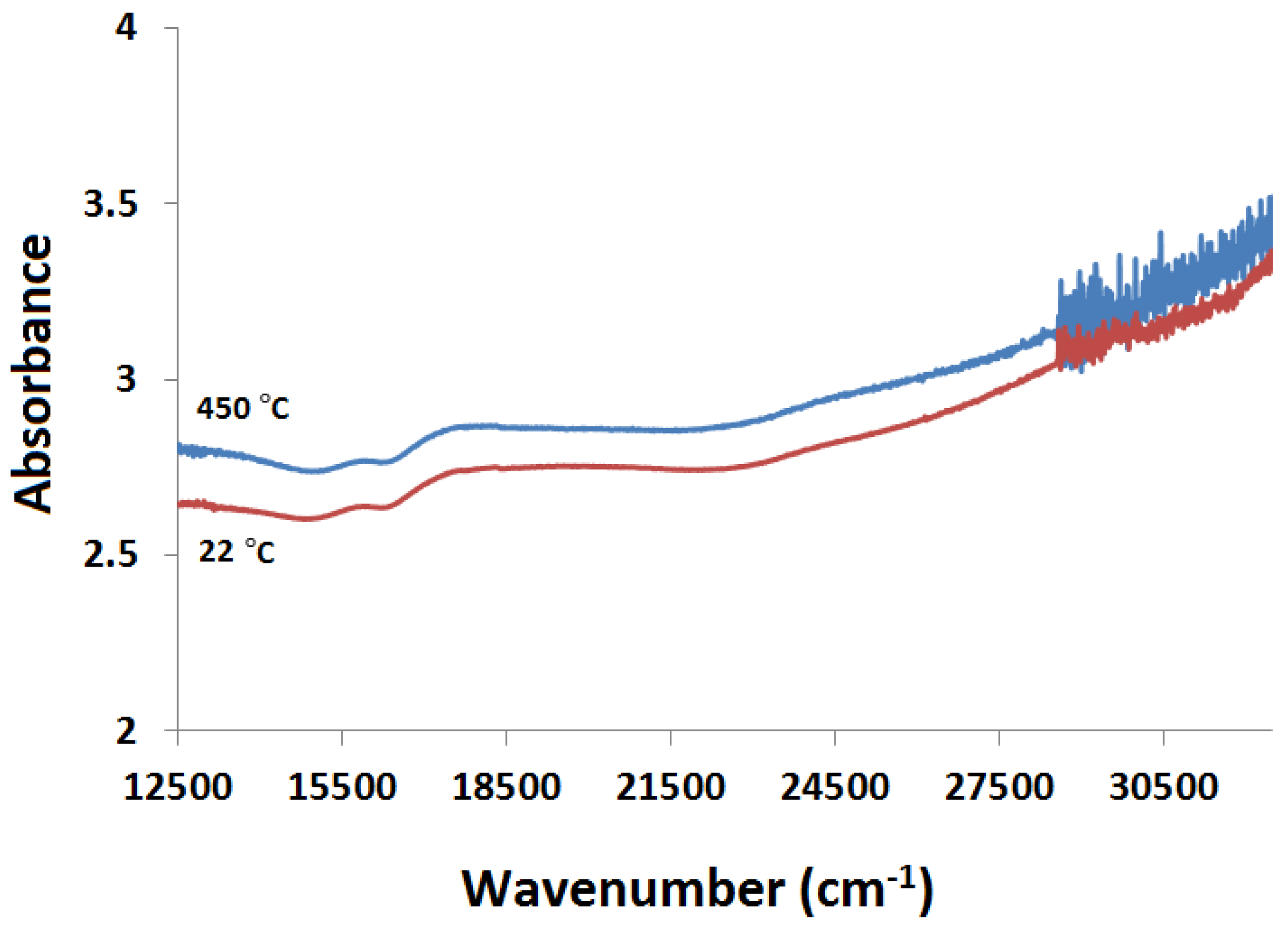
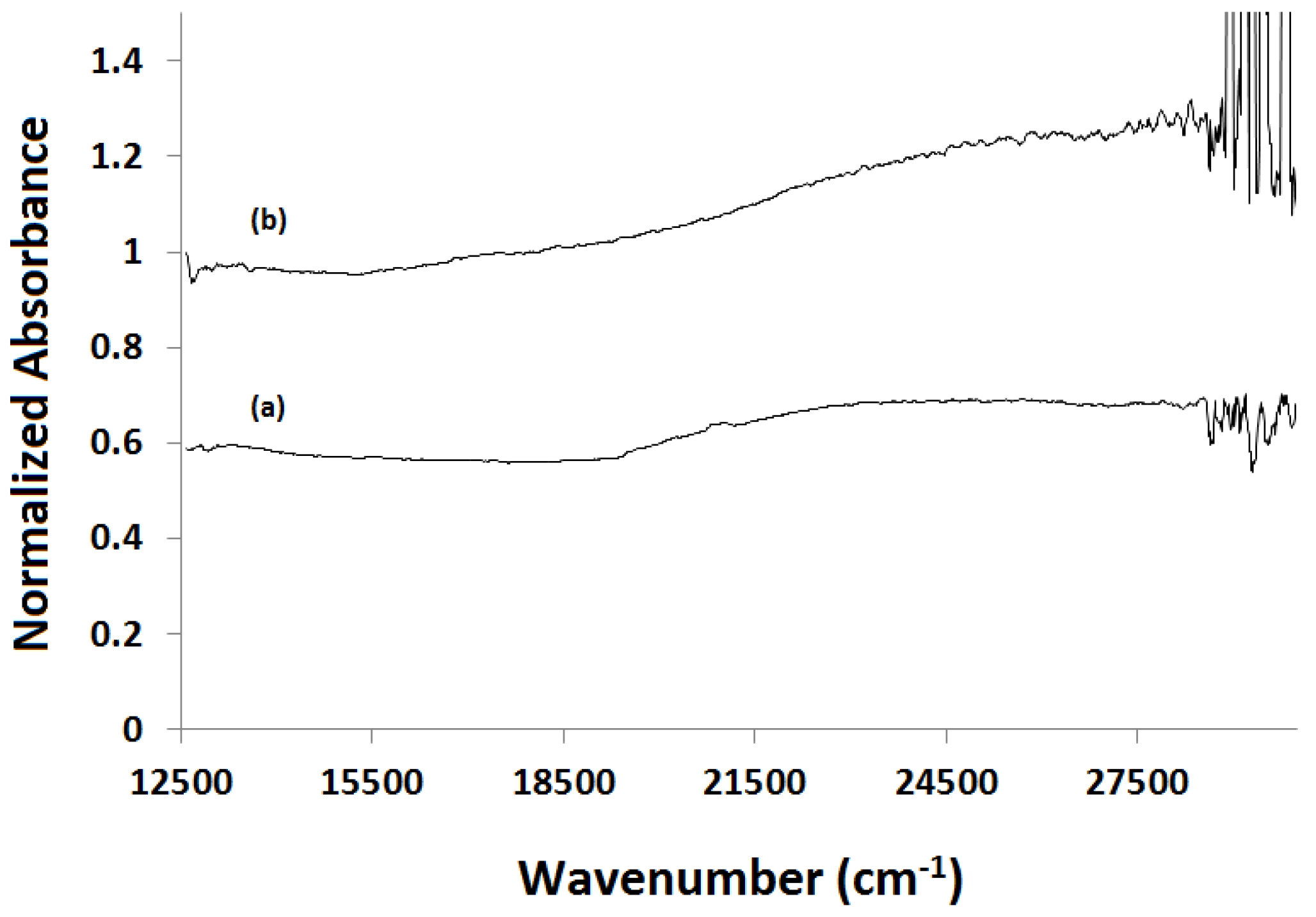
2.3.1. Absorption vs. Time Spectra
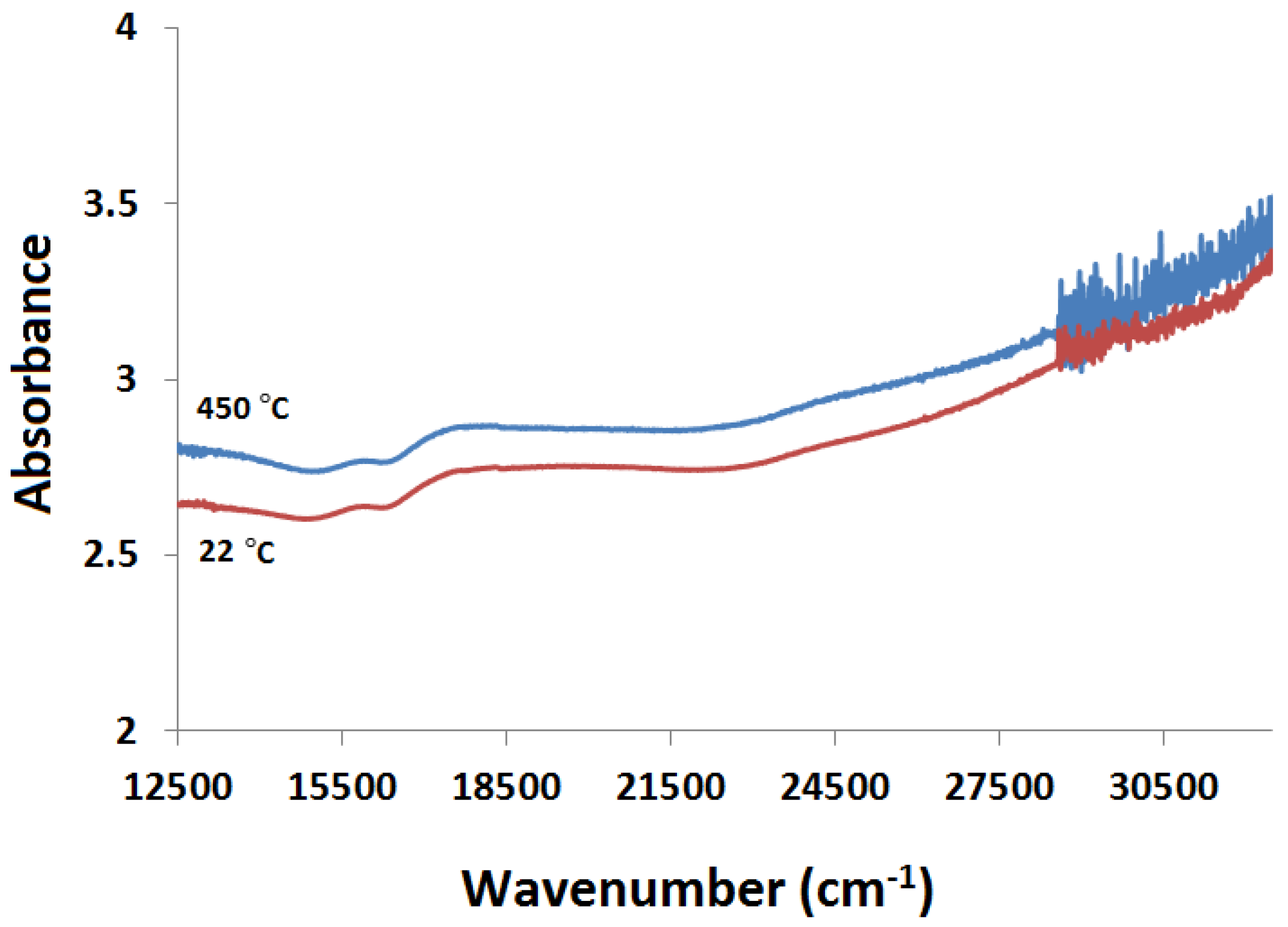
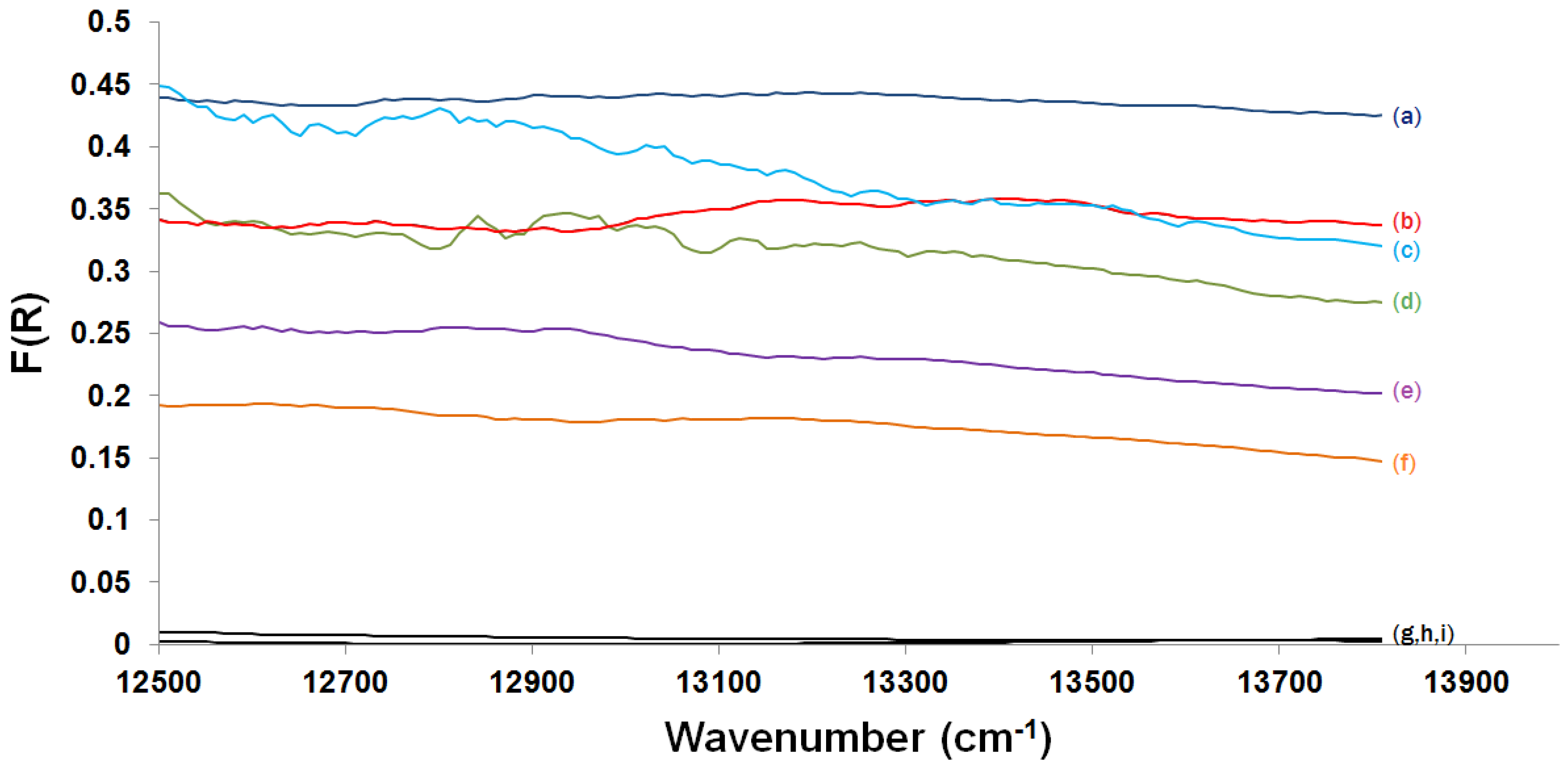
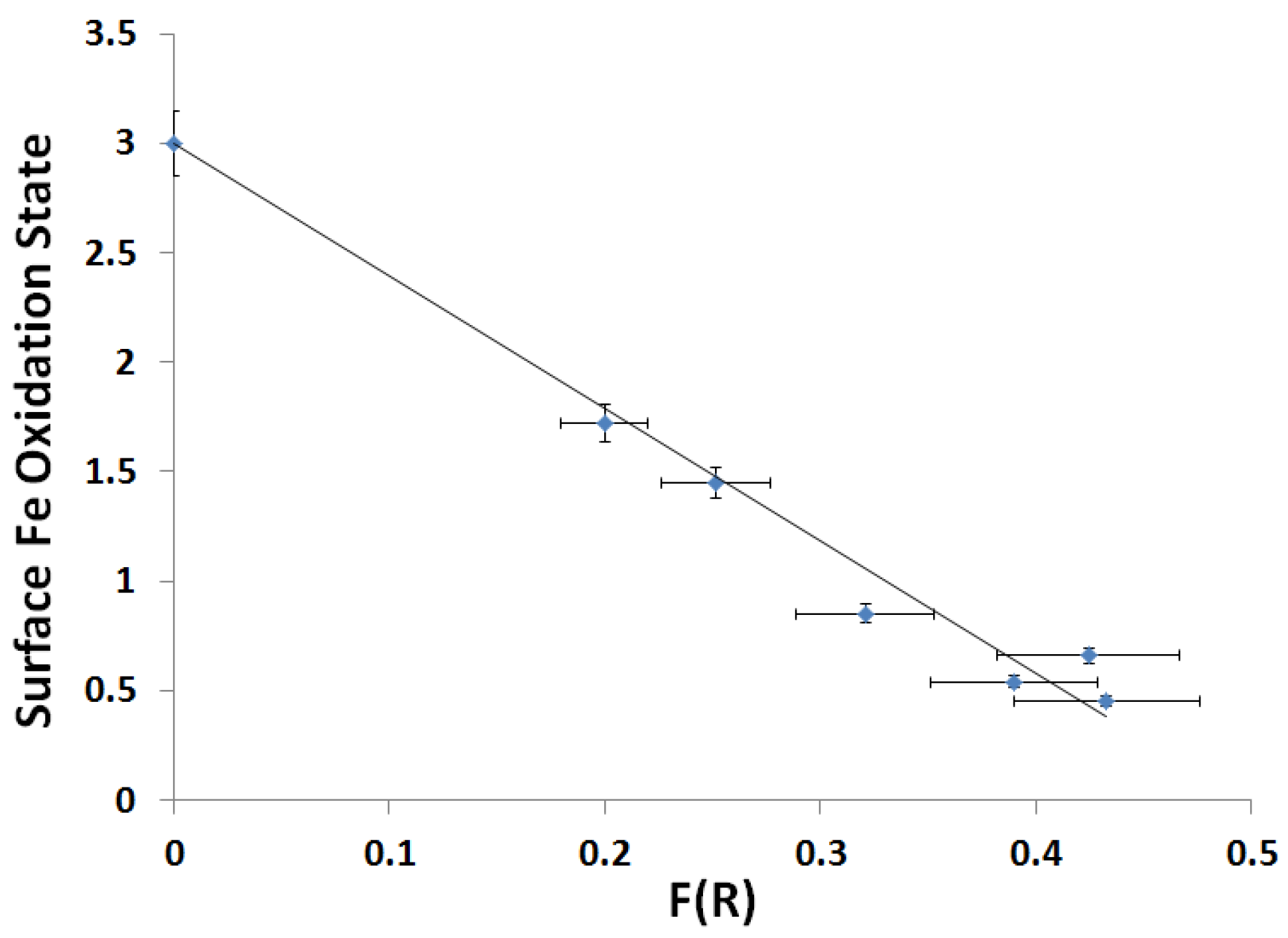
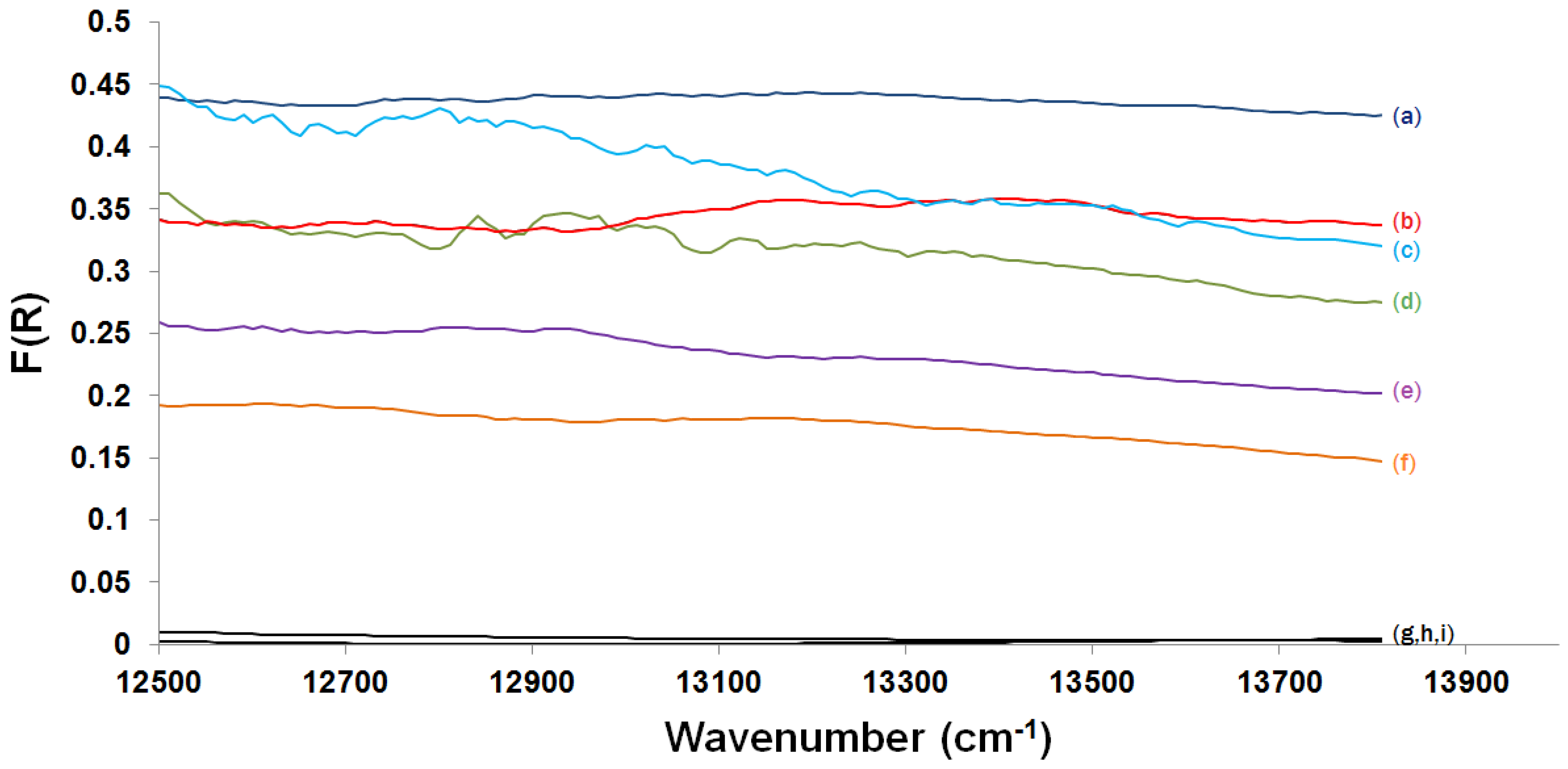
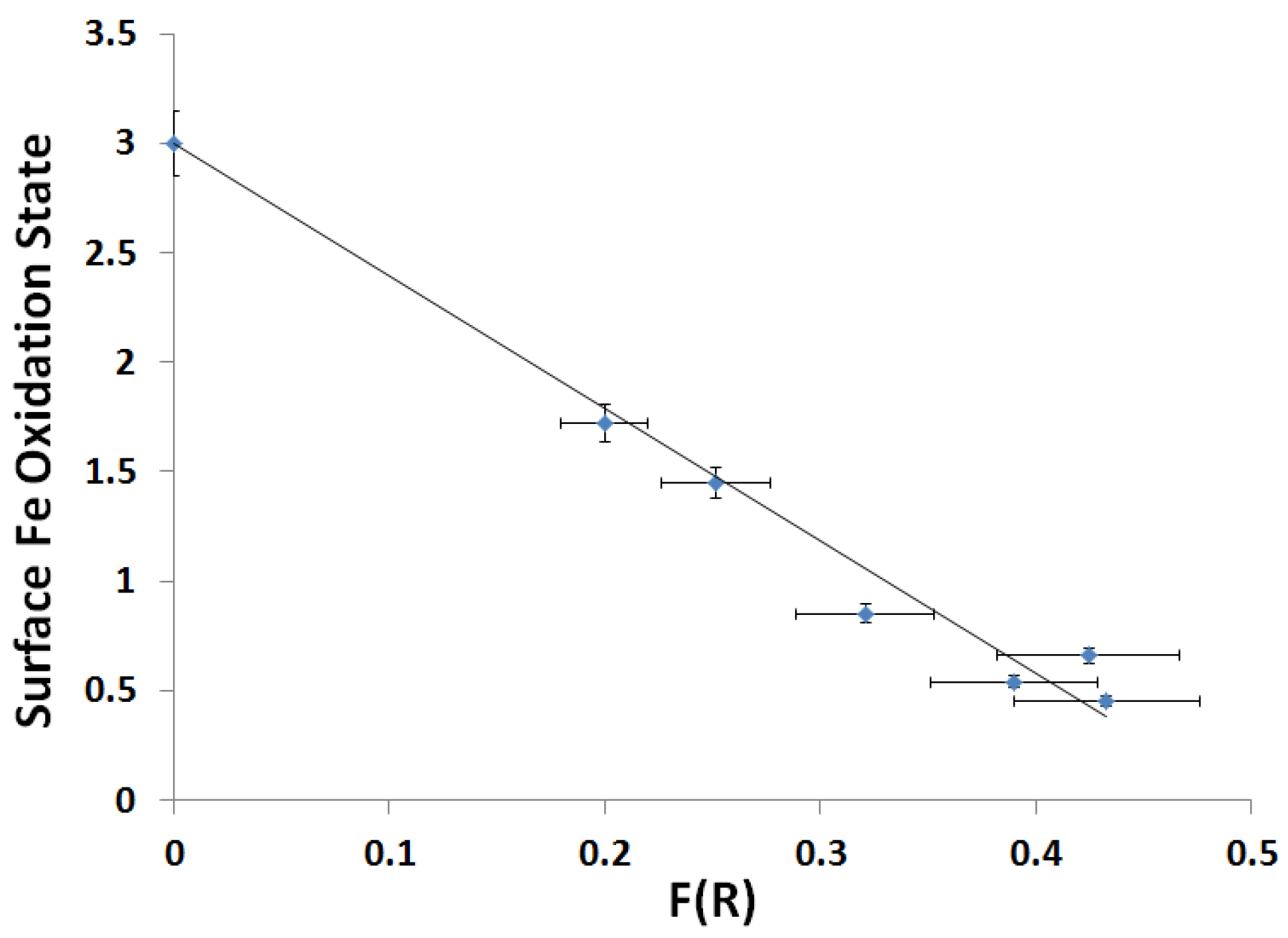
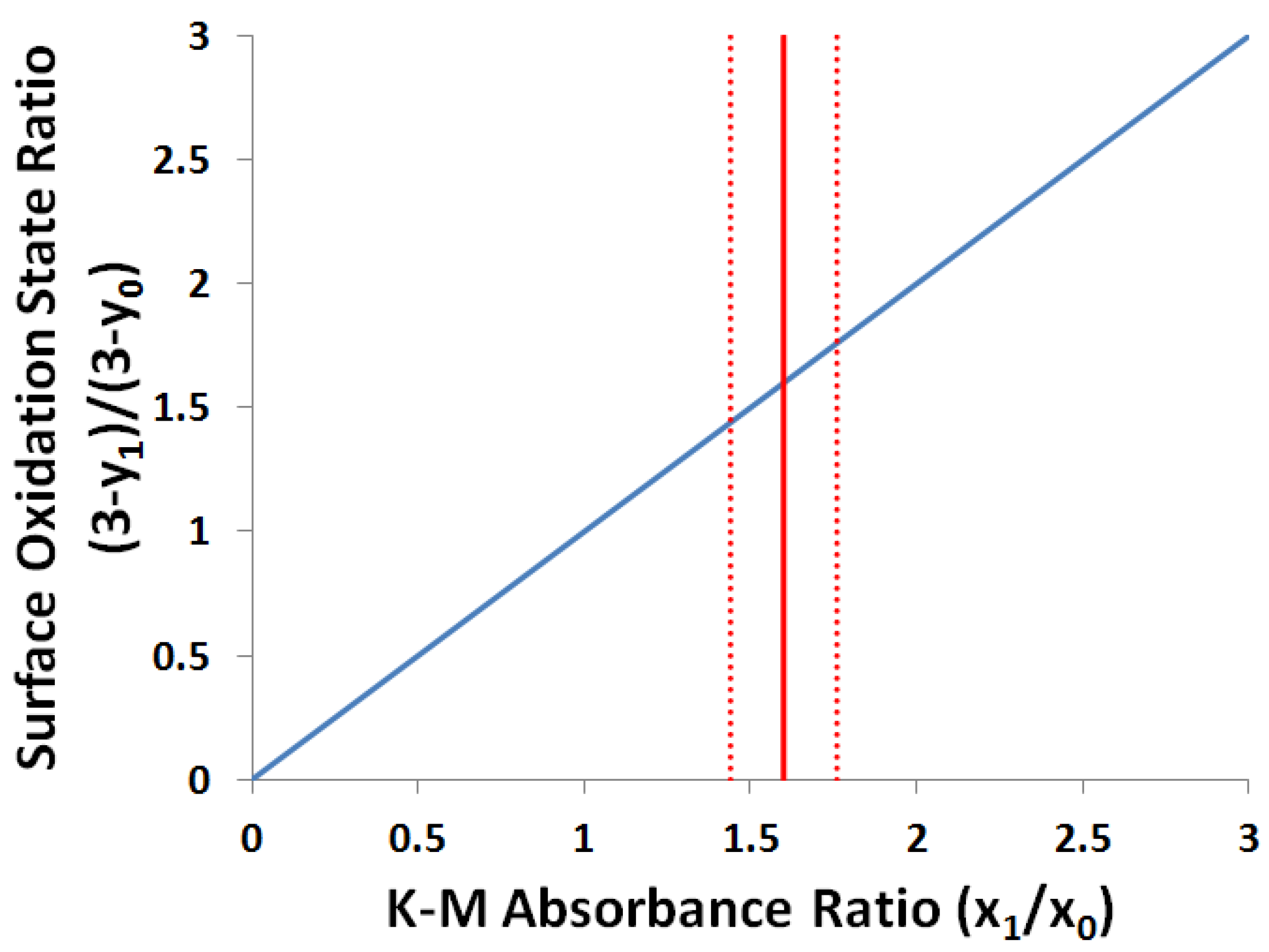
2.3.2. Kubelka-Munk Scans and Calibration
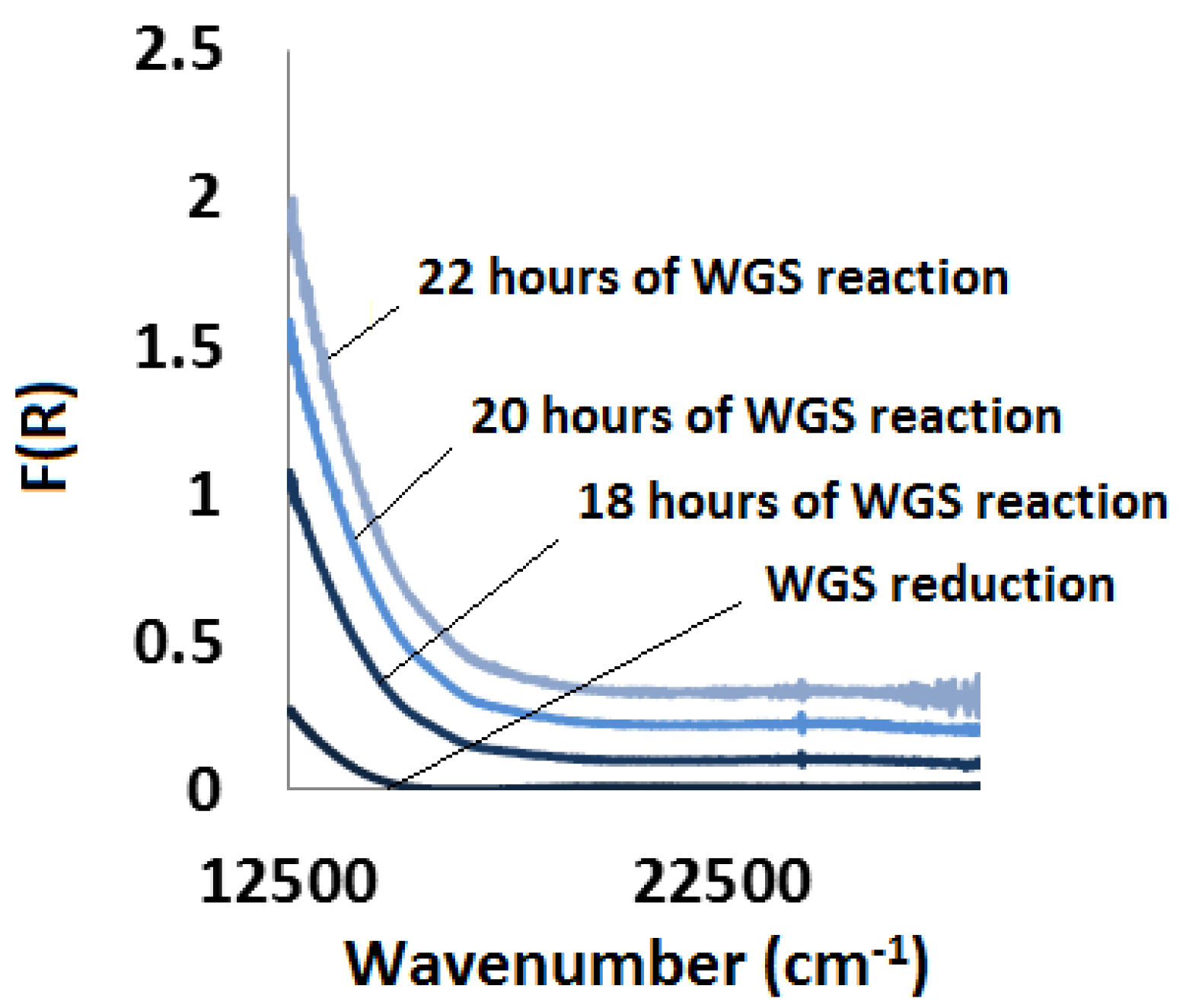
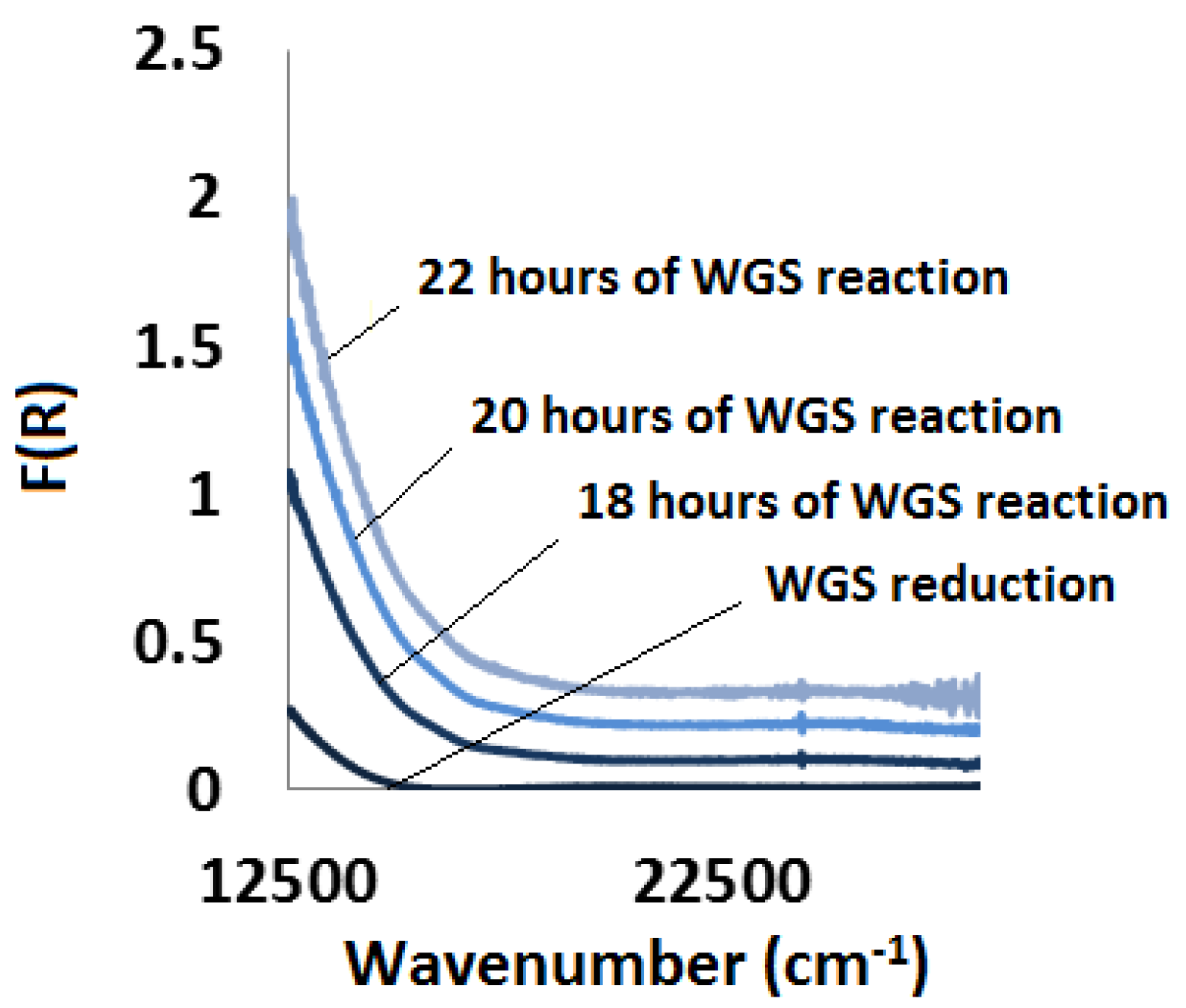
2.3.3. Water-Gas Shift Reaction: In Situ and Ex Situ Analysis
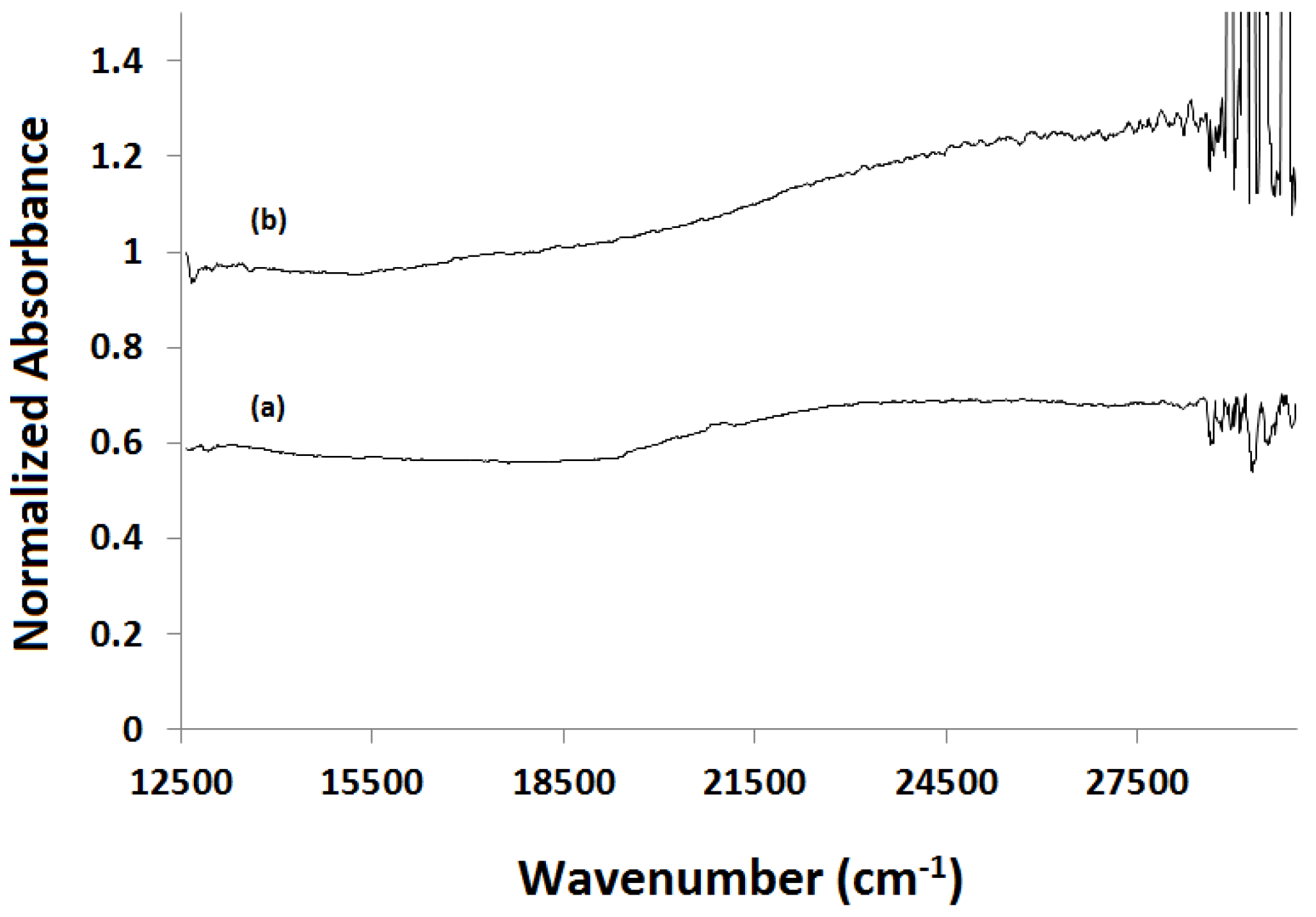
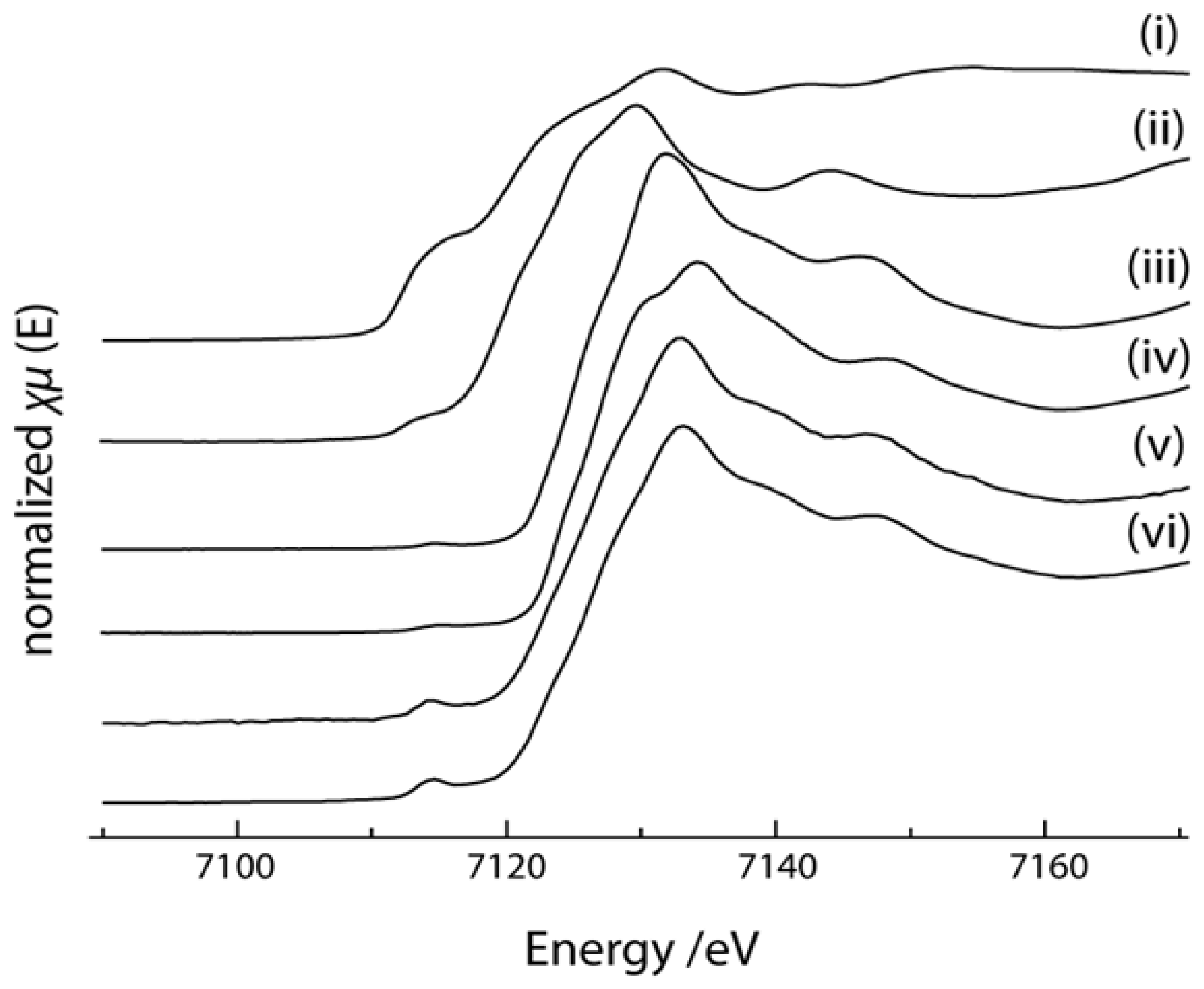
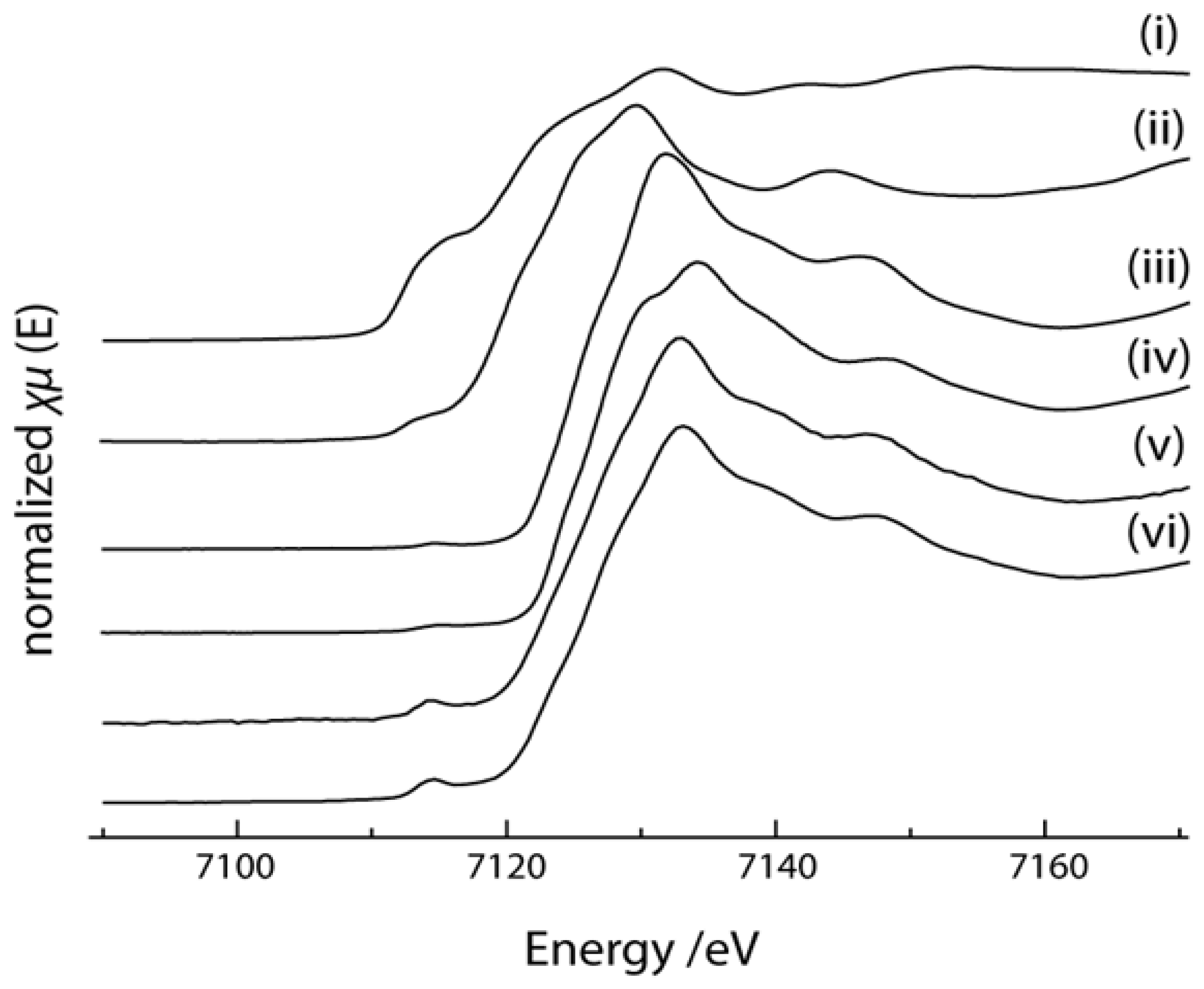
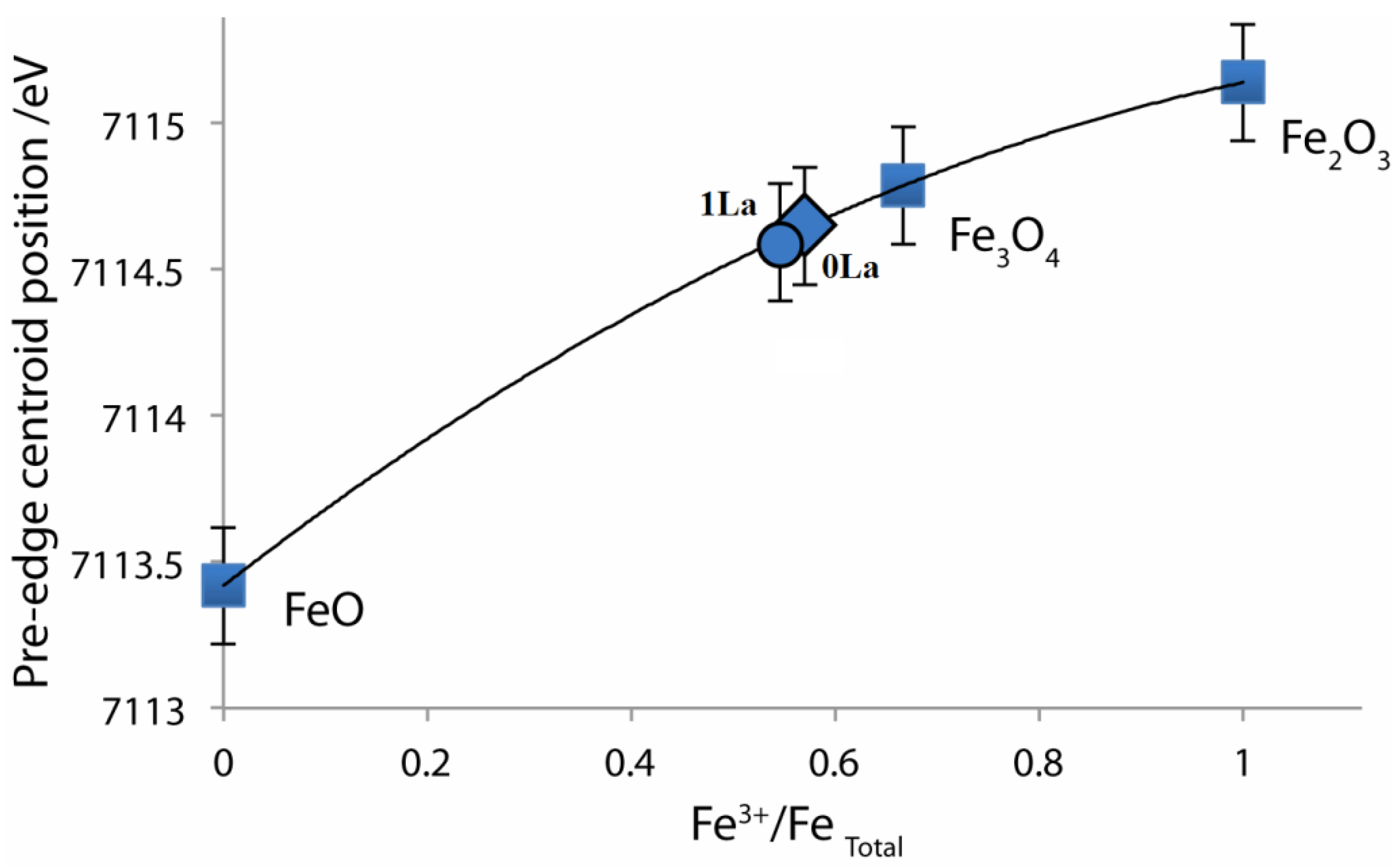
2.4. X-ray Absorption Near Edge Structure (XANES) Spectra Compared to UV-Visible Results
3. Materials and Methods
3.1. Temperature-Programmed Reduction and Oxidation (TPR and TPO)
3.2. In Situ UV-Visible Experiments
3.3. High-Temperature Water-Gas Shift Reaction
3.4. Powder X-ray Diffraction (XRD)
3.5. Energy Dispersive X-ray Spectroscopy (EDS) and Scanning Electron Microscopy (SEM)
3.6. X-ray Absorption Near Edge Structure (XANES)
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Twigg, M.V. Catalyst Handbook, 2nd ed.; Wolfe: London, UK, 1989. [Google Scholar]
- Bartholomew, C.H.; Farrauto, R.J. Fundamentals of Industrial Catalytic Processes, 2nd ed.; Wiley-InterScience: New York, NY, USA, 2005. [Google Scholar]
- Rhodes, C.; Hutchings, G.J. Studies of the Role of the Copper Promoter in the Iron-Chromia High Temperature Water Gas Shift Catalyst. Phys. Chem. Chem. Phys. 2003, 5, 2719–2723. [Google Scholar] [CrossRef]
- Rhodes, C.; Williams, B.P.; King, F.; Hutchings, G.J. Promotion of Fe3O4/Cr2O3 High Temperature Water Gas Shift Catalyst. Catal. Commun. 2002, 3, 381–384. [Google Scholar] [CrossRef]
- Edwards, M.A.; Whittle, D.M.; Rhodes, C.; Ward, A.M.; Rohan, D.; Shannon, M.D.; Hutchings, G.J.; Kiely, C.J. Microstructural Studies of the Copper Promoted Iron Oxide/Chromia Water-Gas Shift Catalyst. Phys. Chem. Chem. Phys. 2002, 4, 3902–3908. [Google Scholar] [CrossRef]
- Lei, Y.; Cant, N.W.; Trimm, D.L. Activity Patterns for the Water Gas Shift Reaction over Supported Precious Metal Catalysts. Catal. Lett. 2005, 103, 133–136. [Google Scholar] [CrossRef]
- Andreev, A.; Idakiev, V.; Mihajlova, D.; Shopov, D. Iron-based Catalysts for the Water-Gas Shift Reaction Promoted by First-Row Transition Metal Oxides. Appl. Catal. 1986, 22, 385–387. [Google Scholar] [CrossRef]
- Júnior, I.; Lima, M.; Jean-Marc, M.; Aouine, M.; Do Carmo, M.R. The Role of Vanadium on the Properties of Iron Based Catalysts for the Water Gas Shift Reaction. Appl. Catal. A Gen. 2005, 283, 91–98. [Google Scholar] [CrossRef]
- Hu, Y.; Jin, H.; Liu, J.; Hao, D. Reactive Behaviors of Iron-Based Shift Catalyst Promoted by Ceria. Chem. Eng. J. 2000, 78, 147–152. [Google Scholar] [CrossRef]
- Hallac, B.B.; Brown, J.C.; Baxter, L.L.; Argyle, M.D. A Kinetic Study on the Structural and Functional Roles of Lanthana in Iron-Based High-Temperature Water-Gas Shift Catalysts. Int. J. Hydrogen Energy 2014, 39, 7306–7317. [Google Scholar] [CrossRef]
- Sun, Y.; Hla, S.S.; Duffy, G.J.; Cousins, A.J.; French, D.; Morpeth, L.D.; Edwards, J.H.; Roberts, D.G. Effect of Ce on the Structural Features and Catalytic Properties of La(0.9−X)CeXFeO3 Perovskite-Like Catalysts for the High Temperature Water-Gas Shift Reaction. Int. J. Hydrogen Energy 2011, 36, 79–86. [Google Scholar] [CrossRef]
- Hla, S.S.; Sun, Y.; Duffy, G.J.; Morpeth, L.D.; Ilyuschechkin, A.; Cousins, A.; Edwards, J.H. Kinetics of the Water-Gas Shift Reaction Over a La0.7Ce0.2FeO3 Perovskite-Like Catalyst Using Simulated Coal-Derived Syngas at High Temperature. Int. J. Hydrogen Energy 2011, 36, 518–527. [Google Scholar] [CrossRef]
- Wang, T.; Ding, Y.; Lü, Y.; Zhu, H.; Lin, L. Influence of Lanthanum on the Performance of Ze-Co/Activated Carbon Catalysts in Fischer-Tropsch Synthesis. J. Nat. Gas Chem. 2008, 17, 153–158. [Google Scholar] [CrossRef]
- Weckhuysen, B.M.; Verberckmoes, A.A.; Debaere, J.; Ooms, K.; Langhans, I.; Schoonheydt, R.A. In Situ UV-Vis Diffuse Reflectance Spectroscopy—On Line Activity Measurements of Supported Chromium Oxide Catalysts: Relating Isobutane Dehydrogenation Activity With Cr-Speciation Via Experimental Design. J. Mol. Catal. A Chem. 2000, 151, 115–131. [Google Scholar] [CrossRef]
- Wachs, I.E. Recent Conceptual Advances in the Catalysis Science of Mixed Metal Oxide Catalytic Materials. Catal. Today 2005, 100, 79–94. [Google Scholar] [CrossRef]
- Argyle, M.D.; Chen, K.; Resini, C.; Krebs, C.; Bell, A.T.; Iglesia, E. In Situ UV-vis Assessment of Extent of Reduction During Oxidation Reactions on Oxide Catalysts. Chem. Commun. 2003, 2082–2083. [Google Scholar] [CrossRef]
- Argyle, M.D.; Chen, K.; Iglesia, E.; Bell, A.T. In Situ UV-vis Spectroscopic Measurements of Kinetic Parameters and Active Sites for Catalytic Oxidation of Alkanes on Vanadium Oxides. J. Phys. Chem. B 2005, 109, 2414–2420. [Google Scholar] [CrossRef] [PubMed]
- Ovsister, O.; Maymol, C.; Angelika, B.; Evgenii, K.V. Dynamics of Redox Behavior of Nano-Sized VOx Species Over Ti–Si-MCM-41 from Time-Resolved in Situ UV/Vis Analysis. J. Catal. 2009, 265, 8–18. [Google Scholar]
- Gao, X.; Jehng, J.M.; Wachs, I.E. In Situ UV-Vis-NIR Diffuse Reflectance and Raman Spectroscopic Studies of Propane Oxidation over ZrO2-Supported Vanadium Oxide Catalysts. J. Catal. 2002, 209, 43–50. [Google Scholar] [CrossRef]
- Sayah, E.; La Fontaine, C.; Briois, V.; Brouri, D.; Massiani, P. Silver Species Reduction upon Exposure of Ag/Al2O3 Catalyst to Gaseous Ethanol: An In Situ Quick-XANES Study. Catal. Today 2012, 189, 55–59. [Google Scholar] [CrossRef]
- Singh, P.; Das, A.K.; Sarkar, B.; Niemeyer, M.; Roncaroli, F.; Olabe, J.A.; Fiedler, J.; Zalis, S.; Kaim, W. Redox Properties of Ruthenium Nitrosyl Porphyrin Complexes with Different Axial Ligation: Structural, Spectroelectrochemical (IR, UV-Visible, and EPR), and Theoretical Studies. Inorg. Chem. 2008, 47, 7106–7113. [Google Scholar] [CrossRef] [PubMed]
- Daughtry, K.D.; Xiao, Y.; Stoner-Ma, D.; Cho, E.; Orville, A.M.; Liu, P.; Allen, K.N. Quaternary Ammonium Oxidative Demethylation: X-ray Crystallographic, Resonance Raman, and UV-Visible Spectroscopic Analysis of a Rieske-Type Demethylase. J. Am. Chem. Soc. 2012, 134, 2823–2834. [Google Scholar] [CrossRef] [PubMed]
- Gunter, K.K.; Miller, L.M.; Aschner, M.; Eliseey, R.; Depuis, D.; Gavin, C.E.; Gunter, T.E. XANES Spectroscopy: A Promising Tool for Toxicology: A Tutorial. Neurotoxicology 2002, 23, 127–146. [Google Scholar] [CrossRef]
- Atkins, P.; Shriver, D. Inorganic Chemistry; Oxford University Press: New York, NY, USA, 2006. [Google Scholar]
- Rao, C.N.R. Ultra-Violet and Visible Spectroscopy: Chemical Applications; Butterworth-Heinemann: London, UK, 1974. [Google Scholar]
- Clark, B.J.; Frost, T.; Russell, M.A. UV Spectroscopy: Techniques, Instrumentation, Data Handling; Chapman & Hall: London, UK, 1993. [Google Scholar]
- Tossell, J.A.; Vaughan, D.; Johnson, K.H. The Electronic Structure of Rutile, Wustite and Hematite from Molecular Orbital Calculations. Am. Mineral. 1974, 59, 319–334. [Google Scholar]
- Cornell, R.M.; Schwertmann, U. The Iron Oxides: Structure, Properties, Reactions, Occurrences and Uses, 2nd ed.; Wiley-VCH: New York, NY, USA, 2003. [Google Scholar]
- Zhang, Z.; Boxall, C.; Kelsall, G.H. Photoelectropheresis of Colloidal Iron Oxides. Part 2.—Magnetite (Fe3O4). J. Chem. Soc. Faraday Trans. 1996, 92, 791–802. [Google Scholar]
- Sherman, D.M. Electronic Structures of Iron(III) and Manganese(IV) (Hydr)oxide Minerals: Thermodynamics of Photochemical Reductive Dissolution in Aquatic Environments. Geochim. Cosmochim. Acta 2005, 69, 3249–3255. [Google Scholar] [CrossRef]
- Sherman, D.M. Electronic Stuctures of Fe3+ Coordination Sites in Iron Oxides: Applications to Spectra, Bonding, and Magnetism. Phys. Chem. Miner. 1985, 12, 161–175. [Google Scholar] [CrossRef]
- Christy, A.A.; Kvalheim, O.M.; Velapoldi, R.A. Quantitative Analysis in Diffuse Reflectance Spectrometry: A Modified Kubelka-Munk Equation. Vib. Spectrosc. 1995, 9, 19–27. [Google Scholar] [CrossRef]
- Džimbeg-Malčić, V.; Barbarić-Mikočević, Ž.; Itrić, K. Kubelka-Munk Theory in Describing Optical Properties of Paper (I). Tech. Gazette 2011, 18, 117–124. [Google Scholar]
- Kubelka, P.; Munk, F. Ein Beitrag zur Optik der Farbanstriche (A Contribution to the Optics of Paint). Z. Tech. Phys. 1931, 12, 593–601. [Google Scholar]
- Schuster, A. Radiation Through a Foggy Atmosphere. Astrophys. J. 1905, 21, 1–22. [Google Scholar] [CrossRef]
- Wyszecki, G.; Stiles, W.S. Color Science, 2nd ed.; John Wiley & Sons: New York, NY, USA, 2000. [Google Scholar]
- Doak, J.; Gupta, R.K.; Manivannan, K.; Ghosh, K.; Kahol, P.K. Effect of Particle Size Distributions on Absorbance Spectra of Gold Nanoparticles. Physica E 2010, 42, 1605–1609. [Google Scholar] [CrossRef]
- Van de Hulst, H.C. Light Scattering by Small Particles; John Wiley & Sons: New York, NY, USA, 1957. [Google Scholar]
- Koningsberger, D.C.; Prins, R. X-ray Absorption: Principles Applications Techniques of EXAFS SEXAFS and XANES, Chemical Analysis; John Wiley & Sons: New York, NY, USA, 1988. [Google Scholar]
- Puig-Molina, A.; Cano, F.M.; Janssens, T.V.W. The Cu Promoter in an Iron-Chromium-Oxide Based Water-Gas Shift Catalyst under Industrial Conditions Studied by In-Situ XAFS. J. Phys. Chem. C 2010, 114, 15410–15416. [Google Scholar] [CrossRef]
- Gonzalez, J.C.; Gronzalez, M.G.; Laborde, M.A.; Moreno, N. Effect of Temperature and Reduction on the Activity of High Temperature Water Gas Shift Catalysts. Appl. Catal. 1986, 20, 3–13. [Google Scholar] [CrossRef]
- Kundakovic, L.; Flytzani-Stephanopoulos, M. Reduction Characteristics of Copper Oxide in Cerium and Zirconium Oxide Systems. Appl. Catal. A 1998, 171, 13–29. [Google Scholar] [CrossRef]
- Greenwood, N.N.; Earnshaw, A. Chemistry of the Elements; Butterworth-Heinemann: London, UK, 1997. [Google Scholar]
- Li, X.; Wang, Y.; Xu, P.; Zhang, Q.; Nie, K.; Hu, X.; Kong, B.; Li, L.; Chen, J. Effects of Temperature and Wavelength Choice on In-Situ Dissolution Test of Cimetidine Tablets. J. Pharm. Anal. 2013, 3, 71–74. [Google Scholar] [CrossRef]
- Zhu, Y.; Sun, D.; Huang, Q.; Jin, X.; Liu, H. UV-vis Spectra of Perovskite Iron-Doped Ba0.72Sr0.28TiO3. Mater. Lett. 2008, 62, 407–409. [Google Scholar] [CrossRef]
- Kim, J.Y.; Magesh, G.; Youn, D.H.; Jang, J.; Kubota, J.; Domen, K.; Lee, J.S. Single-Crystalline, Wormlike Hematite Photoanodes for Efficient Solar Water Splitting. Sci. Rep. 2013, 3, 2681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherman, D.M.; Waite, T.D. Electronic Spectra of Fe3+ Oxides and Oxide Hydroxides in the Near-IR to Near-UV. Am. Mineral. 1985, 70, 1262–1269. [Google Scholar]
- Popa, T.; Xu, G.; Barton, T.F.; Argyle, M.D. High Temperature Water Gas Shift Catalysts with Alumina. Appl. Catal. A Gen. 2010, 379, 15–23. [Google Scholar] [CrossRef]
- Patlolla, A.; Carino, E.V.; Ehrlich, S.N.; Stavitski, E.; Frenkel, A.I. Application of Operando XAS, XRD, and Raman Spectroscopy for Phase Speciation in Water Gas Shift Reaction Catalysts. ACS Catal. 2012, 2, 2216–2223. [Google Scholar] [CrossRef]
- Wilke, M.; Hahn, O.; Woodland, A.B.; Rickers, K. The Oxidation State of Iron Determined by Fe K-Edge XANES—Application to Iron Gall Ink in Historical Manuscripts. J. Anal. At. Spectrom. 2009, 24, 1364–1372. [Google Scholar] [CrossRef]
- Labinger, J.A.; Ott, K.C. Is There a Difference Between Surface and Bulk Oxidation Levels in Partially Reduced Metal Oxide Catalysts? Evidence from Methane Oxidative Coupling Kinetics. Catal. Lett. 1990, 4, 245–250. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst ID | TPR 1 (First Peak) % EOR to Fe3O4 a | TPR 1 (Both Peaks) % EOR to Fe a | TPR 2 % EOR to Fe |
|---|---|---|---|
| 0La | 42 | 82 | 42 |
| 0.5La | 53 | 78 | 72 |
| 1La | 67 | 85 | 53 |
| 2La | 79 | 89 | N/A |
| 5La | 82 | 89 | 57 |
| Slope | Intercept | r2 |
|---|---|---|
| −6.05 ± 0.39 | 3.00 ± 0.13 | 0.977 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hallac, B.B.; Brown, J.C.; Stavitski, E.; Harrison, R.G.; Argyle, M.D. In Situ UV-Visible Assessment of Iron-Based High-Temperature Water-Gas Shift Catalysts Promoted with Lanthana: An Extent of Reduction Study. Catalysts 2018, 8, 63. https://doi.org/10.3390/catal8020063
Hallac BB, Brown JC, Stavitski E, Harrison RG, Argyle MD. In Situ UV-Visible Assessment of Iron-Based High-Temperature Water-Gas Shift Catalysts Promoted with Lanthana: An Extent of Reduction Study. Catalysts. 2018; 8(2):63. https://doi.org/10.3390/catal8020063
Chicago/Turabian StyleHallac, Basseem B., Jared C. Brown, Eli Stavitski, Roger G. Harrison, and Morris D. Argyle. 2018. "In Situ UV-Visible Assessment of Iron-Based High-Temperature Water-Gas Shift Catalysts Promoted with Lanthana: An Extent of Reduction Study" Catalysts 8, no. 2: 63. https://doi.org/10.3390/catal8020063
APA StyleHallac, B. B., Brown, J. C., Stavitski, E., Harrison, R. G., & Argyle, M. D. (2018). In Situ UV-Visible Assessment of Iron-Based High-Temperature Water-Gas Shift Catalysts Promoted with Lanthana: An Extent of Reduction Study. Catalysts, 8(2), 63. https://doi.org/10.3390/catal8020063