1. Introduction
In the 1980s, it was reported that tungstated zirconia (WO
x/ZrO
2) could be used as a solid acid catalyst; however, there are still many questions and apparent contradictions regarding the nature and strength of the acid sites in this material, as demonstrated in the recent review by Zhou et al. [
1]. A major difficulty with tungstated-zirconia catalysts is that their properties depend on how they are made. It is likely, for this reason, that some studies indicated that the structure of the underlying ZrO
2, whether amorphous or crystalline (tetragonal or monoclinic), influences the activity of the sites [
2], while other workers reported that the crystallographic structure of the zirconia is not important [
3,
4]. It was also suggested that the sites may be of varying strength, depending on the WO
x cluster size, with one theoretical study reporting that the Brønsted sites not only depend on cluster size, but can also approach super-acid strength [
1]. However, the observations that it is necessary to add Pt in order for tungstated zirconia to exhibit alkane-isomerization activity [
5,
6] would suggest that the sites are less strong, in addition to tungstated zirconia being selective to ether formation in the dehydration of alcohols [
7], without forming olefins, unlike with protonic zeolites.
Most researchers prepared their tungstated-zirconia catalysts via the aqueous impregnation of ammonium metatungstate ((NH
4)
10W
12O
41·5H
2O), followed by calcination [
1]. In addition to the fact that this precursor consists of a relatively large number of tungsten atoms, the drying process itself can result in multiple clusters coming together before decomposing upon calcination [
8]. While the WO
x species may spread over the support surface during calcination, the implications of there being so many tungsten atoms together in the initial state implies that there will likely be large clusters on the support in all cases. This is important because mono-tungstate species are not believed to be the active component [
1]. Furthermore, the fact that the catalytic properties are a function of the tungsten-oxide coverage and pretreatment conditions [
9,
10] implies that discrepancies in the results from different studies are likely due to differences in the detailed nature of the clusters that are formed in the catalysts.
In the presented work, we set out to investigate the catalytic sites in WO
x/ZrO
2 catalysts prepared using atomic layer deposition (ALD). ALD is a self-limiting process in which WO
x is deposited through repeated, cyclic exposures of W(CO)
6 and oxidants [
11,
12,
13]. Because the amount of WO
x deposited per cycle in ALD is low and the deposition is uniform, ALD ensures the formation of uniform, atomic-scale layers, which maximize the interfacial contact between WO
x and ZrO
2. This allows the formation of isolated WO
x species, as well as much more control over the tungsten cluster sizes by increasing tungsten coverage. A complicating factor in the characterization is that zirconia itself exhibits activity in the reaction of amines and alcohols, and contributes to the activity of catalysts for some reactions. Fortunately, as demonstrated in this work, the chemistries on bare zirconia are oftentimes distinguishable from those that occur on WO
x/ZrO
2 interfaces, and thus, provide additional information on coverage and dispersion.
We also set out to test claims that tungstated-zirconia could show super-acidic properties. The quantification of Brønsted-acid strength in solid acids is difficult, and some common measures, such as heats of adsorption for ammonia or pyridine, were shown to be uninformative [
14]. A simple reaction that appears to depend only on the ability of the solid to protonate a weak base involves the H–D exchange between an aromatic molecule and deuterated acid sites [
15]. In our case, we examined light-off curves for H–D exchange between D
2O and either toluene (C
7H
8) or chlorobenzene (C
6H
5Cl) [
15]. Essentially, all hydroxides on solids exchange readily with D
2O [
14,
16], meaning that the presence of D
2O ensures a high concentration of deuterated acid sites. However, the deuteration of toluene (proton affinity = 784 kJ/mol) or chlorobenzene (proton affinity = 753 kJ/mol) requires the formation of a carbenium ion, so that the temperature at which exchange becomes rapid should be a reasonable measure of the acid strength. In agreement with expectations based on proton affinities, higher temperatures are required for H–D exchange with chlorobenzene compared to that with toluene; however, the Brønsted sites in tungstated zirconia appear to be significantly weaker than those in H-ZSM-5.
2. Results
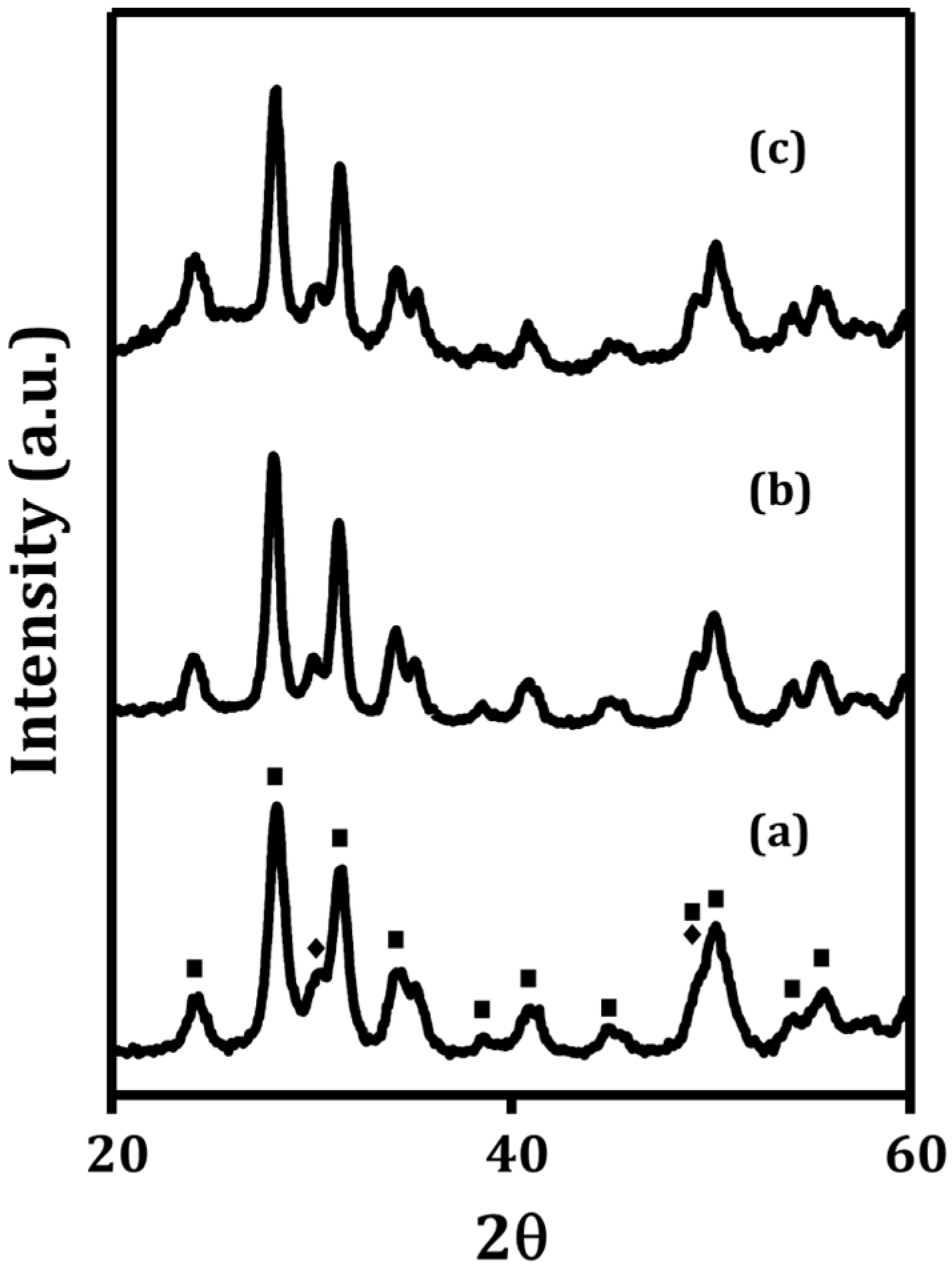
The ZrO
2 substrate prepared for this study had an initial BET surface area of 65 m
2/g. As shown by the X-ray diffraction (XRD) pattern in
Figure 1a, its phase was primarily monoclinic with a small amount of the tetragonal phase. The WO
x loadings were then determined as a function of the number of ALD cycles by measuring the sample weights.
Table 1 shows these loadings, together with BET surface areas. In the first five ALD cycles, the sample weight increased almost linearly at 22 mg WO
x/g ZrO
2 per cycle. Assuming an O:W stoichiometry of three and uniform film growth over the entire ZrO
2 surface, this corresponds to a growth rate of 0.048 nm/cycle, a value that is reasonable for the size of the W(CO)
6 precursor, but that is somewhat larger than the value reported in the literature for the growth of WO
3 films on flat surfaces (0.023 nm/cycle) for similar growth conditions [
11,
13]. It is interesting to notice that the growth rate of WO
x on the 140-m
2/g SiO
2 support, calculated from the amount deposited after five ALD cycles, was only 0.025 nm/cycle, implying that the substrate does influence the initial deposition rate. Finally, it is useful to consider that deposition of 22 mg of WO
3 onto the 65-m
2/g ZrO
2 sample corresponds to 8.8 × 10
17 W atoms/m
2. This is a fraction of an oxide monolayer, which implies that, in the absence of surface migration, WO
x species are likely spatially isolated after a single ALD cycle.
After five ALD cycles, the BET surface area decreased to 46 m
2/g. Some of the decrease was due to the increase in sample mass; however, the majority of the loss in surface area per mass of sample must be associated with the narrowing or blocking of pores. The WO
x added with five ALD cycles did not cause any changes in the XRD pattern (
Figure 1b). Because of the decreasing surface area of the sample, the rate at which the weight changed decreased somewhat with the number of ALD cycles. After 40 ALD cycles, the WO
x loading was 42 wt%, or 0.72 g WO
x/g ZrO
2. Assuming that the film was uniform with a stoichiometry and density of bulk WO
3, the film thickness after 40 cycles was 1.5 nm, a thickness significantly greater than that of a monolayer. However, even with this relatively thick film, the XRD pattern (
Figure 1c) showed no evidence of a well-defined crystalline WO
x phase. The only change in the XRD pattern with the addition of this large amount of WO
x was a slightly elevated baseline between 25 and 35 degrees 2θ, and again above 50 degrees 2θ. These broad features may be associated with a very small fraction of mixed WO
x phases. However, if large, three-dimensional, crystalline clusters were being formed, it would have been apparent in the diffraction pattern.
Scanning transmission electron microscopy (STEM) imaging and energy-dispersive spectroscopy (EDS) elemental mapping on the 40W-Zr sample (
Figure 2) demonstrate that the WO
x ALD films deposited uniformly over the ZrO
2 surface. While there were no obvious features in the image, despite the high WO
x coverage, the EDS mapping of W and Zr indicates the co-existence of W and Zr elements in the sample. The overlap in the signals reveals their uniform distribution.
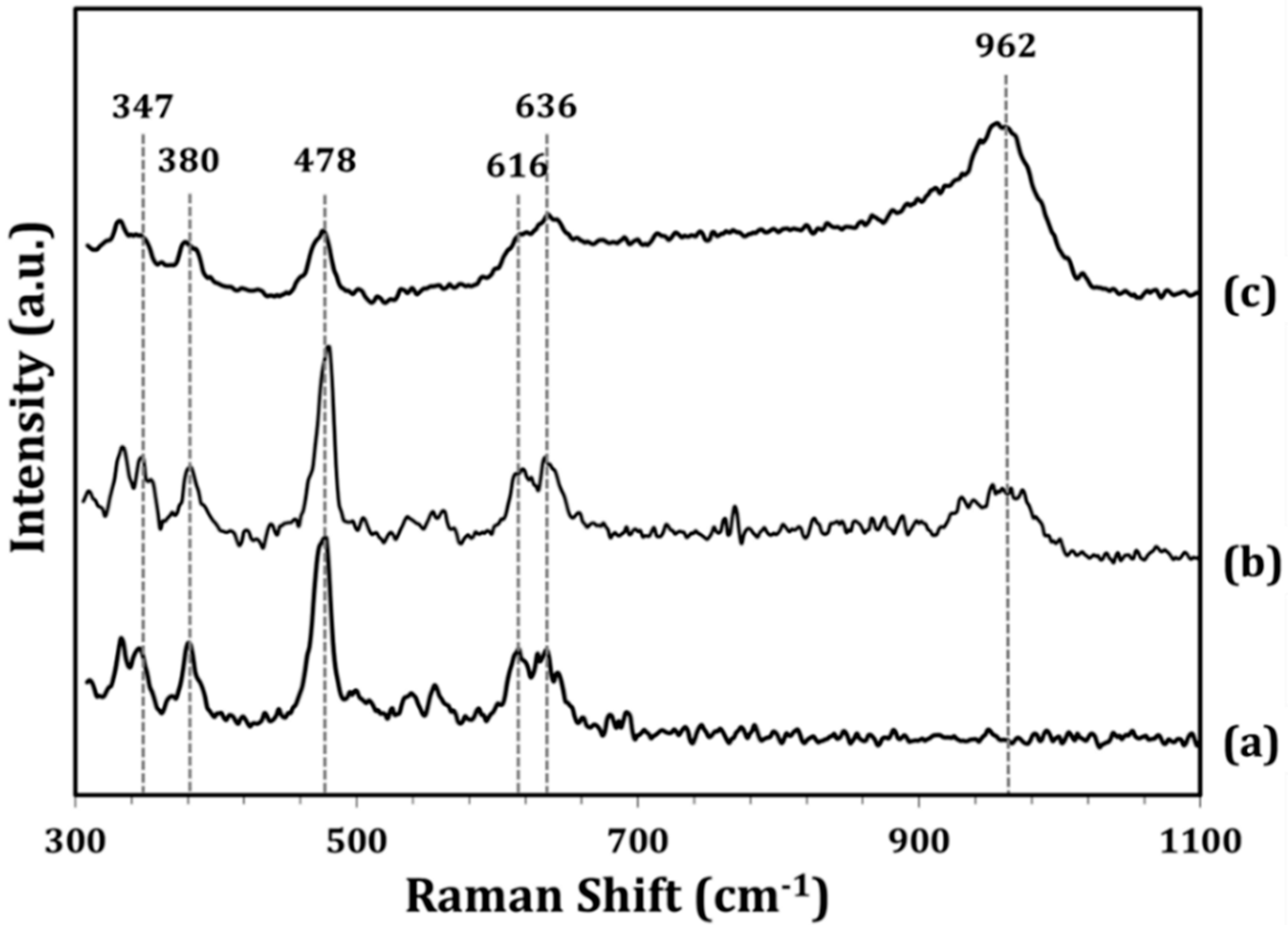
To characterize the structure of the WO
x ALD films, Raman spectra of the ZrO
2 support and of the impW-Zr (10-wt% WO
x) and 5W-Zr samples were measured, both of which had WO
x loadings that were close to 10 wt%. The data for ZrO
2 (
Figure 3a) show vibrational bands at ~347, 380, 478, 616, and 636 cm
−1, which are well known to be due to ZrO
2 [
17]. The spectrum for the impW-Zr sample (
Figure 3b) exhibits the same bands, but with a new, broad feature centered at ~962 cm
−1, which was previously assigned to the symmetric stretching mode of a terminal W=O bond [
17]. This broad feature may also include a contribution from a bridging W–O–Zr bond at 915 cm
−1. The spectrum of the 5W-Zr sample (
Figure 3c) is similar, except that the vibrational features associated with WO
x relative to those with ZrO
2 are significantly more intense, despite having the same WO
x loading. This is likely due to the WO
x being spread more uniformly over the ZrO
2 surface in the ALD-prepared sample.
A further indication that the WO
x layer was more uniform on the ALD-prepared sample is demonstrated in
Figure 4, which shows the temperature-programmed desorption/thermogravimetric analysis (TPD-TGA) results for 2-propanol on the ZrO
2, 5W-Zr, and impW-Zr samples. The TGA data show that the initial coverages following room-temperature adsorption and 1-h evacuation were slightly higher on the ZrO
2 and impW-Zr samples (between 250 and 300 µmol/g) compared to the 5W-Zr (200 µmol/g). However, this is almost certainly due to the lower surface area of the 5W-Zr sample. The initial specific coverages were roughly 2.7 × 10
18 molecules/m
2 for each of the samples. While some of the 2-propanol (
m/
e = 45) desorbed unreacted from all three samples, significant fractions of the 2-propanol desorbed from the samples as propene (
m/
e = 41) and H
2O due to reactions on either Lewis- or Brønsted-acid sites [
18]. Water is not shown because it tends to desorb over a broad temperature range. What is more interesting is that the dehydration reaction occurred at very different temperatures on ZrO
2 and on WO
x/ZrO
x sites. On the ZrO
2 and 5W-Zr samples, the dehydration reaction occurred over narrow temperature ranges in peaks centered at 560 K on ZrO
2, and 405 K on 5W-Zr. The TPD result of impW-Zr shows similarly sized peaks at both temperatures, suggesting that impW-Zr has regions of bare ZrO
2 and regions covered by WO
x. Even though the WO
x loadings were the same for 5W-Zr and impW-Zr, the distributions of WO
x were clearly different, with tungsten distributed much more uniformly on the sample prepared using ALD.
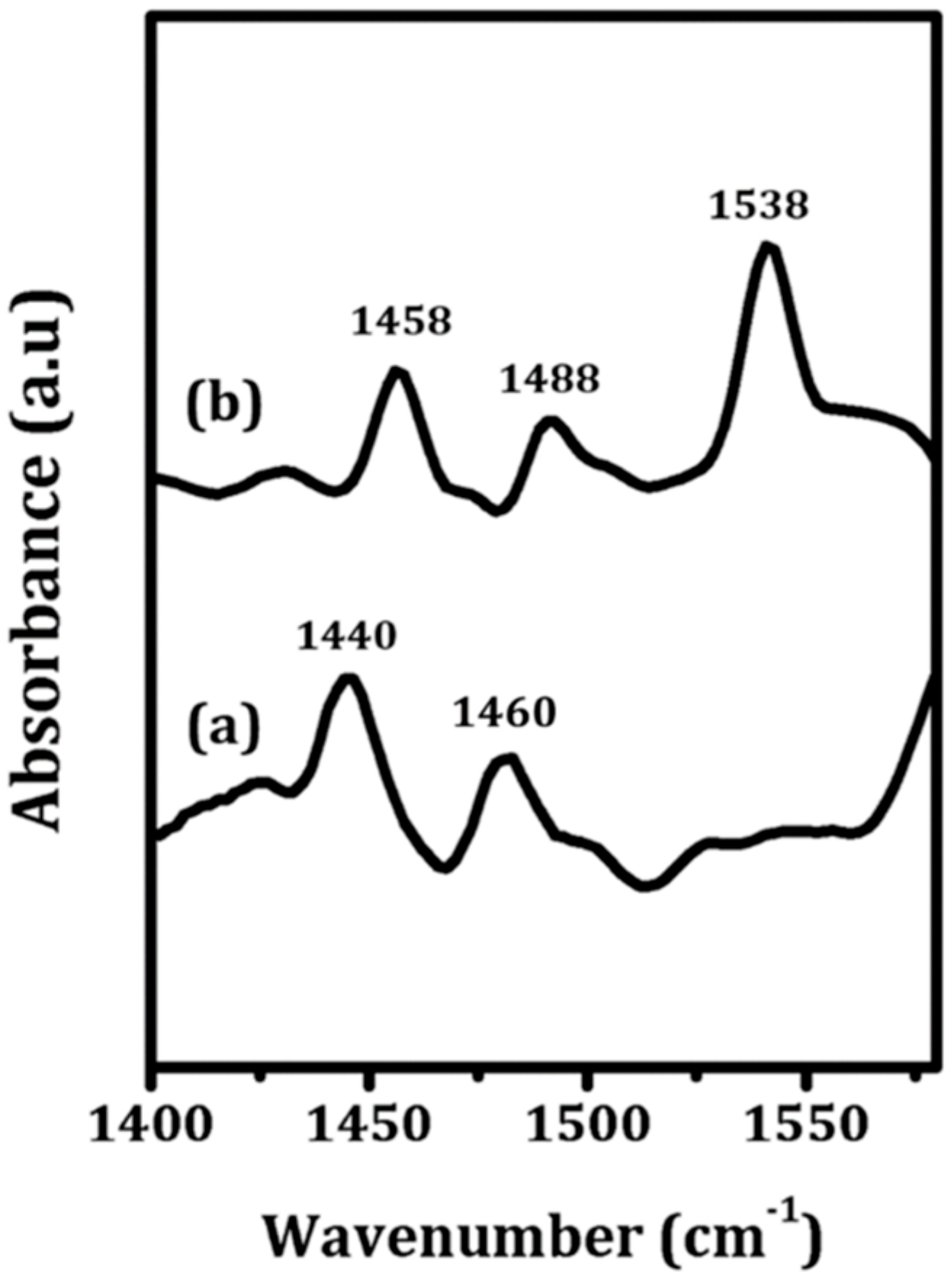
To qualitatively assess the nature of the sites on the ZrO
2 and 5W-Zr samples, FTIR measurements were performed following adsorption of pyridine, with results shown in
Figure 5. The spectrum for ZrO
2 (
Figure 5a) shows only bands at 1440 and 1460 cm
−1, which are characteristic of adsorption at Lewis sites. In contrast to this, the spectrum of pyridine on the ALD-prepared 5W-Zr (
Figure 5b) also exhibits a band near 1540 cm
−1, which can be assigned to adsorbed pyridinium ions, implying 5W-Zr contains a significant concentration of Brønsted sites.
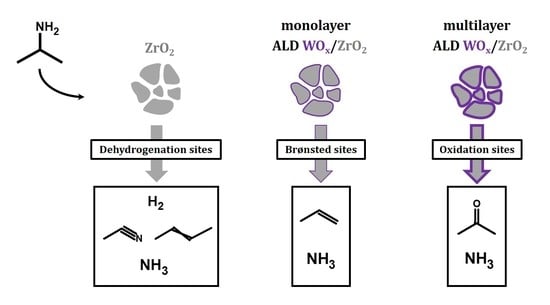
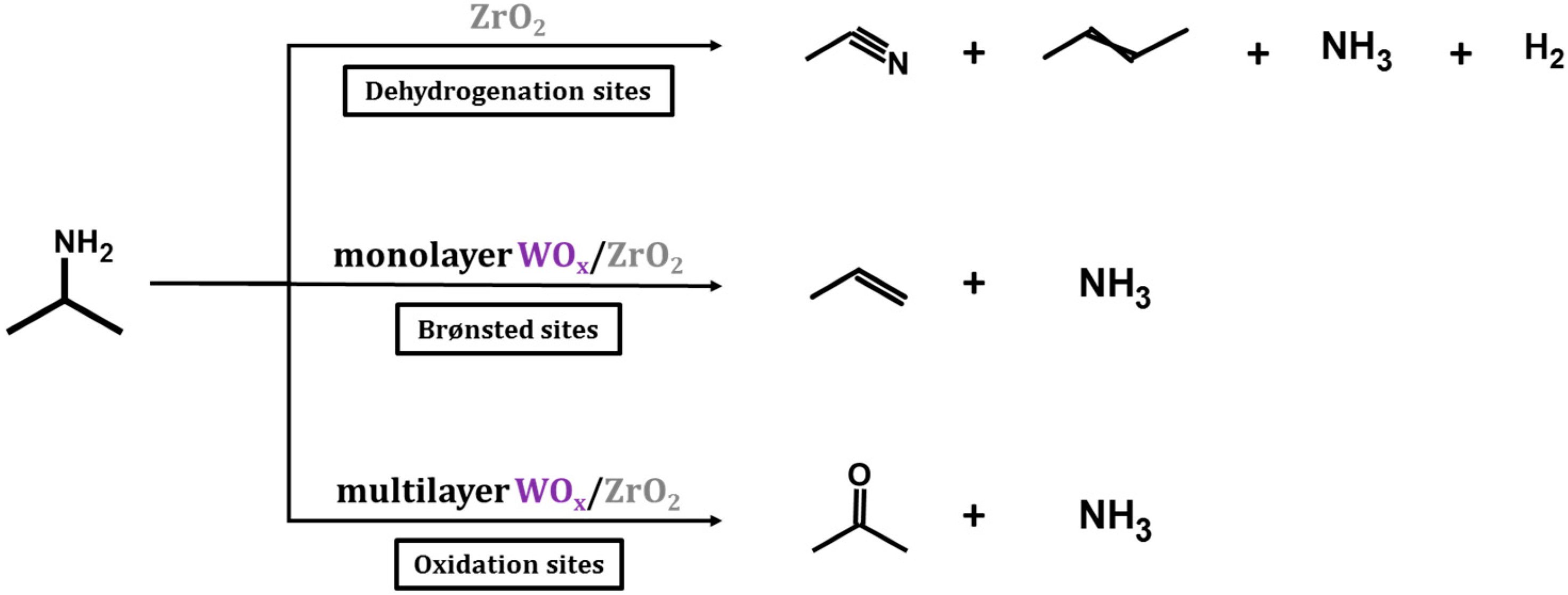
To quantify the concentration of the sites, TPD-TGA measurements were performed using 2-propanamine as the probe molecule. On Brønsted sites, 2-propanamine forms 2-propylammonium which decompose during TPD to form ammonia and propylene between 573 and 650 K due to a Hoffman Elimination reaction [
19,
20], allowing site concentrations to be determined from this decomposition feature. Reaction of the amines can occur on catalytic sites other than Brønsted sites; however, the products that are formed and the temperature range in which the reactions occur are different and depend on the nature of the site [
21]. The TPD-TGA results for the WO
x/ZrO
2 samples show evidence for the presence of three different types of reactive sites, with concentrations of those sites dependent on the WO
x coverages. A summary of the three reaction pathways is illustrated in
Scheme 1.
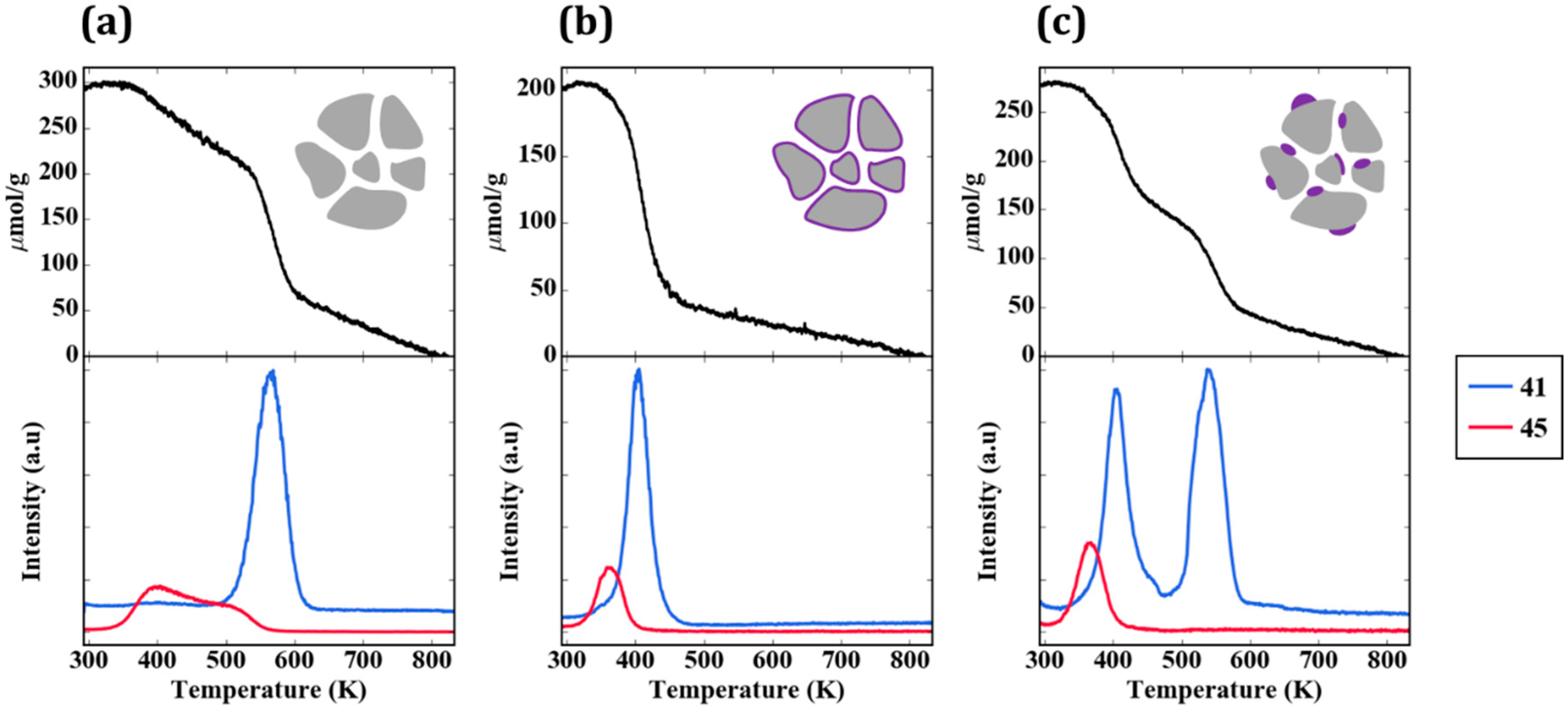
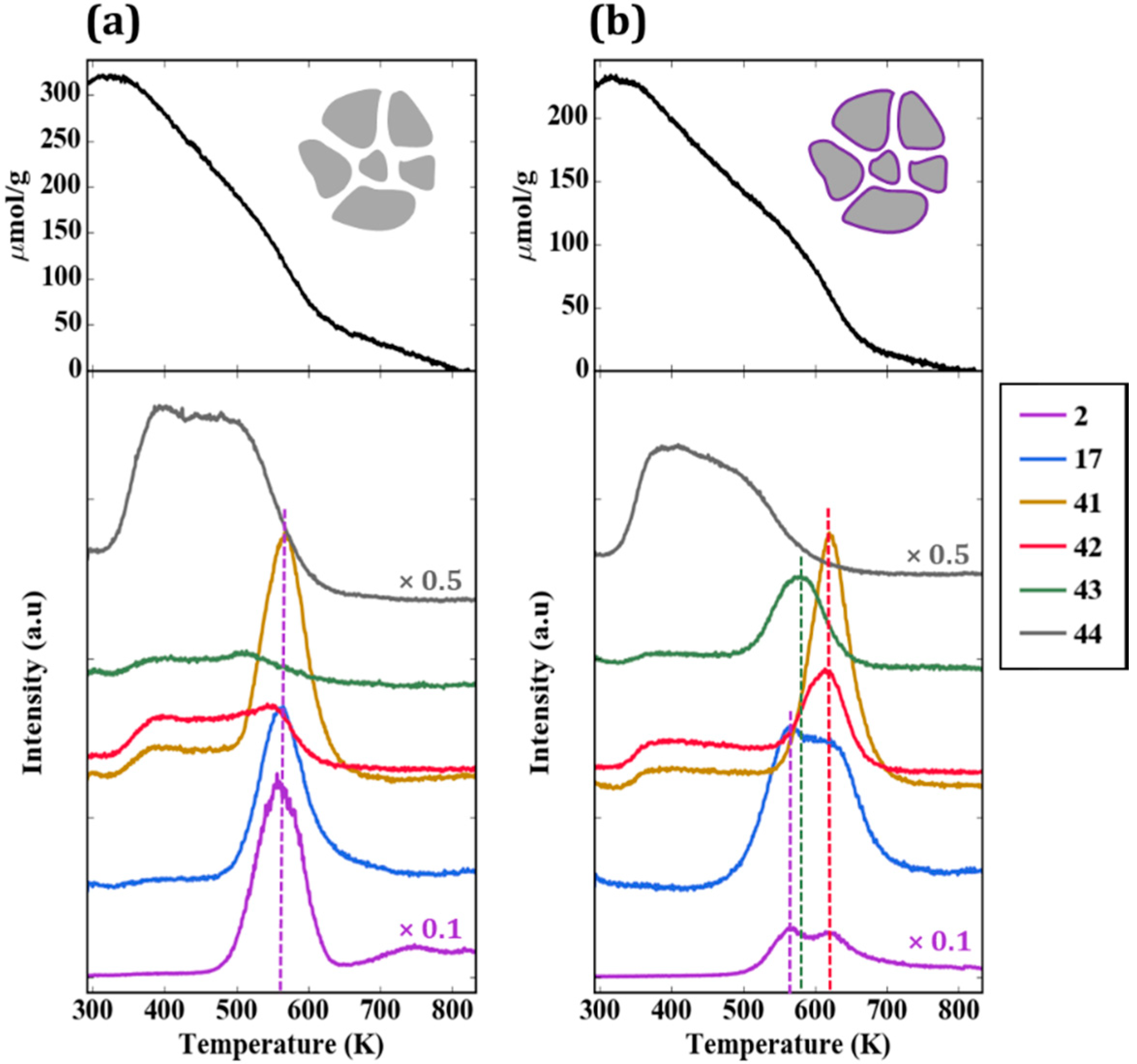
Figure 6a provides the TPD-TGA result for 2-propanamine with pure ZrO
2. Approximately half of the 300 µmol/g that remained on the sample after evacuation desorbed as unreacted amine (
m/
e = 42, 43, 44) below 500 K. Above that temperature, most of the amine molecules reacted in a feature centered at approximately 565 K to form H
2 (
m/
e = 2), a mixture of acetonitrile and butenes (
m/
e = 41), and ammonia (
m/
e = 17). The absence of a peak at
m/
e = 42 is particularly important for demonstrating that the high temperature peak at
m/
e = 41 is not propene, the product that would be formed on Brønsted sites. The identification of acetonitrile and propene as products was achieved by a more complete analysis of the mass spectra and was confirmed in steady-state flow-reactor measurements over ZrO
2 at 673 K. The same products were observed in those measurements, with the addition of small amounts of dipropylamine. Apparently, ZrO
2 catalyzes the dehydrogenation of 2-propanamine to form the imine, which is unstable and reacts to form the smaller nitrile and butenes. The reaction of adsorbed 1-propanamine on ZrO
2, shown in
Figure S1, was simpler, forming primarily propionitrile and H
2. However, because the products formed by 1-propanamine on sites formed by WO
x were more difficult to distinguish from those formed on ZrO
2, most of our work focused on using 2-propanamine to characterize the samples. It is noteworthy that ZrO
2 was previously demonstrated to exhibit dehydrogenation chemistry under some conditions, and it is this functionality that is apparently responsible for the amine reactions [
22].
The TPD-TGA result for the 5W-Zr sample, shown in
Figure 6b, differs significantly from that obtained on ZrO
2. As in the case of 2-propanol, the initial coverage was slightly lower due to the lower specific surface area; similar to the case of ZrO
2, unreacted amine desorbed from the sample below 500 K. However, on the 5W-Zr sample, there are two distinct reaction features at ~585 K and ~620 K, and the products formed in both peaks differ from that observed on ZrO
2. The 620-K peak is characteristic of Brønsted-acid sites. The major products are propene (
m/
e = 41 and 42, in the correct ratio for propene) and ammonia (
m/
e = 17), and these were formed via the Hoffman elimination at exactly the same temperature reported for the reaction of adsorbed 2-propanamine on acidic zeolites [
19]. The lower-temperature peak at ~585 K may have a small contribution from the bare ZrO
2, since the temperature is similar to the reaction temperature on ZrO
2; however, the major products formed in this case were very different. The major products determined from a complete analysis of the mass spectra in this temperature range were acetone (
m/
e = 43) and ammonia. Minimal amounts of acetonitrile and much less H
2 were formed, suggesting that most of the ZrO
2 was covered. The formation of acetone implies that partial oxidation took place. The total amount of 2-propanamine that reacted on the 5W-Z sample was ~100 µmol/g. By integrating the product peaks, we estimate that this sample had less than 10 µmol/g of dehydrogenation sites (from the amount of H
2 and nitrile that formed), 80 µmol/g of Brønsted sites (from propene and ammonia), and 15 µmol/g of oxidation sites (acetone). These results are summarized in
Table 2.
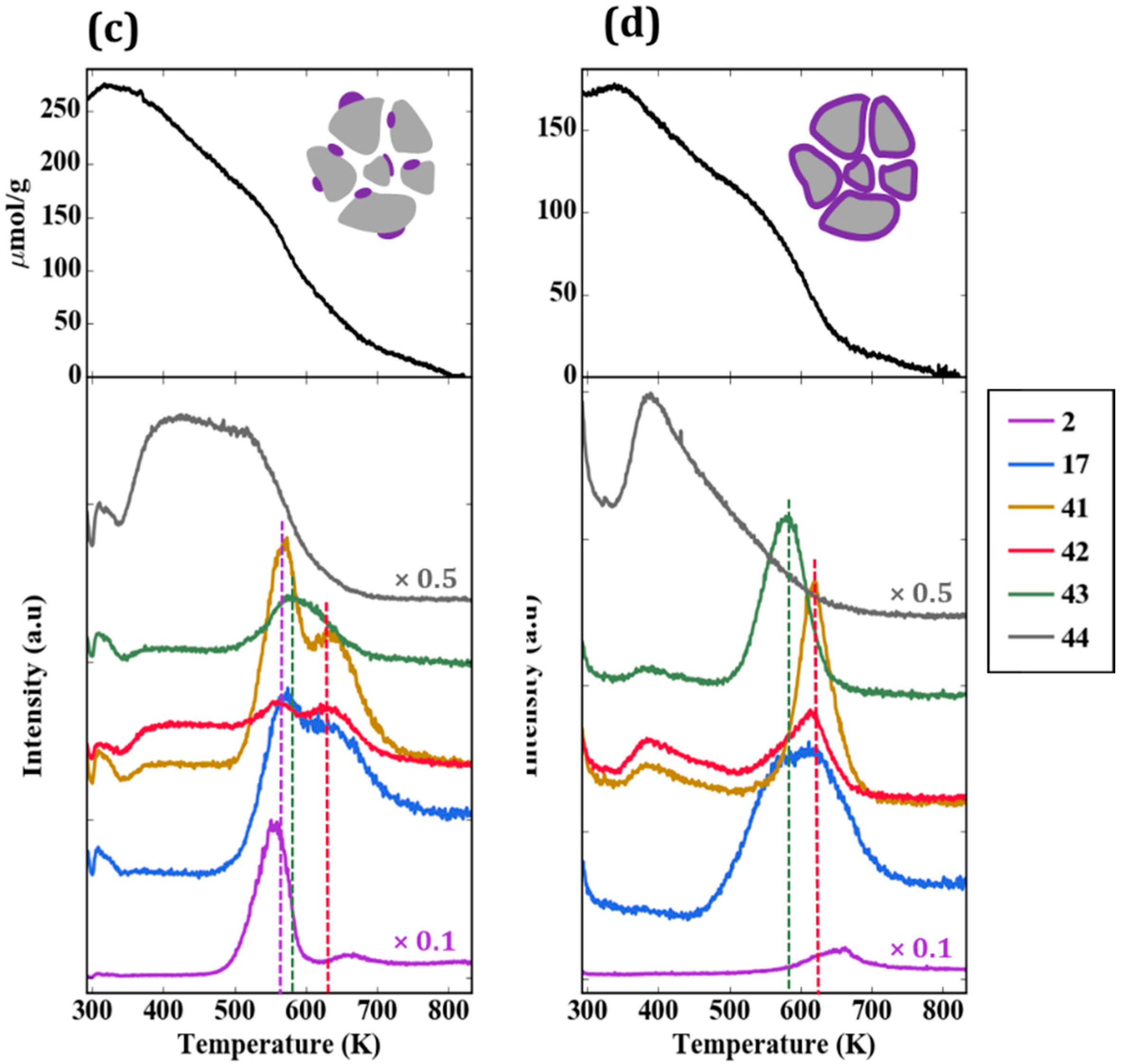
TPD-TGA measurements were also performed for the impW-Zr sample (10-wt% WO
x), with the result shown in
Figure 6c. This sample exhibited all three types of reaction features, with a clearly defined H
2 and acetonitrile/butene feature at 565 K, associated with ZrO
2. An estimate of the site densities gave 60 µmol/g of dehydrogenation sites, 40 µmol/g of Brønsted sites, and 10 µmol/g of oxidation sites.
Figure 6d is the TPD-TGA result obtained for 20W-Zr, the sample prepared using ALD which had a 30-wt% WO
x loading, corresponding to a uniform film of ~1 nm. In comparison to the near-monolayer 5W-Zr sample, the TPD trace shows more intense features associated with oxidation sites at ~585 K, and smaller features associated with Brønsted sites at 620 K.
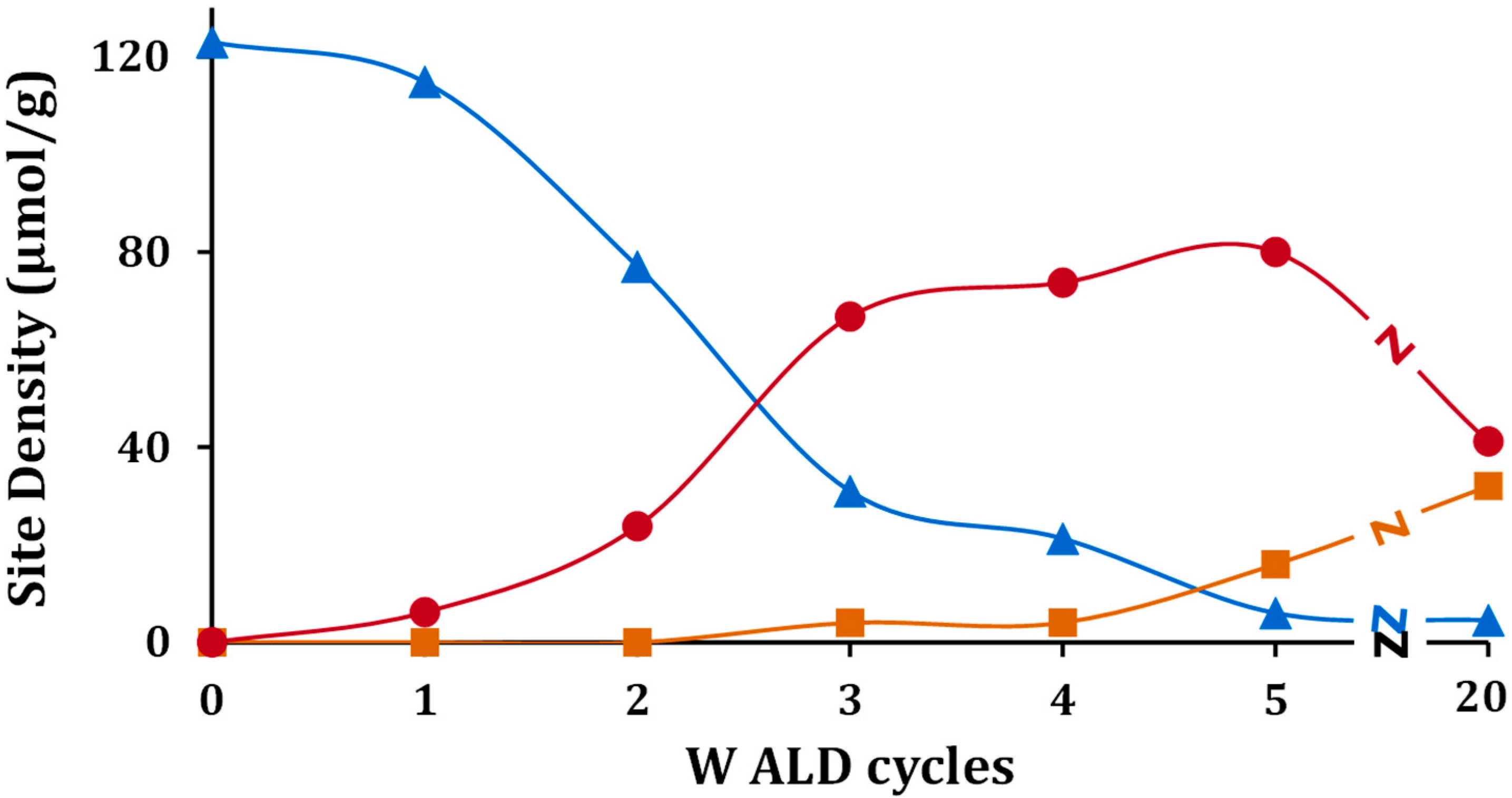
To understand the effect of WO
x coverage, TPD-TGA measurements of 2-propanamine were performed on samples exposed to a varying number of ALD cycles. All of the samples exhibited the same three features in the TPD-TGA but at different concentrations. A summary of the calculated site concentrations is reported in
Table 2 and
Figure 7. The concentration of accessible dehydrogenation sites decreased dramatically with the number of ALD cycles, and essentially disappeared after five ALD cycles. Using the deposition rate of 8.8 × 10
17 W atoms/m
2 per cycle reported earlier, five ALD cycles corresponds to 4.4 × 10
18 W atoms/m
2, a reasonable value for an oxide monolayer. In agreement with previous reports that individual W atoms do not form Brønsted sites, the Brønsted-site concentration was negligible after one cycle, and increased non-linearly thereafter, converging to a maximum after about five cycles. At higher WO
x coverages, the Brønsted-site concentration decreased, implying that contact with the ZrO
2 substrate was essential for forming these sites. Confirmation of the importance of ZrO
2 came from the fact that the silica-supported sample, 5W-Si, showed a low Brønsted-site acidity. Finally, the concentration of oxidation sites, those responsible for forming acetone, were negligible below five ALD rounds, the point at which the dehydrogenation sites disappeared. This implies that the oxidation sites are associated with bulk WO
x that is not in contact with the ZrO
2 surface. It is also confirmed by the fact that oxidation-site density reached a maxima for 20W-Zr when multilayer WO
x was formed. However, the decrease in oxidation-site density for 40W-Zr was only due to a loss of surface area.
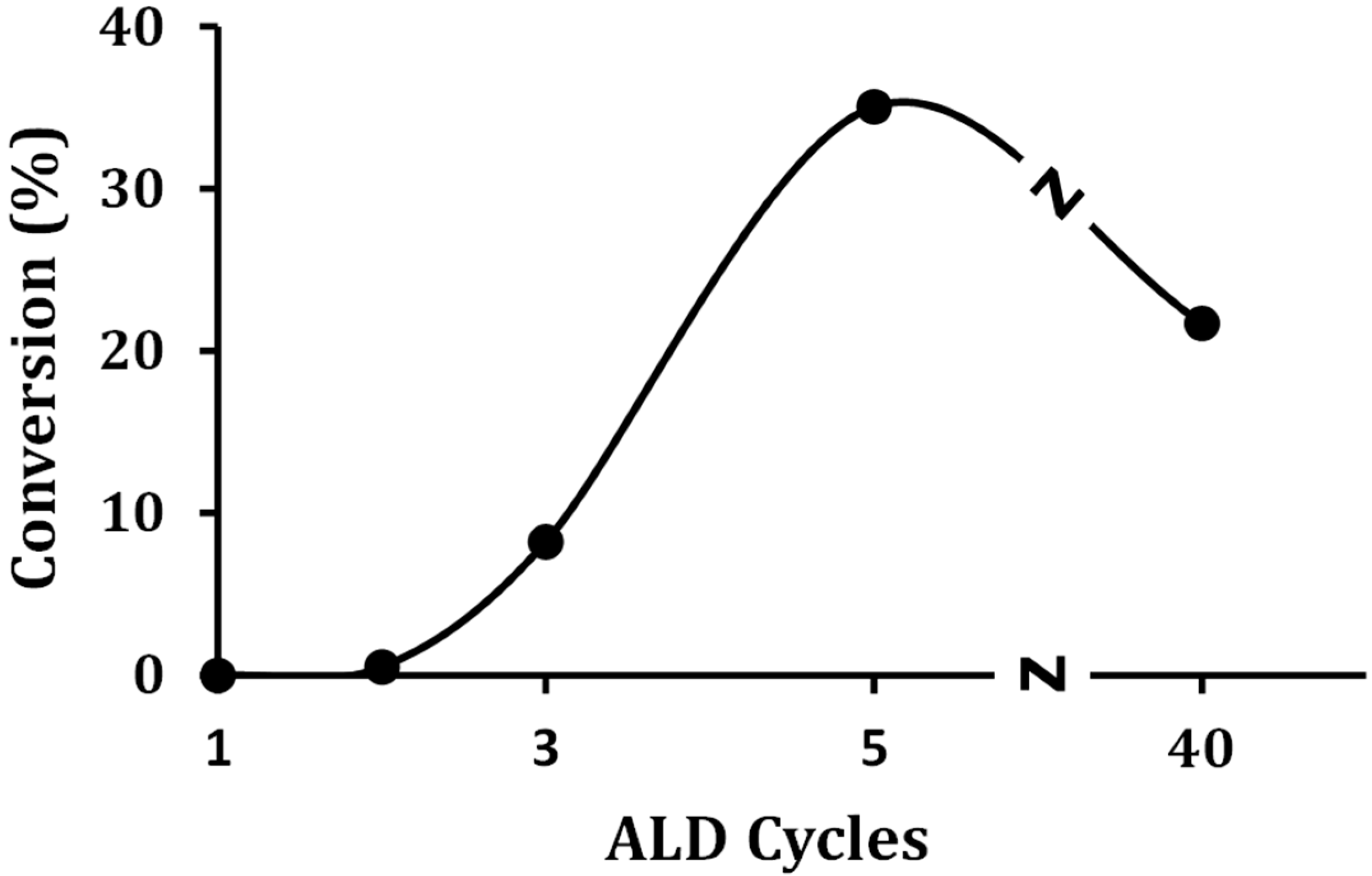
To determine the relationship between catalytic activity and the presence of Brønsted sites, the steady-state conversion of 2-propanol was performed on a series of ALD-prepared WO
x/ZrO
2 catalysts at 403 K and a Weight Hourly Space Velocity (WHSV) of 0.40 h
−1. Under these conditions, pure ZrO
2 was not catalytically active. As shown in
Figure 8, there was no significant conversion over catalysts prepared with one or two WO
x ALD cycles. With higher WO
x concentrations, the propanol reacted to a mixture of water with dipropyl ether (~60%) and propene (40%). Conversions were maximized after five ALD rounds, and there was a reasonable correspondence between the concentration of Brønsted sites on these samples and the conversions. The fact that the 40W-Zr sample showed a higher conversion than would be expected based on its Brønsted-site density suggests that the bulk WO
x also has some activity for alcohol dehydration.
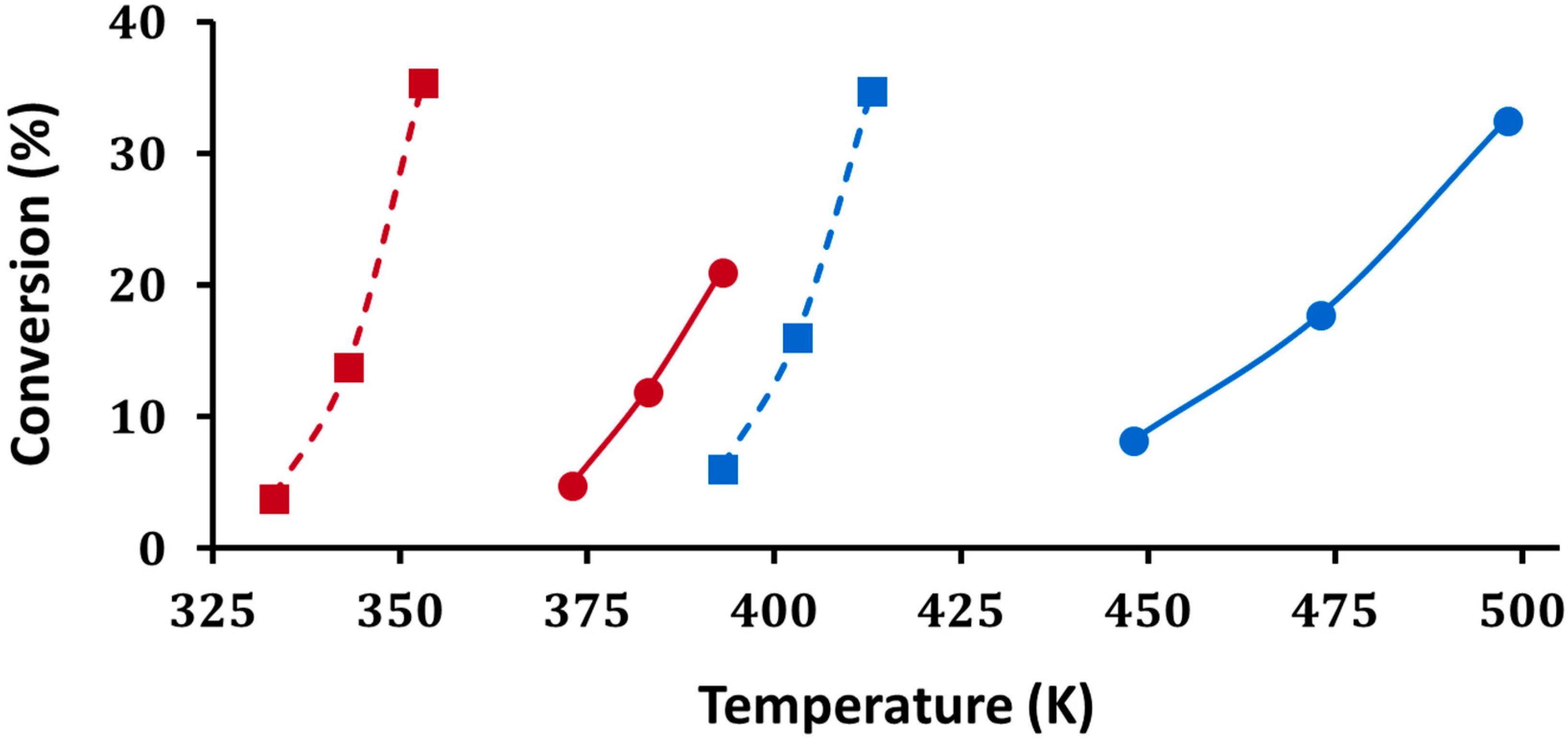
As discussed in the introduction, the quantification of Brønsted-acid strength in solid acids is difficult. Here, we measured light-off curves for H–D exchange between D
2O and either toluene (C
7H
8) or chlorobenzene (C
6H
5Cl) on the 5W-Zr sample, and compared the results to those obtained on an H-ZSM-5 zeolite which had a similar Brønsted-site density of 80 µmol/g. Measurements began at room temperature, and were carried out in a steady-state reaction environment after each increment of 10 K. The results are shown in
Figure 9. Firstly, on both H-ZSM-5 and 5W-Zr, the light-off temperatures for chlorobenzene occurred at higher temperatures, roughly 60 degrees higher on H-ZSM-5 and 75 degrees higher on 5W-Zr. This is consistent with a lower barrier for the reaction of toluene due to its significantly higher proton affinity. Secondly, the temperatures at which H–D exchange occurred were significantly higher on the tungstated zirconia. Sorption effects and other factors can play a role in the observed rates on solid acids [
23]; however, the fact that the reaction occurred at higher temperatures for both adsorbates suggests that tungstated zirconia has much weaker acid sites than does H-ZSM-5.
3. Discussion
In agreement with previous reports [
1,
17], our data here indicate that tungstated zirconia is a complex material that can exhibit Brønsted acidity, Lewis acidity, and oxidation activity, along with dehydrogenation activity on ZrO
2 [
22]. The concentrations of the various types of sites depend on how the material is made, as shown by the differences we observed in materials synthesized via impregnation and using ALD. This complexity almost certainly accounts for at least some of the differences reported by the various groups who have worked on tungstated zirconia. In the presented study, we focused on controlling the composition and structure by synthesizing catalysts using ALD because of the uniformity that this approach provides.
The TPD results also demonstrate the power of using reactive probe molecules, rather than simple bases like ammonia or pyridine, for characterizing the types of sites that are present on these materials. The application of TPD in this manner is certainly not new, and 2-propanol [
18] and 2-propanamine [
19] were previously used to characterize site concentrations for Lewis and Brønsted acids. However, the results demonstrate that additional information can also be obtained. For example, the comparison of 2-propanol results for WO
x/ZrO
2 samples prepared via impregnation gave a strong indication that the WO
x did not uniformly cover the ZrO
2. The 2-propanamine results were also able to delineate concentrations of the various types of sites that were formed with increasing WO
x coverage.
Based on our reactivity studies, the Brønsted-acid sites formed in tungstated zirconia are relatively weak, certainly compared to the Brønsted-acid sites formed in zeolites. While site strengths may well depend on the detailed structure and method of synthesis, it is worth noting that others reported that WO
x/ZrO
2 is selective for the etherification of alcohols [
7], without forming olefins as with zeolites, which would again imply the sites are relatively weak. We suggest that the high activity sometimes observed at low temperatures in tungstated zirconia is due to the combination of oxidation sites and/or dehydrogenation sites, together with Brønsted sites, similar to that which was proposed for sulfated zirconia [
24]. With sulfated zirconia, the oxidation sites were shown to transform alkanes to olefins, which were then more easily activated by the acid sites. A similar combination of sites likely explains at least some of the catalytic properties of tungstated zirconia. The combination of sites allows isomerization reactions to occur at low temperatures without the need for especially strong Brønsted sites.
It is interesting that there were essentially no Brønsted sites formed after the deposition of a single WO
x ALD cycle on the ZrO
2. One would expect to form isolated WO
x species under these conditions, and the lack of Brønsted sites is interesting. The fact that more than one WO
x species must be present in order to form a Brønsted site was stated by others [
1], who argued that the W:Brønsted-site ratio is about four. The value of four is indeed the approximate minimum value observed in
Table 2, where the Brønsted-site concentrations are listed as a function of the number of ALD cycles added to the ZrO
2. While this ratio suggests that a (WO
x)
n cluster is required for forming Brønsted sites, it is worth noting that the ratio of four occurs after three cycles, at which point it is expected that most of the tungsten is deposited in the first monolayer, implying that the clusters would be two-dimensional and still in contact with ZrO
2. The importance of zirconia is confirmed by the fact that we do not form significant quantities of Brønsted sites when WO
x is deposited on silica.
Finally, our H–D exchange results imply that the Brønsted sites in WO
x/ZrO
2 are relatively weak compared to the sites in high-silica zeolites. The early work by Hino and Arata on the isomerization of small alkanes at low temperatures argued that these materials are very strong Brønsted acids [
25]; similarly, the calculated deprotonation energies that were reported imply that tungstated zirconia should have much stronger sites than those present in zeolites [
1]. However, the recent publication on dodecanol etherification showed that WO
x/ZrO
2 was more selective than acidic zeolites for forming ethers, and produced less olefin and oligomerization products [
7], a result that would imply weaker Brønsted sites. We suggest that the high activity reported for alkane activation at low temperatures could be the result of a combination of oxidation sites and Brønsted sites. For example, the low-temperature activity of sulfated zirconia was associated in part with oxidation sites which form olefins that then go on to react over the Brønsted sites [
9]. Since the tungsten clusters on zirconia were shown to be reducible, both in this study and previously [
26], the low-temperature, isomerization activity may be the result of a combination of acidic and oxidation sites. Similarly, the presence of Pt in low-temperature isomerization catalysts likely leads to the formation of alkenes, which are much more easily protonated [
27,
28].
Obviously, there is still much to learn about the nature and properties of the catalytic sites in tungstated zirconia. We believe that the combination of controlled synthesis using ALD with careful adsorption studies of the type performed here can help elucidate the nature of these sites.
4. Experimental
The ZrO
2 support was prepared by titrating (0.2 mL/s) 30% aqueous NH
4OH (Fischer) to a 5-wt% aqueous solution of ZrO(NO
3)
2•xH
2O (99%, Sigma Aldrich, St. Louis, MO, USA) with vigorous stirring. The precipitate was then dried at 333 K for 12 h before being calcined in a Muffle furnace at 773 K for 5 h. ALD was performed in a custom-built, static system that could be evacuated by a mechanical pump, and was described in previous studies [
12,
29]. In a typical ALD cycle, the evacuated 300 mg of zirconia powder was first exposed to the vapor of W(CO)
6 (99%, Strem, Newburyport, MA, USA) at 473 K for 3 min, followed by a 5-min evacuation. The samples were then exposed to air at the same temperature for 6 min, before once again being evacuated to complete the cycle. We used the nomenclature
xW-Zr to refer to a sample exposed to
x ALD cycles. For comparison purposes, a sample with 10-wt% WO
x/ZrO
2 (impW-Zr) was prepared via conventional aqueous incipient wetness of (NH
4)
10W
12O
41•5H
2O (99.999%, Alfa Aesar, Haverhill, MA, USA), followed by a 4-h calcination at 773 K. To determine the effect of the support, a WO
x/SiO
2 sample (5W-Si) was prepared by depositing five ALD cycles onto a stabilized SiO
2 (Degussa AG, Essen, Germany, Ultrasil VN 3 SP, 140 m
2/g).
The temperature-programmed desorption/thermogravimetric analysis (TPD-TGA) measurements were performed on samples held in a system consisting of an evacuated CAHN 2000 microbalance, equipped with an SRI quadrupole mass spectrometer (RGA100), that is described elsewhere [
30]. The 50-mg samples were first heated in a vacuum to 823 K, before being cooled to room temperature in a vacuum, and then exposed to the vapor of the probe adsorbate, either 2-propanol (99.9%, Fisher, Hampton, NH, USA), or 1-propanamine (99+%, Alfa Aesar), or 2-propanamine (99%, Alfa Aesar). After 1 h of evacuation, the TPD and TGA measurements were obtained while ramping the temperature at 10 K/min.
Scanning transmission electron microscopy (STEM) and elemental mapping via energy-dispersive X-ray spectroscopy (EDS) were performed with a JEOL 2010F field-emission scanning transmission electron microscope (JEOL, Tokyo, Japan), operated at an accelerating voltage of 200 kV with a 0.7 nm STEM probe. Infrared spectra of adsorbed pyridine were performed on a Mattson Galaxy FTIR (Madison Instruments Inc., Middleton, WI, USA) with a diffuse-reflectance attachment (Collector II™) in order to confirm the presence of Brønsted sites [
31]. In the FTIR cell, the samples were initially heated to 373 K in flowing He at 60 mL/min to remove any adsorbed water for 10 min. After cooling the samples to room temperature, pyridine vapors were exposed to the sample for 10 min. The samples were then flushed with flowing He at 60 mL/min for 10 min. Raman spectra were obtained with an NTEGRA Spectra system (NT-MDT) with an excitation laser wavelength of 532 nm. The experiments were carried out with a laser power of 15 mW (10% of 150 W from the natural-density filter setting) at the samples and a collection time of 60 s. Powder X-ray Diffraction (XRD) patterns were collected from a Rigaku Smartlab diffractometer equipped with a Cu Kα source (The Woodlands, TX, USA).
The steady-state reaction rates for various reactions (2-propanol dehydration, H–D exchange between toluene (C7H8) and D2O, and H–D exchange between chlorobenzene (C6H5Cl) and D2O) were measured in a flow reactor that consisted of a 200-mm long, 4.6-mm ID stainless-steel tube, packed with 100 mg of catalyst that was held in place by two quartz-wool plugs and an inert tube from the back end. Products were monitored using an online GC-MS (QP-5000, Shimadzu, Kyoto, Japan) and the results were compared to reaction measurements over an H-ZSM-5 catalyst (Si/Al2 = 280; Zeolyst, CBV 28014, Conshohocken, PA, USA). For the dehydration of 2-propanol, the inlet flow to the reactor was 5% 2-propanol, achieved by feeding 0.9 µL/min liquid 2-propanol with a syringe pump (Harvard Apparatus, PHD 2000, Holliston, MA, USA) in a 5 mL/min He flow. In the H–D exchange measurements, an equal molar fraction of 1% toluene (or chlorobenzene) and D2O were co-fed with a syringe pump in a 20 mL/min He flow. Prior to the reaction, the catalysts were pretreated in flowing He at 523 K for 30 min before measuring steady-state reaction rates using an online GC-MS (QP-5000, Shimadzu). The conversions of H–D exchange were quantified via the deconvolution of mass fragmentations from m/e 91 to 97 for toluene, and from m/e 112 to 118 for chlorobenzene.

 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}