Recent Advances in ω-Transaminase-Mediated Biocatalysis for the Enantioselective Synthesis of Chiral Amines




Abstract
:1. Introduction
2. Synthetic Approaches for Chiral Amines and Equilibrium Shift for ω-TA-Mediated Biocatalysis
2.1. Kinetic Resolution
2.2. Asymmetric Synthesis
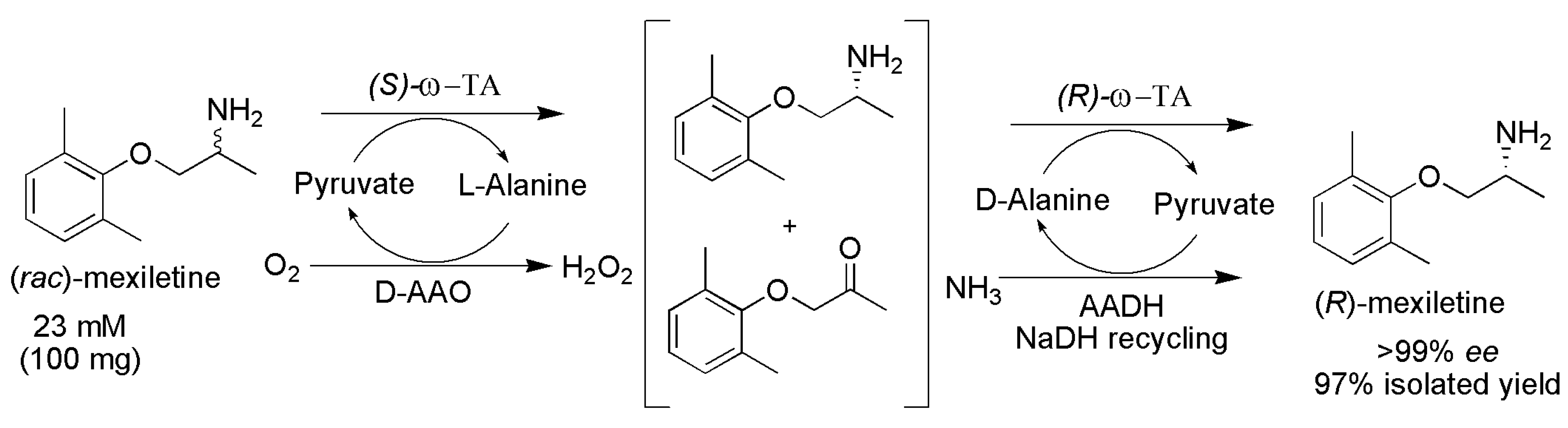
2.3. Deracemization
3. Protein Engineering Aspects for Improved Substrate Scope of ω-Transaminase (ω-TA) Biocatalysts
4. Establishing Stable ω-TA Biocatalysts for Synthetic Applications
4.1. Finding Thermostable ω-TAs
4.2. Protein Engineering
4.3. Enzyme Immobilization
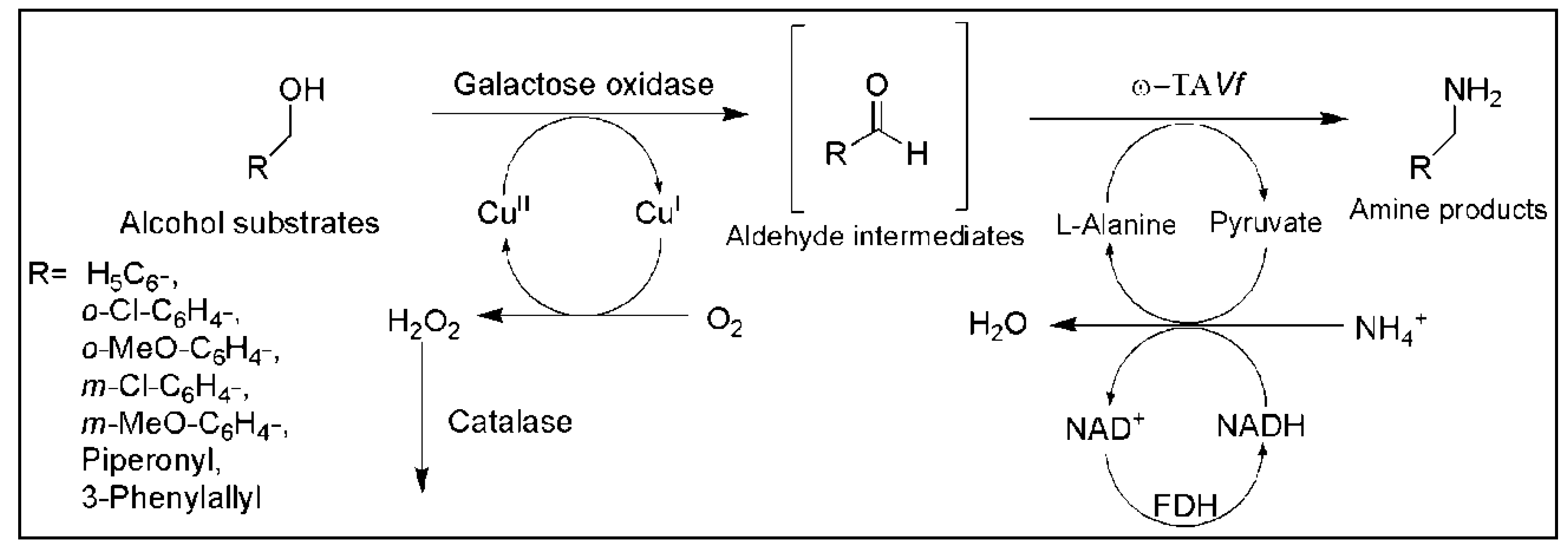
5. Cascade Reactions Involving ω-TAs
6. Conclusions and Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Abaházi, E.; Sátorhelyi, P.; Erdélyi, B.; Vértessy, B.G.; Land, H.; Paizs, C.; Berglund, P.; Poppe, L. Covalently immobilized Trp60Cys mutant of ω-transaminase from Chromobacterium violaceum for kinetic resolution of racemic amines in batch and continuous-flow modes. Biochem. Eng. J. 2018, 132, 270–278. [Google Scholar] [CrossRef]
- Zheng, G.W.; Xu, J.H. New opportunities for biocatalysis: Driving the synthesis of chiral chemicals. Curr. Opin. Biotechnol. 2011, 22, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Solano, D.M.; Hoyos, P.; Hernáiz, M.J.; Alcántara, A.R.; Sánchez-Montero, J.M. Industrial biotransformations in the synthesis of building blocks leading to enantiopure drugs. Bioresour. Technol. 2012, 115, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Albarran-Velo, J.; Gonzalez-Martinez, D.; Gotor-Fernandez, V. Stereoselective biocatalysis: A mature technology for the asymmetric synthesis of pharmaceutical building blocks. Biocatal. Biotransform. 2018, 36, 102–130. [Google Scholar] [CrossRef]
- Yang, G.; Ding, Y. Recent advances in biocatalyst discovery, development and applications. Bioorg. Med. Chem. 2014, 22, 5604–5612. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.C.; Mutti, F.G.; Kroutil, W. Biocatalytic synthesis of enantiopure building blocks for pharmaceuticals. Drug Discov. Today Technol. 2013, 10, e37–e44. [Google Scholar] [CrossRef] [PubMed]
- Truppo, M.D. Biocatalysis in the Pharmaceutical Industry—The Need for Speed. ACS Med. Chem. Lett. 2017, 8, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Bornscheuer, U.T. The fourth wave of biocatalysis is approaching. Phil. Trans. R. Soc. A 2018, 376, 20170063. [Google Scholar] [CrossRef] [PubMed]
- Ghislieri, D.; Turner, N.J. Biocatalytic Approaches to the Synthesis of Enantiomerically Pure Chiral Amines. Top. Catal. 2014, 57, 284–300. [Google Scholar] [CrossRef]
- Gomm, A.; O’Reilly, E. Transaminases for chiral amine synthesis. Curr. Opin. Chem. Biol. 2018, 43, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Slabu, I.; Galman, J.L.; Lloyd, R.C.; Turner, N.J. Discovery, Engineering, and Synthetic Application of Transaminase Biocatalysts. ACS Catal. 2017, 7, 8263–8284. [Google Scholar] [CrossRef]
- Guo, F.; Berglund, P. Transaminase biocatalysis: Optimization and application. Green Chem. 2017, 19, 333–360. [Google Scholar] [CrossRef]
- Höhne, M.; Bornscheuer, U.T. Biocatalytic routes to optically active amines. ChemCatChem 2009, 1, 42–51. [Google Scholar] [CrossRef]
- Koszelewski, D.; Tauber, K.; Faber, K.; Kroutil, W. ω-Transaminases for the synthesis of non-racemic α-chiral primary amines. Trends Biotechnol. 2010, 28, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Brundiek, H.; Höhne, M. Transaminases—A Biosynthetic Route for Chiral Amines. In Applied Biocatalysis: from Fundamental Science to Industrial Applications; Hilterhaus, L., Liese, A., Kettling, U., Antranikian, G., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2016; pp. 199–218. [Google Scholar]
- Ferrandi, E.E.; Monti, D. Amine transaminases in chiral amines synthesis: Recent advances and challenges. World J. Microbiol. Biotechnol. 2018, 34, 13. [Google Scholar] [CrossRef] [PubMed]
- Busto, E.; Simon, R.C.; Richter, N.; Kroutil, W. Enzymatic Synthesis of Chiral Amines using ω-Transaminases, Amine Oxidases, and the Berberine Bridge Enzyme. In Green Biocatalysis; Patel, R.N., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; pp. 17–57. [Google Scholar]
- Mathew, S.; Yun, H. ω-Transaminases for the production of optically pure amines and unnatural amino acids. ACS Catal. 2012, 2, 993–1001. [Google Scholar] [CrossRef]
- Szmejda, K.; Florczak, T.; Jodłowska, I.; Turkiewicz, M. Extremophilic and modified aminotransferases as a versatile tool for the synthesis of optically pure building blocks for pharmaceutical industry. Biotechnol. Food Sci. 2017, 81, 23–34. [Google Scholar]
- Kelly, S.A.; Pohle, S.; Wharry, S.; Mix, S.; Allen, C.C.; Moody, T.S.; Gilmore, B.F. Application of ω-Transaminases in the Pharmaceutical Industry. Chem. Rev. 2018, 118, 349–367. [Google Scholar] [CrossRef] [PubMed]
- Berglund, P.; Humble, M.S.; Branneby, C. Comprehensive Chirality; Carreira, E.M., Yamamoto, H., Eds.; Elsevier: Amsterdam, The Nertherland, 2012; Volume 7, pp. 390–401. [Google Scholar]
- Kohls, H.; Steffen-Munsberg, F.; Höhne, M. Recent achievements in developing the biocatalytic toolbox for chiral amine synthesis. Curr. Opin. Chem. Biol. 2014, 19, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Matcham, G.W.; Stirling, D.I.; Zeitlin, A.L. Enantiomeric Enrichment and Stereoselective Synthesis of Chiral amines. U.S. Patent 4950606 A, 5 April 1994. [Google Scholar]
- Wu, H.L.; Zhang, J.D.; Zhang, C.F.; Fan, X.J.; Chang, H.H.; Wei, W.L. Characterization of Four New Distinct ω-Transaminases from Pseudomonas putida NBRC 14164 for Kinetic Resolution of Racemic Amines and Amino Alcohols. Appl. Biochem. Biotechnol. 2017, 181, 972–985. [Google Scholar] [CrossRef] [PubMed]
- Höhne, M.; Robins, K.; Bornscheuer, U.T. A Protection Strategy Substantially Enhances Rate and Enantioselectivity in ω-Transaminase-Catalyzed Kinetic Resolutions. Adv. Synth. Catal. 2008, 350, 807–812. [Google Scholar] [CrossRef]
- Kaulmann, U.; Smithies, K.; Smith, M.E.; Hailes, H.C.; Ward, J.M. Substrate spectrum of ω-transaminase from Chromobacterium violaceum DSM30191 and its potential for biocatalysis. Enzyme Microb. Technol. 2007, 41, 628–637. [Google Scholar] [CrossRef]
- Hanson, R.L.; Davis, B.L.; Chen, Y.; Goldberg, S.L.; Parker, W.L.; Tully, T.P.; Montana, M.A.; Patel, R.N. Preparation of (R)-Amines from Racemic Amines with an (S)-Amine Transaminase from Bacillus megaterium. Adv. Synth. Catal. 2008, 350, 1367–1375. [Google Scholar] [CrossRef]
- Malik, M.S.; Park, E.S.; Shin, J.S. ω-Transaminase-catalyzed kinetic resolution of chiral amines using L-threonine as an amino acceptor precursor. Green Chem. 2012, 14, 2137–2140. [Google Scholar] [CrossRef]
- Savile, C.K.; Janey, J.M.; Mundorff, E.C.; Moore, J.C.; Tam, S.; Jarvis, W.R.; Colbeck, J.C.; Krebber, A.; Fleitz, F.J.; Brands, J.; et al. Biocatalytic asymmetric synthesis of chiral amines from ketones applied to Sitagliptin manufacture. Science 2010, 329, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Gomm, A.; Lewis, W.; Green, A.P.; O’Reilly, E. A New Generation of Smart Amine Donors for Transaminase-Mediated Biotransformations. Chem. Eur. J. 2016, 22, 12692–12695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stekhanova, T.N.; Rakitin, A.L.; Mardanov, A.V.; Bezsudnova, E.Y.; Popov, V.O. A Novel highly thermostable branched-chain amino acid aminotransferase from the crenarchaeon Vulcanisaeta moutnovskia. Enzyme Microb. Technol. 2017, 96, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Sayer, C.; Martinez-Torres, R.J.; Richter, N.; Isupov, M.N.; Hailes, H.C.; Littlechild, J.A.; Ward, J.M. The substrate specificity, enantioselectivity and structure of the (R)-selective amine: Pyruvate transaminase from Nectria haematococca. FEBS J. 2014, 281, 2240–2253. [Google Scholar] [CrossRef] [PubMed]
- Mathew, S.; Nadarajan, S.P.; Chung, T.; Park, H.H.; Yun, H. Biochemical characterization of thermostable ω-transaminase from Sphaerobacter thermophilus and its application for producing aromatic β-and γ-amino acids. Enzyme Microb. Technol. 2016, 87, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, C.; Panizza, P.; Giordano, S.R. Identification, expression and characterization of an R-ω-transaminase from Caproniasemi immersa. Appl. Microbiol. Biotechnol. 2017, 101, 5677–5687. [Google Scholar] [CrossRef] [PubMed]
- Baud, D.; Jeffries, J.W.; Moody, T.S.; Ward, J.M.; Hailes, H.C. A metagenomics approach for new biocatalyst discovery: Application to transaminases and the synthesis of allylic amines. Green Chem. 2017, 19, 1134–1143. [Google Scholar] [CrossRef]
- Gao, S.; Su, Y.; Zhao, L.; Li, G.; Zheng, G. Characterization of a (R)-selective amine transaminase from Fusarium oxysporum. Process Biochem. 2017, 63, 130–136. [Google Scholar] [CrossRef]
- Yun, H.; Cho, B.K.; Kim, B.G. Kinetic resolution of (R, S)-sec-butylamine using omega-transaminase from Vibrio fluvialis JS17 under reduced pressure. Biotechnol. Bioeng. 2004, 87, 772–778. [Google Scholar] [CrossRef] [PubMed]
- Tufvesson, P.; Bach, C.; Woodley, J.M. A model to assess the feasibility of shifting reaction equilibrium by acetone removal in the transamination of ketones using 2-propylamine. Biotechnol. Bioeng. 2014, 111, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Hou, A.; Deng, Z.; Ma, H.; Liu, T. Substrate screening of amino transaminase for the synthesis of a sitagliptin intermediate. Tetrahedron 2016, 72, 4660–4664. [Google Scholar] [CrossRef]
- Park, E.S.; Shin, J.S. Biocatalytic cascade reactions for asymmetric synthesis of aliphatic amino acids in a biphasic reaction system. J. Mol. Catal. B Enzym. 2015, 121, 9–14. [Google Scholar] [CrossRef]
- Neto, W.; Schurmann, M.; Panella, L.; Vogel, A.; Woodley, J.M. Immobilization of-transaminase for industrial application: Screening and characterization of commercial ready to use enzyme carriers. J. Mol. Catal. B Enzym. 2015, 117, 54–61. [Google Scholar] [CrossRef]
- Rehn, G.; Grey, C.; Branneby, C.; Lindberg, L.; Adlercreutz, P. Activity and stability of different immobilized preparations of recombinant E. coli cells containing ω-transaminase. Process Biochem. 2012, 47, 1129–1134. [Google Scholar] [CrossRef]
- Bajić, M.; Plazl, I.; Stloukal, R.; Žnidaršič-Plazl, P. Development of a miniaturized packed bed reactor with ω-transaminase immobilized in LentiKats®. Process Biochem. 2017, 52, 63–72. [Google Scholar] [CrossRef]
- Satyawali, Y.; Ehimen, E.; Cauwenberghs, L.; Maesen, M.; Vandezande, P.; Dejonghe, W. Asymmetric synthesis of chiral amine in organic solvent and in-situ product recovery for process intensification: A case study. Biochem. Eng. J. 2017, 117, 97–104. [Google Scholar] [CrossRef]
- Lalonde, J. Highly engineered biocatalysts for efficient small molecule pharmaceutical synthesis. Curr. Opin. Biotechnol. 2016, 42, 152–158. [Google Scholar] [CrossRef] [PubMed]
- López-Iglesias, M.; González-Martínez, D.; Gotor, V.; Busto, E.; Kroutil, W.; Gotor-Fernández, V. Biocatalytic Transamination for the Asymmetric Synthesis of Pyridylalkylamines. Structural and Activity Features in the Reactivity of Transaminases. ACS Catal. 2016, 6, 4003–4009. [Google Scholar] [CrossRef] [Green Version]
- Busto, E.; Simon, R.C.; Grischek, B.; Gotor-Fernández, V.; Kroutil, W. Cutting short the asymmetric synthesis of the Ramatroban precursor by employing ω-Transaminases. Adv. Synth. Catal. 2014, 356, 1937–1942. [Google Scholar] [CrossRef]
- Costa, I.C.; De Souza, R.O.; Bornscheuer, U.T. Asymmetric synthesis of serinol-monoesters catalyzed by amine transaminases. Tetrahedron Asymmetry 2017, 28, 1183–1187. [Google Scholar] [CrossRef]
- Martin, A.R.; Shonnard, D.; Pannuri, S.; Kamat, S. Characterization ofa High Activity (S)-Aminotransferase for Substituted (S)-Aminotetralin Production: Properties and Kinetics. J. Bioprocess. Biotech. 2011, 1, 107. [Google Scholar] [CrossRef]
- Ito, N.; Kawano, S.; Hasegawa, J.; Yasohara, Y. Purification and Characterization of a Novel (S)-Enantioselective Transaminase from Pseudomonas fluorescens KNK08-18 for the Synthesis of Optically Active Amines. Biosci. Biotechnol. Biochem. 2011, 75, 2093–2098. [Google Scholar] [CrossRef] [PubMed]
- Voges, M.; Schmidt, F.; Wolff, D.; Sadowski, G.; Held, C. Thermodynamics of the alanine aminotransferase reaction. Fluid Ph. Equilibria 2016, 422, 87–98. [Google Scholar] [CrossRef]
- Seo, J.H.; Kyung, D.; Joo, K.; Lee, J.; Kim, B.G. Necessary and sufficient conditions for the asymmetric synthesis of chiral amines using ω-aminotransferases. Biotechnol. Bioeng. 2011, 108, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Green, A.P.; Turner, N.J.; O’Reilly, E. Chiral amine synthesis using ω-transaminases: An amine donor that displaces equilibria and enables high-throughput screening. Angew. Chem. Int. Ed. 2014, 53, 10714–10717. [Google Scholar] [CrossRef] [PubMed]
- Richter, N.; Farnberger, J.E.; Pressnitz, D.; Lechner, H.; Zepeck, F.; Kroutil, W. A system for ω-transaminase mediated (R)-amination using L-alanine as an amine donor. Green Chem. 2015, 17, 2952–2958. [Google Scholar] [CrossRef]
- Martínez-Montero, L.; Gotor, V.; Gotor-Fernández, V.; Lavandera, I. Stereoselective amination of racemic sec-alcohols through sequential application of laccases and transaminases. Green Chem. 2017, 19, 474–480. [Google Scholar] [CrossRef] [Green Version]
- Galman, J.L.; Slabu, I.; Weise, N.J.; Iglesias, C.; Parmeggiani, F.; Lloyd, R.C.; Turner, N.J. Biocatalytic transamination with near-stoichiometric inexpensive amine donors mediated by bifunctional mono-and di-amine transaminases. Green Chem. 2017, 19, 361–366. [Google Scholar] [CrossRef]
- Mathew, S.; Nadarajan, S.P.; Sundaramoorthy, U.; Jeon, H.; Chung, T.; Yun, H. Biotransformation of β-keto nitriles to chiral (S)-β-amino acids using nitrilase and ω-transaminase. Biotechnol. Lett. 2017, 39, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Rehn, G.; Ayres, B.; Adlercreutz, P.; Grey, C. An improved process for biocatalytic asymmetric amine synthesis by in situ product removal using a supported liquid membrane. J. Mol. Catal. B Enzym. 2016, 123, 1–7. [Google Scholar] [CrossRef]
- Rudroff, F.; Mihovilovic, M.D.; Gröger, H.; Snajdrova, R.; Iding, H.; Bornscheuer, U.T. Opportunities and challenges for combining chemo-and biocatalysis. Nat. Catal. 2018, 1, 12–22. [Google Scholar] [CrossRef]
- Kroutil, W.; Fischereder, E.M.; Fuchs, C.S.; Lechner, H.; Mutti, F.G.; Pressnitz, D.; Rajagopalan, A.; Sattler, J.H.; Simon, R.C.; Siirola, E. Asymmetric preparation of prim, sec-, and tert-amines employing selected biocatalysts. Org. Proc. Res. Dev. 2013, 17, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Koszelewski, D.; Pressnitz, D.; Clay, D.; Kroutil, W. Deracemization of mexiletine biocatalyzed by ω-transaminases. Org. Lett. 2009, 11, 4810–4812. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.K.; Bulger, P.G.; Kosjek, B.; Belyk, K.M.; Rivera, N.; Scott, M.E.; Humphrey, G.R.; Limanto, J.; Bachert, D.C.; Emerson, K.M. Process development of C–N cross-coupling and enantioselective biocatalytic reactions for the asymmetric synthesis of niraparib. Org. Proc. Res. Dev. 2013, 18, 215–227. [Google Scholar] [CrossRef]
- Peng, Z.; Wong, J.W.; Hansen, E.C.; Puchlopek-Dermenci, A.L.; Clarke, H.J. Development of a concise, asymmetric synthesis of a smoothened receptor (SMO) inhibitor: Enzymatic transamination of a 4-piperidinone with dynamic kinetic resolution. Org. Lett. 2014, 16, 860–863. [Google Scholar] [CrossRef] [PubMed]
- Koszelewski, D.; Clay, D.; Rozzell, D.; Kroutil, W. Deracemisation of α-chiral primary amines by a one-pot, two-step cascade reaction catalysed by ω-transaminases. Eur. J. Org. Chem. 2009, 14, 2289–2292. [Google Scholar] [CrossRef]
- Shin, G.; Mathew, S.; Shon, M.; Kim, B.G.; Yun, H. One-pot one-step deracemization of amines using ω-transaminases. Chem. Commun. 2013, 49, 8629–8631. [Google Scholar] [CrossRef] [PubMed]
- Steffen-Munsberg, F.; Vickers, C.; Thontowi, A.; Schätzle, S.; Tumlirsch, T.; Svedendahl-Humble, M.; Land, H.; Berglund, P.; Bornscheuer, U.T.; Höhne, M. Connecting unexplored protein crystal structures to enzymatic function. ChemCatChem 2013, 5, 150–153. [Google Scholar] [CrossRef]
- Shin, J.S.; Kim, B.G. Exploring the Active Site of Amine: Pyruvate Aminotransferase on the Basis of the Substrate Structure-Reactivity Relationship: How the Enzyme Controls Substrate Specificity and Stereoselectivity. J. Org. Chem. 2002, 67, 2848–2853. [Google Scholar] [CrossRef] [PubMed]
- Nobili, A.; Steffen-Munsberg, F.; Kohls, H.; Trentin, I.; Schulzke, C.; Höhne, M.; Bornscheuer, U.T. Engineering the active site of the amine transaminase from Vibrio fluvialis for the asymmetric synthesis of aryl–alkyl amines and amino alcohols. ChemCatChem 2015, 7, 757–760. [Google Scholar] [CrossRef]
- Steffen-Munsberg, F.; Vickers, C.; Thontowi, A.; Schätzle, S.; Meinhardt, T.; Svedendahl-Humble, M.; Land, H.; Berglund, P.; Bornscheuer, U.T.; Höhne, M. Revealing the structural basis of promiscuous amine transaminase activity. ChemCatChem 2013, 5, 154–157. [Google Scholar] [CrossRef]
- Desai, A.A. Sitagliptin manufacture: A compelling tale of green chemistry, process intensification, and industrial asymmetric catalysis. Angew. Chem. Int. Ed. 2011, 50, 1974–1976. [Google Scholar] [CrossRef] [PubMed]
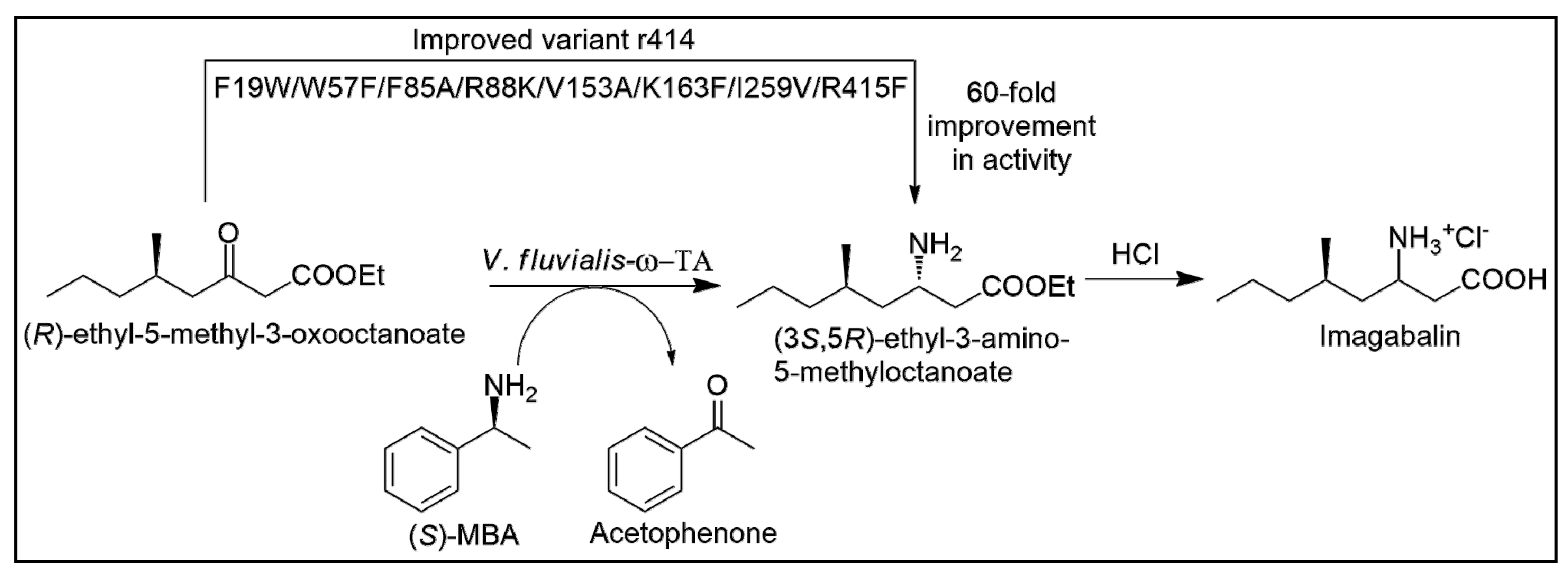
- Midelfort, K.S.; Kumar, R.; Han, S.; Karmilowicz, M.J.; McConnell, K.; Gehlhaar, D.K.; Mistry, A.; Chang, J.S.; Anderson, M.; Villalobos, A.; et al. Redesigning and characterizing the substrate specificity and activity of Vibrio fluvialis aminotransferase for the synthesis of Imagabalin. Protein Eng. Des. Sel. 2012, 26, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Genz, M.; Melse, O.; Schmidt, S.; Vickers, C.; Dörr, M.; Van den Bergh, T.; Joosten, H.J.; Bornscheuer, U.T. Engineering the Amine Transaminase from Vibrio fluvialis towards Branched-Chain Substrates. ChemCatChem 2016, 8, 3199–3202. [Google Scholar] [CrossRef]
- Dourado, D.F.; Pohle, S.; Carvalho, A.T.; Dheeman, D.S.; Caswell, J.M.; Skvortsov, T.; Miskelly, I.; Brown, R.T.; Quinn, D.J.; Allen, C.C.; et al. Rational Design of a (S)-Selective-Transaminase for Asymmetric Synthesis of (1S)-1-(1, 1′-biphenyl-2-yl) ethanamine. ACS Catal. 2016, 6, 7749–7759. [Google Scholar] [CrossRef]
- Han, S.W.; Park, E.S.; Dong, J.Y.; Shin, J.S. Active-Site Engineering of ω-Transaminase for Production of Unnatural Amino Acids Carrying a Side Chain Bulkier than an Ethyl Substituent. Appl. Environ. Microbiol. 2015, 81, 6994–7002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlidis, I.V.; Weiß, M.S.; Genz, M.; Spurr, P.; Hanlon, S.P.; Wirz, B.; Iding, H.; Bornscheuer, U.T. Identification of (S)-selective transaminases for the asymmetric synthesis of bulky chiral amines. Nat. Chem. 2016, 8, 1076–1082. [Google Scholar] [CrossRef] [PubMed]
- Weiß, M.S.; Pavlidis, I.V.; Spurr, P.; Hanlon, S.P.; Wirz, B.; Iding, H.; Bornscheuer, U.T. Protein-engineering of an amine transaminase for the stereoselective synthesis of a pharmaceutically relevant bicyclic amine. Org. Biomol. Chem. 2016, 14, 10249–10254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiß, M.S.; Pavlidis, I.V.; Spurr, P.; Hanlon, S.P.; Wirz, B.; Iding, H.; Bornscheuer, U.T. Amine Transaminase Engineering for Spatially Bulky Substrate Acceptance. ChemBioChem 2017, 18, 1022–1026. [Google Scholar] [CrossRef] [PubMed]
- Polizzi, K.M.; Bommarius, A.S.; Broering, J.M.; Chaparro-Riggers, J.F. Stability of biocatalysts. Curr. Opin. Chem. Biol. 2007, 11, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Stepankova, V.; Bidmanova, S.; Koudelakova, T.; Prokop, Z.; Chaloupkova, R.; Damborsky, J. Strategies for stabilization of enzymes in organic solvents. ACS Catal. 2013, 3, 2823–2836. [Google Scholar] [CrossRef]
- Littlechild, J.A. Enzymes from extreme environments and their industrial applications. Front. Bioeng. Biotechnol. 2015, 3, 161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerioli, L.; Planchestainer, M.; Cassidy, J.; Tessaro, D.; Paradisi, F. Characterization of a novel amine transaminase from Halomonas elongata. J. Mol. Catal. B Enzym. 2015, 120, 141–150. [Google Scholar] [CrossRef]
- Chen, S.; Land, H.; Berglund, P.; Humble, M.S. Stabilization of an amine transaminase for biocatalysis. J. Mol. Catal. B Enzym. 2016, 124, 20–28. [Google Scholar] [CrossRef]
- Mathew, S.; Deepankumar, K.; Shin, G.; Hong, E.Y.; Kim, B.G.; Chung, T.; Yun, H. Identification of novel thermostable ω-transaminase and its application for enzymatic synthesis of chiral amines at high temperature. RSC Adv. 2016, 6, 69257–69260. [Google Scholar] [CrossRef]
- Chen, Y.; Yi, D.; Jiang, S.; Wei, D. Identification of novel thermostable taurine–pyruvate transaminase from Geobacillus thermodenitrificans for chiral amine synthesis. Appl. Microbiol. Biotechnol. 2016, 100, 3101–3111. [Google Scholar] [CrossRef] [PubMed]
- Ferrandi, E.E.; Previdi, A.; Bassanini, I.; Riva, S.; Peng, X.; Monti, D. Novel thermostable amine transferases from hot spring metagenomes. Appl. Microbiol. Biotechnol. 2017, 101, 4963–4979. [Google Scholar] [CrossRef] [PubMed]
- Pannuri, S.; Kamat, S.; Venkatesh; Garcia, A.; Rogelio, M. Thermostable Omega-Transaminases. PCT Application No. WO/2006/063336.15 June 2006.
- Sayer, C.; Isupov, M.N.; Westlake, A.; Littlechild, J.A. Structural studies of Pseudomonas and Chromobacterium ω-aminotransferases provide insights into their differing substrate specificity. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 564–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Börner, T.; Rämisch, S.; Reddem, E.R.; Bartsch, S.; Vogel, A.; Thunnissen, A.M.; Adlercreutz, P.; Grey, C. Explaining Operational Instability of Amine Transaminases: Substrate-Induced Inactivation Mechanism and Influence of Quaternary Structure on Enzyme–Cofactor Intermediate Stability. ACS Catal. 2017, 7, 1259–1269. [Google Scholar] [CrossRef]
- Börner, T.; Rämisch, S.; Bartsch, S.; Vogel, A.; Adlercreutz, P.; Grey, C. Three In One Go: Thermo-, Solvent and Catalytic Stability by Engineering the Cofactor-Binding Element of Amine Transaminases. ChemBioChem 2017, 18, 1482–1486. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Xie, D.F.; Feng, Y. Engineering thermostable (R)-selective amine transaminase from Aspergillus terreus through in silico design employing B-factor and folding free energy calculations. Biochem. Biophys. Res. Commun. 2017, 483, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Deepankumar, K.; Nadarajan, S.P.; Mathew, S.; Lee, S.G.; Yoo, T.H.; Hong, E.Y.; Kim, B.G.; Yun, H. Engineering Transaminase for Stability Enhancement and Site-Specific Immobilization through Multiple Noncanonical Amino Acids Incorporation. ChemCatChem 2015, 7, 417–421. [Google Scholar] [CrossRef]
- Deepankumar, K.; Shon, M.; Nadarajan, S.P.; Shin, G.; Mathew, S.; Ayyadurai, N.; Kim, B.G.; Choi, S.H.; Lee, S.H.; Yun, H. Enhancing Thermostability and Organic Solvent Tolerance of ω-Transaminase through Global Incorporation of Fluorotyrosine. Adv. Synth. Catal. 2014, 356, 993–998. [Google Scholar] [CrossRef]
- Patil, M.D.; Dev, M.J.; Shinde, A.S.; Bhilare, K.D.; Patel, G.; Chisti, Y.; Banerjee, U.C. Surfactant-mediated permeabilization of Pseudomonas putida KT2440 and use of the immobilized permeabilized cells in biotransformation. Process Biochem. 2017, 63, 113–121. [Google Scholar] [CrossRef]
- Yewale, T.; Singhal, R.S.; Vaidya, A.A. Immobilization of inulinase from Aspergillus niger NCIM 945 on chitosan and its application in continuous inulin hydrolysis. Biocatal. Agric. Biotechnol. 2013, 2, 96–101. [Google Scholar] [CrossRef]
- Koszelewski, D.; Müller, N.; Schrittwieser, J.H.; Faber, K.; Kroutil, W. Immobilization of ω-transaminases by encapsulation in a sol–gel/celite matrix. J. Mol. Catal. B Enzym. 2010, 63, 39–44. [Google Scholar] [CrossRef]
- Mallin, H.; Menyes, U.; Vorhaben, T.; Höhne, M.; Bornscheuer, U.T. Immobilization of two (R)-Amine Transaminases on an Optimized Chitosan Support for the Enzymatic Synthesis of Optically Pure Amines. ChemCatChem 2013, 5, 588–593. [Google Scholar] [CrossRef]
- Mallin, H.; Höhne, M.; Bornscheuer, U.T. Immobilization of (R)-and (S)-amine transaminases on chitosan support and their application for amine synthesis using isopropylamine as donor. J. Biotechnol. 2014, 191, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Truppo, M.D.; Strotman, H.; Hughes, G. Development of an immobilized transaminase capable of operating in organic solvent. ChemCatChem 2012, 4, 1071–1074. [Google Scholar] [CrossRef]
- Yi, S.S.; Lee, C.W.; Kim, J.; Kyung, D.; Kim, B.G.; Lee, Y.S. Covalent immobilization of ω-transaminase from Vibrio fluvialis JS17 on chitosan beads. Process Biochem. 2007, 42, 895–898. [Google Scholar] [CrossRef]
- De Souza, S.P., II; Silva, G.M.; Miranda, L.S.; Santiago, M.F.; Lam, F.L.; Dawood, A.; Bornscheuer, U.T.; De Souza, R.O. Cellulose as an efficient matrix for lipase and transaminase immobilization. RSC Adv. 2016, 6, 6665–6671. [Google Scholar] [CrossRef]
- Cárdenas-Fernández, M.; Neto, W.; López, C.; Alvaro, G.; Tufvesson, P.; Woodley, J.M. Immobilization of Escherichia coli containing ω-transaminase activity in LentiKats®. Biotechnol. Prog. 2012, 28, 693–698. [Google Scholar] [CrossRef] [PubMed]
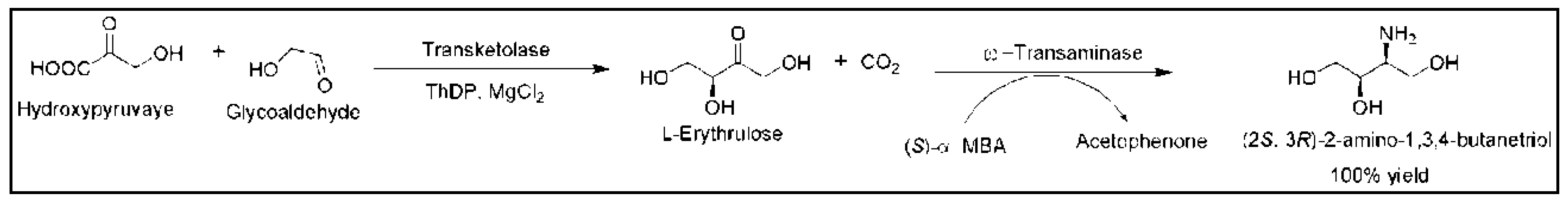
- Matosevic, S.; Lye, G.J.; Baganz, F. Immobilised enzyme microreactor for screening of multi-step bioconversions: Characterisation of a de novo transketolase-ω-transaminase pathway to synthesise chiral amino alcohols. J. Biotechnol. 2011, 155, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Halim, A.A.; Szita, N.; Baganz, F. Characterization and multi-step transketolase-ω-transaminase bioconversions in an immobilized enzyme microreactor (IEMR) with packed tube. J. Biotechnol. 2013, 168, 567–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Ni, K.; Wei, D.; Ren, Y. One-step purification and immobilization of his-tagged protein via Ni2+-functionalized Fe3O4@ polydopamine magnetic nanoparticles. Biotechnol. Bioprocess Eng. 2015, 20, 901–907. [Google Scholar] [CrossRef]
- Jia, H.; Huang, F.; Gao, Z.; Zhong, C.; Zhou, H.; Jiang, M.; Wei, P. Immobilization of ω-transaminase by magnetic PVA-Fe3O4 nanoparticles. Biotechnol. Rep. 2016, 10, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Cui, W.H.; Du, K.; Gao, Q.; Du, M.; Ji, P.; Feng, W. Immobilization of R-ω-transaminase on MnO2 nanorods for catalyzing the conversion of (R)-1-phenylethylamine. J. Biotechnol. 2017, 245, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Sperl, J.M.; Sieber, V. Multienzyme Cascade Reactions- Status and Recent Advances. ACS Catal. 2018, 8, 2385–2396. [Google Scholar] [CrossRef]
- Ahsan, M.M.; Jeon, H.; Nadarajan, S.P.; Chung, T.; Yoo, H.W.; Kim, B.G.; Patil, M.D.; Yun, H. Biosynthesis of the Nylon 12 Monomer, ω-Aminododecanoic Acid with Novel CYP153A, AlkJ, and ω-TA Enzymes. Biotechnol. J. 2018, 13, 1700562. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, M.M.; Sung, S.; Jeon, H.; Patil, M.D.; Chung T Yun, H. Biosynthesis of Medium-to Long-Chain α, ω-Diols from Free Fatty Acids Using CYP153A Monooxygenase, Carboxylic Acid Reductase, and E. coli Endogenous Aldehyde Reductases. Catalysts 2017, 8, 4. [Google Scholar] [CrossRef]
- Simon, R.C.; Richter, N.; Busto, E.; Kroutil, W. Recent developments of cascade reactions involving ω-transaminases. ACS Catal. 2013, 4, 129–143. [Google Scholar] [CrossRef]
- Hepworth, L.J.; France, S.P.; Hussain, S.; Both, P.; Turner, N.J.; Flitsch, S.L. Enzyme cascades in whole cells for the synthesis of chiral cyclic amines. ACS Catal. 2017, 7, 2920–2925. [Google Scholar] [CrossRef]
- Fuchs, M.; Farnberger, J.E.; Kroutil, W. The industrial age of biocatalytic transamination. Eur. J. Org. Chem. 2015, 32, 6965–6982. [Google Scholar] [CrossRef] [PubMed]
- France, S.P.; Hepworth, L.J.; Turner, N.J.; Flitsch, S.L. Constructing biocatalytic cascades: In vitro and in vivo approaches to de novo multi-enzyme pathways. ACS Catal. 2016, 7, 710–724. [Google Scholar] [CrossRef]
- Fuchs, M.; Tauber, K.; Sattler, J.; Lechner, H.; Pfeffer, J.; Kroutil, W.; Faber, K. Amination of benzylic and cinnamic alcohols via a biocatalytic, aerobic, oxidation–transamination cascade. RSC Adv. 2012, 2, 6262–6265. [Google Scholar] [CrossRef]
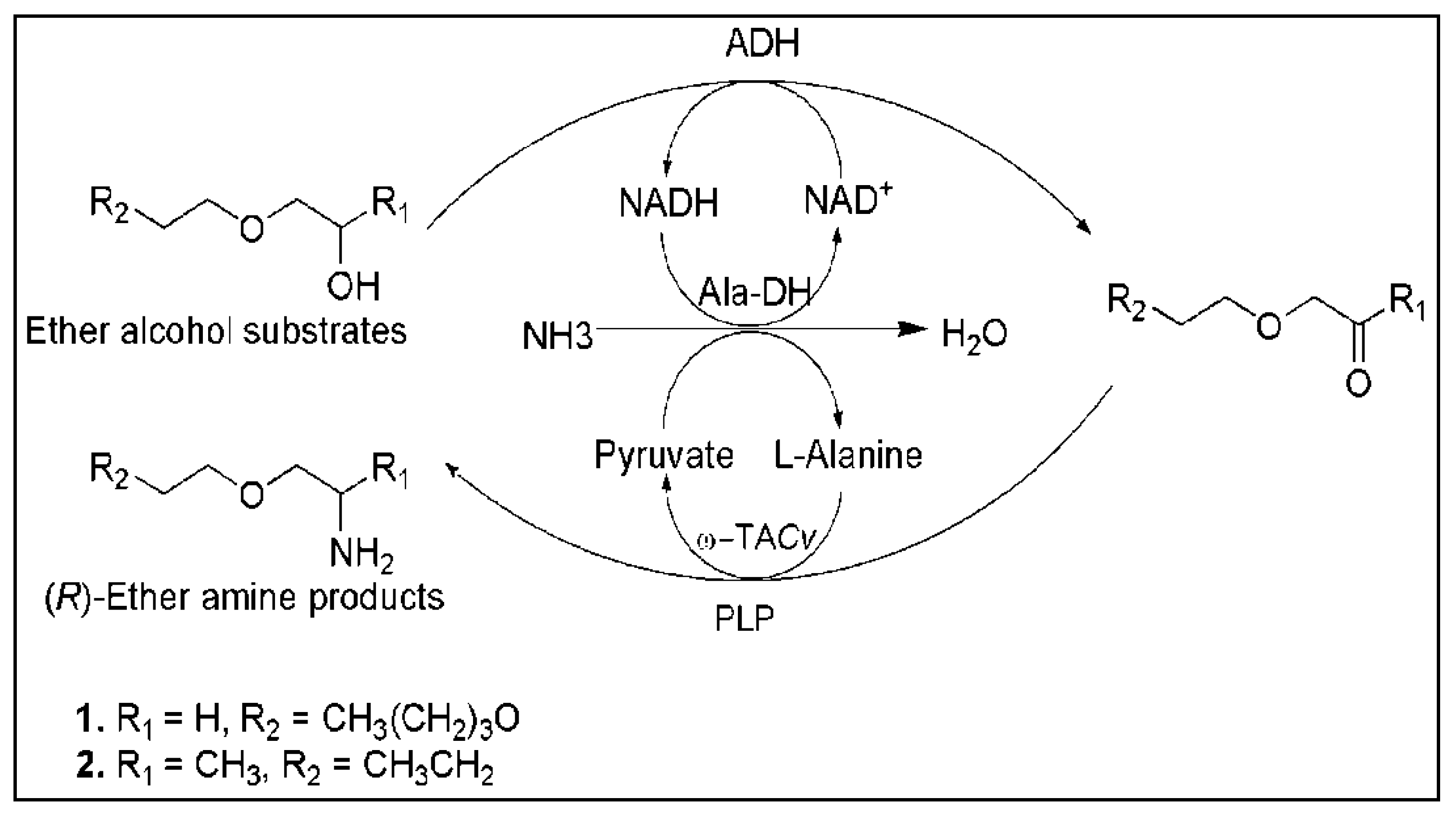
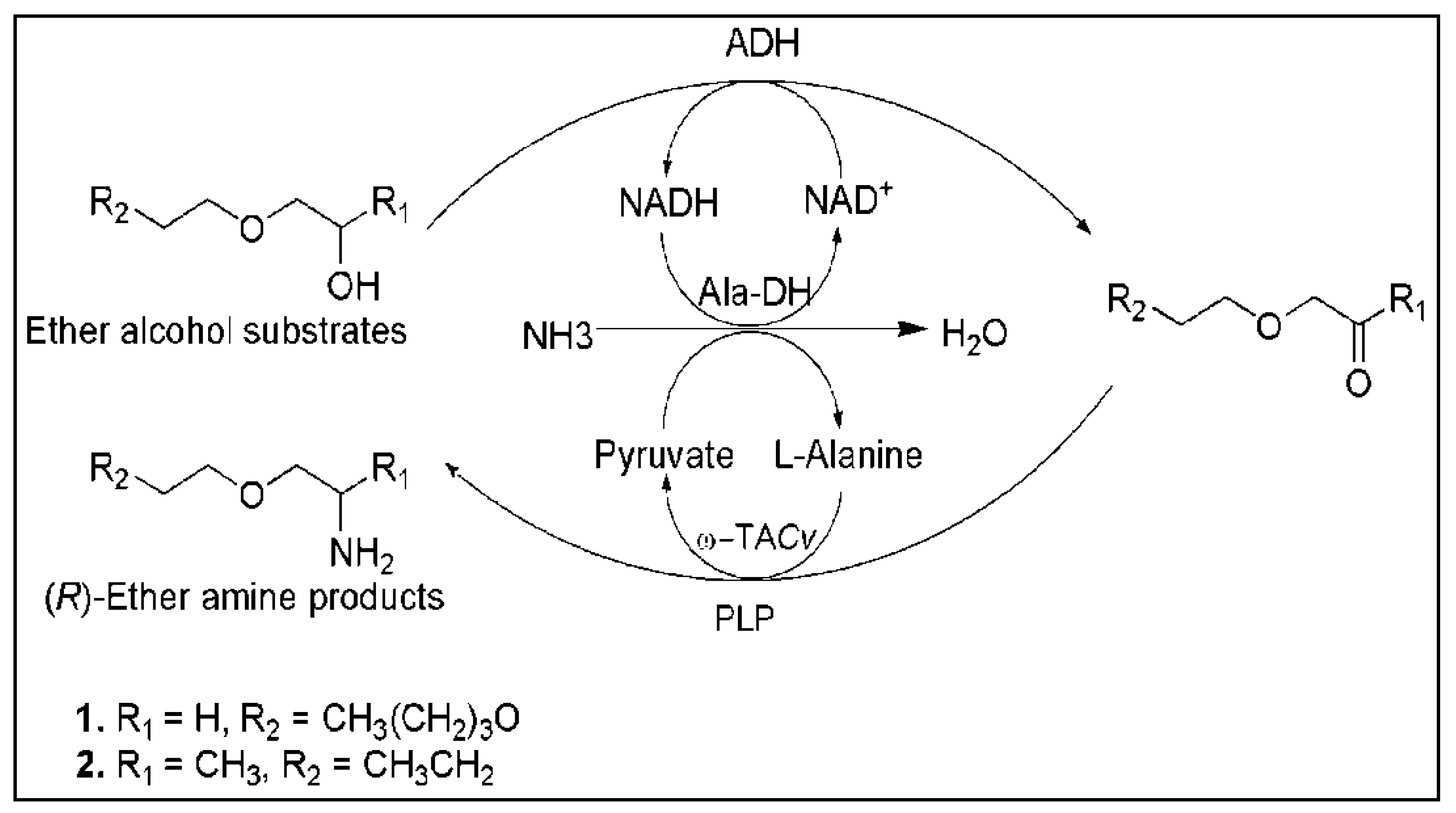
- Palacio, C.M.; Crismaru, C.G.; Bartsch, S.; Navickas, V.; Ditrich, K.; Breuer, M.; Abu, R.; Woodley, J.M.; Baldenius, K.; Wu, B.; et al. Enzymatic network for production of ether amines from alcohols. Biotechnol. Bioeng. 2016, 113, 1853–1861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birrell, J.A.; Jacobsen, E.N. A Practical method for the synthesis of highly enantioenriched trans-1, 2-amino alcohols. Org. Lett. 2013, 15, 2895–2897. [Google Scholar] [CrossRef] [PubMed]
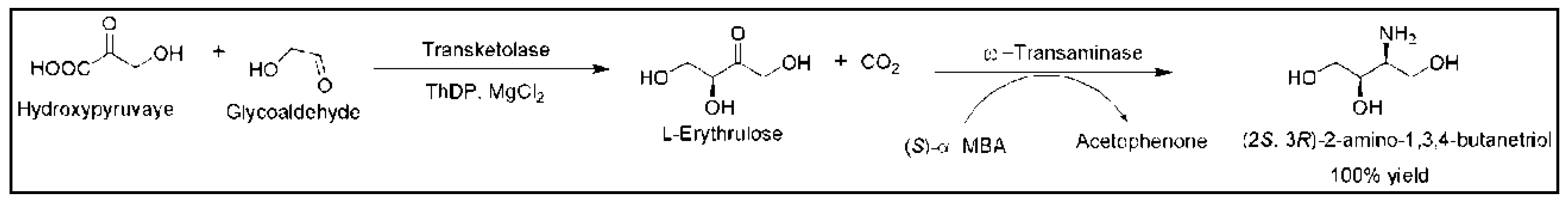
- Gruber, P.; Carvalho, F.; Marques, M.P.; O’Sullivan, B.; Subrizi, F.; Dobrijevic, D.; Ward, J.; Hailes, H.C.; Fernandes, P.; Wohlgemuth, R.; et al. Enzymatic synthesis of chiral amino-alcohols by coupling transketolase and transaminase-catalyzed reactions in a cascading continuous-flow microreactor system. Biotechnol. Bioeng. 2018, 115, 586–596. [Google Scholar] [CrossRef] [PubMed]
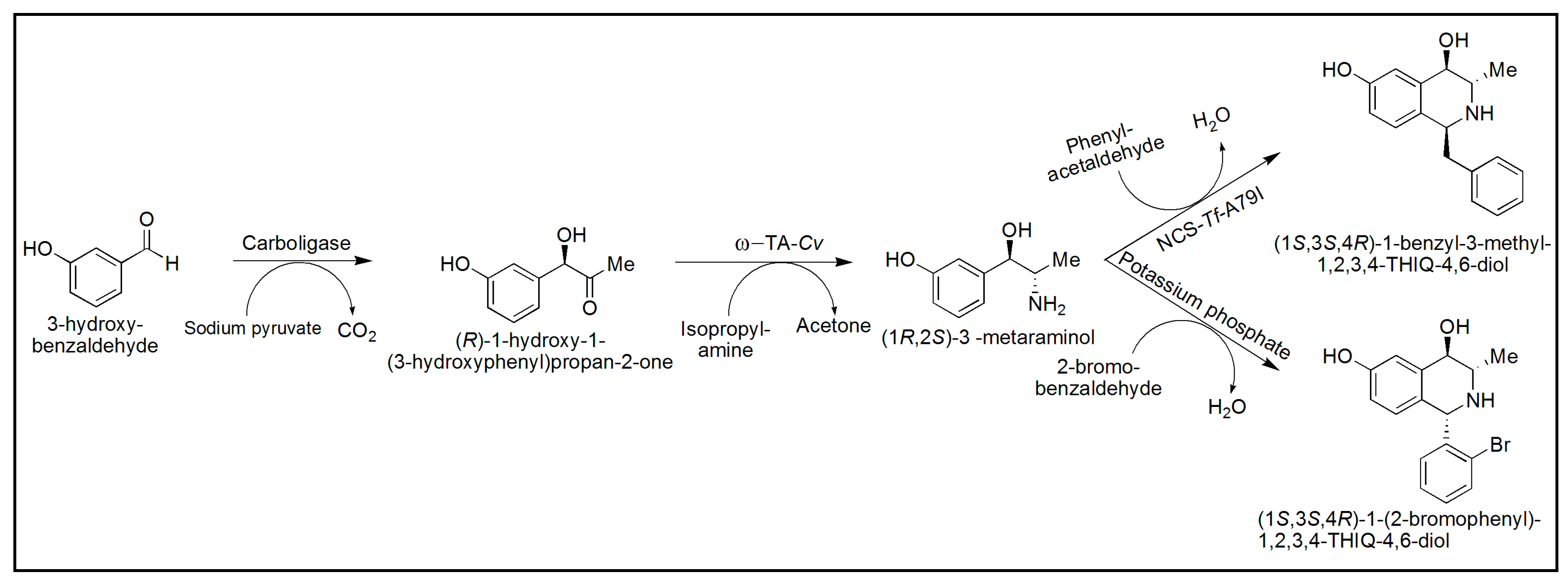
- Erdmann, V.; Lichman, B.R.; Zhao, J.; Simon, R.C.; Kroutil, W.; Ward, J.M.; Hailes, H.C.; Rother, D. Enzymatic and Chemoenzymatic Three-Step Cascades for the Synthesis of Stereochemically Complementary Trisubstituted Tetrahydroisoquinolines. Angew. Chem. Int. Ed. 2017, 56, 12503–12507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischereder, E.M.; Pressnitz, D.; Kroutil, W. Stereoselective Cascade to C3-methylated strictosidine derivatives employing transaminases and strictosidine synthases. ACS Catal. 2015, 6, 23–30. [Google Scholar] [CrossRef]
- Turner, N.J.; Kumar, R. Editorial overview: Biocatalysis and biotransformation: The golden age of biocatalysis. Curr. Opin. Chem. Biol. 2018, 43, A1–A3. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
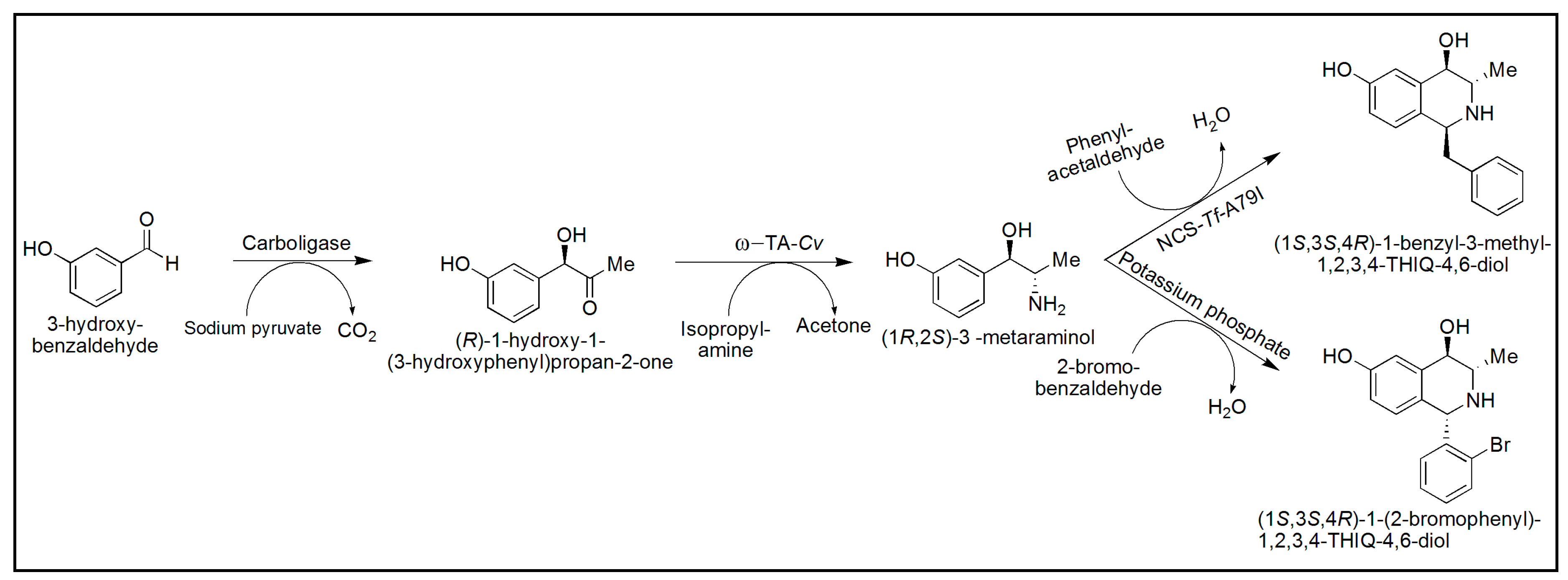{kind=link}
{kind=link}
| Sr. No. | Source | Enzyme Selectivity | Strategy Used | Optimum Temperature (°C) | Remarks | Reference |
|---|---|---|---|---|---|---|
| 1 | Halomonas elongata DSM 2581 | (S)-selective | Conserved domain analysis | 50 | First study reported on ω-TA from the moderate halophile bacterium H. elongate. The stability was unaffected in the presence of organic solvents. | [81] |
| 2 | Chromobacterium violaceum | (S)-selective | 65 | The enzyme performance was improved 5-fold by a co-lyophilization with surfactants | [82] | |
| 3 | Sphaerobacter thermophilus | (S)-selective | BLAST search against protein sequences from thermophiles | 60 | The enzyme was utilized for the stereoselective synthesis of β- and γ- amino acids | [33] |
| 4 | Thermomicrobium roseum | (S)-selective | BLAST search against protein sequences from thermophiles | 87 | Volatile inhibitory byproducts were removed by performing asymmetric synthesis and kinetic resolution at high temperature | [83] |
| 5 | Geobacillus thermodenitrificans | (S)-selective | BLAST search against protein sequences from thermophiles | 65 | The enzyme showed relatively good activity toward ketoses, suggesting its potential for catalyzing the asymmetric synthesis of chiral amino alcohols. | [84] |
| 6 | Hot spring metagenomes | (S)-selective | Metagenomics | 88 | The most thermostable natural ω-TA known till date. | [85] |
| Sr. No. | Source | Enzyme Selectivity | Strategy Used | Remarks | Reference |
|---|---|---|---|---|---|
| 1 | Arthrobacter sp. | (R)-selective | Substrate walking, modeling, and mutation approach | 11 rounds of evolution generated the optimized biocatalyst, which could afford a successful transamination of in the presence of 50% DMSO as a co-solvent | [29] |
| 2 | Arthobacter citreus | (S)-selective | Structure-guided enzyme mutagenesis | The enzyme was used for the industrial production of substituted aminotetraline | [86] |
| 3 | Novel enzyme from c-LEcta’smetagenomic library | Semi-rational mutagenesis | 32 amino acid residues were targeted around the active site including the cofactor-ring motif for superior operational, thermo- and solvent stability | [89] | |
| 4 | Aspergillus terreus | (R)-selective | In silico design employing B-factor and folding free energy calculations | The optimum catalytic temperature of a mutant T130F was increased by 10 °C | [90] |
| 5 | Sphaerobacter thermophilus | (S)-selective | Residue-specific andsite-specific incorporation of unnatural amino acids | The residue-specific incorporation showed 2-fold enhancement in the half life at 70 °C | [91] |
| 6 | Vibrio fluvialis JS17 | (S)-selective | Global incorporation of unnatural amino acids | Mutant exhibited ~2-fold higher tolerance towards 20% DMSO compared to wild-type | [92] |
| Sr. No. | Source | Type of Immobilization | Support Used for Immobilization | Comment | Reference |
|---|---|---|---|---|---|
| 1 | Chromobacterium violaceum (Trp60Cys mutant) | Covalent binding | bisepoxide-activated polymeric resins | Immobilized preparation was used for 19 consecutive reaction cycles and in media containing up to 50% (v/v) DMSO as co-solvent in batch mode reactions. | [1] |
| 2 | (S)-selective ATA-47 | Ionic interaction | Relizyme HA403 (commercial material) | Immobilized preparation retained more than 50% initial activity for 8 cycles (each of 24 h; corresponding to more than 250 h of operation) | [41] |
| 3 | (R)-selective Ate-TA | Hydrophobic | Supabeads EC-HA (commercial material) | The immobilized preparation increased the thermal stability, allowing storing the enzyme for more than 60 days | [41] |
| 4 | ω-TA 117 | Sol–gel entrapment | Celite 545 | The immobilized enzyme preparation could be recycledeight times with only moderate decrease of activity for each cycle | [95] |
| 5 | Gibberella zeae | Covalent binding | Modified chitosan | Significant improvement in pH and temperature stability | [96] |
| 6 | Neosartorya fischeri | Covalent binding | Modified chitosan | Significant improvement in pH and temperature stability | [96] |
| 7 | Aspergillus fumigatus | Covalent binding | Chitosan | The immobilized enzyme showed higher activity at 70 °C compared to free enzyme | [97] |
| 8 | Ruegeria pomeroyi | Covalent binding | Chitosan | The immobilized enzyme retained activity after four batches | [97] |
| 9 | Vibrio fluvialis JS17 | Covalent binding | Chitosan | Significant improvement in stability over broad range of pH and temperature | [99] |
| 10 | Vibrio fluvialis | Covalent binding | Functionalized cellulose | The immobilized enzyme retained activity for four cycles | [100] |
| 11 | Bradyrhizobium japonicum | Affinity | Ni2+-functionalized polydopamine magneticna noparticles | Simultaneous purification and immobilization of the his-tagged protein could be achieved | [104] |
| 12 | Mycobacterium vanbaalenii | Covalent binding | magnetic PVA-Fe3O4 nanoparticles | The immobilized enzyme could be successfully reused for 13 times in biotransformation | [105] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D. Patil, M.; Grogan, G.; Bommarius, A.; Yun, H. Recent Advances in ω-Transaminase-Mediated Biocatalysis for the Enantioselective Synthesis of Chiral Amines. Catalysts 2018, 8, 254. https://doi.org/10.3390/catal8070254
D. Patil M, Grogan G, Bommarius A, Yun H. Recent Advances in ω-Transaminase-Mediated Biocatalysis for the Enantioselective Synthesis of Chiral Amines. Catalysts. 2018; 8(7):254. https://doi.org/10.3390/catal8070254
Chicago/Turabian StyleD. Patil, Mahesh, Gideon Grogan, Andreas Bommarius, and Hyungdon Yun. 2018. "Recent Advances in ω-Transaminase-Mediated Biocatalysis for the Enantioselective Synthesis of Chiral Amines" Catalysts 8, no. 7: 254. https://doi.org/10.3390/catal8070254
APA StyleD. Patil, M., Grogan, G., Bommarius, A., & Yun, H. (2018). Recent Advances in ω-Transaminase-Mediated Biocatalysis for the Enantioselective Synthesis of Chiral Amines. Catalysts, 8(7), 254. https://doi.org/10.3390/catal8070254