Metallosupramolecular Polymer Precursor Design for Multi-Element Co-Doped Carbon Shells with Improved Oxygen Reduction Reaction Catalytic Activity

 and
and 
Abstract
:
1. Introduction
2. Results and Discussion
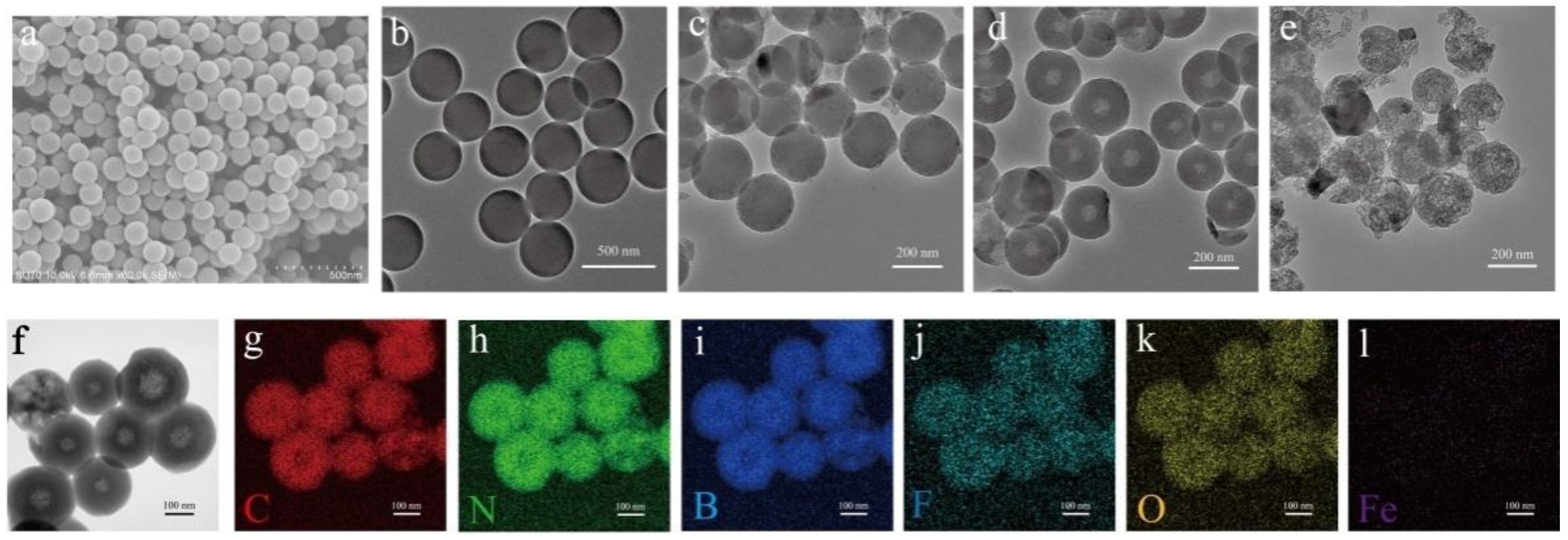
2.1. Morphology Evolution
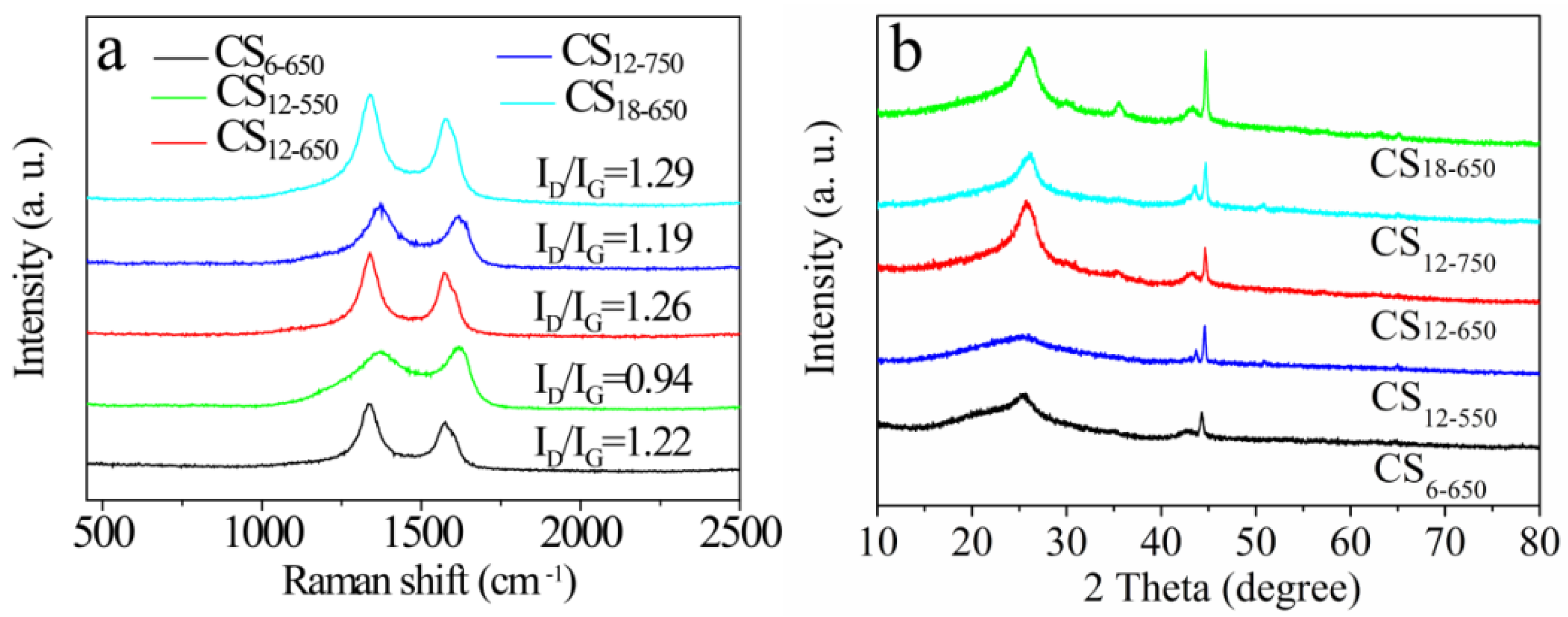
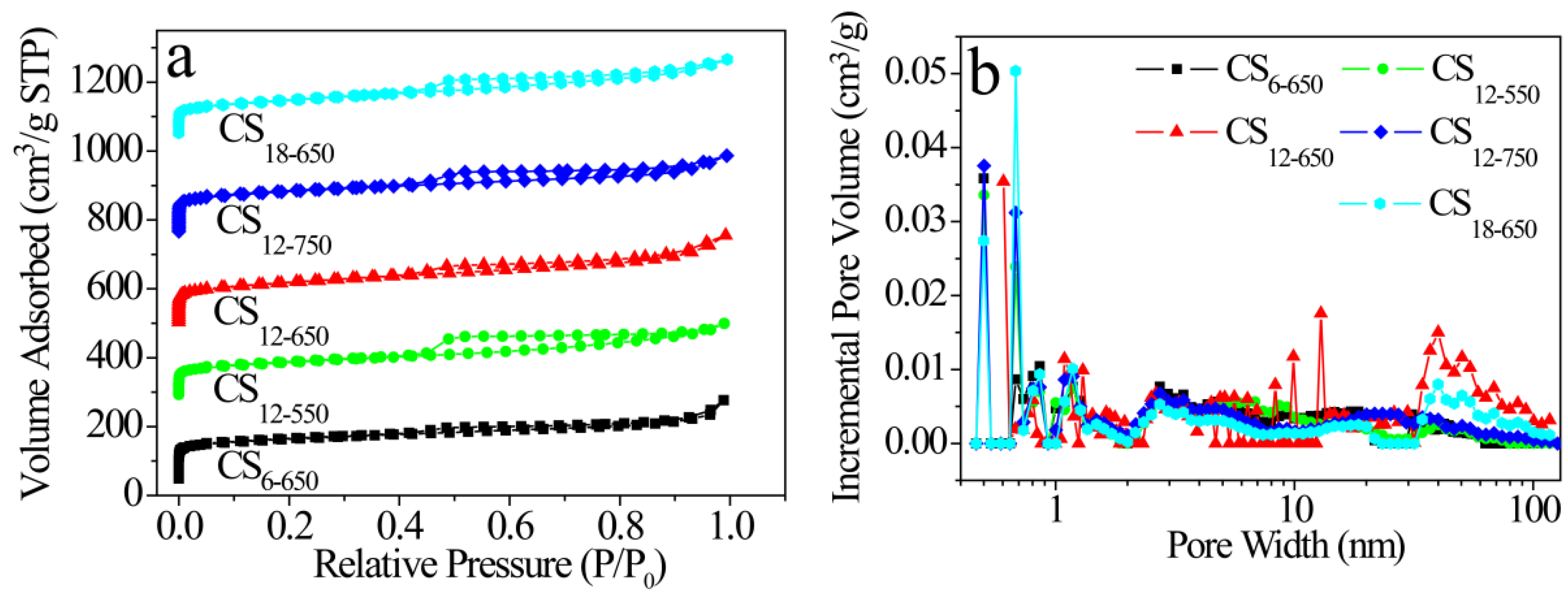
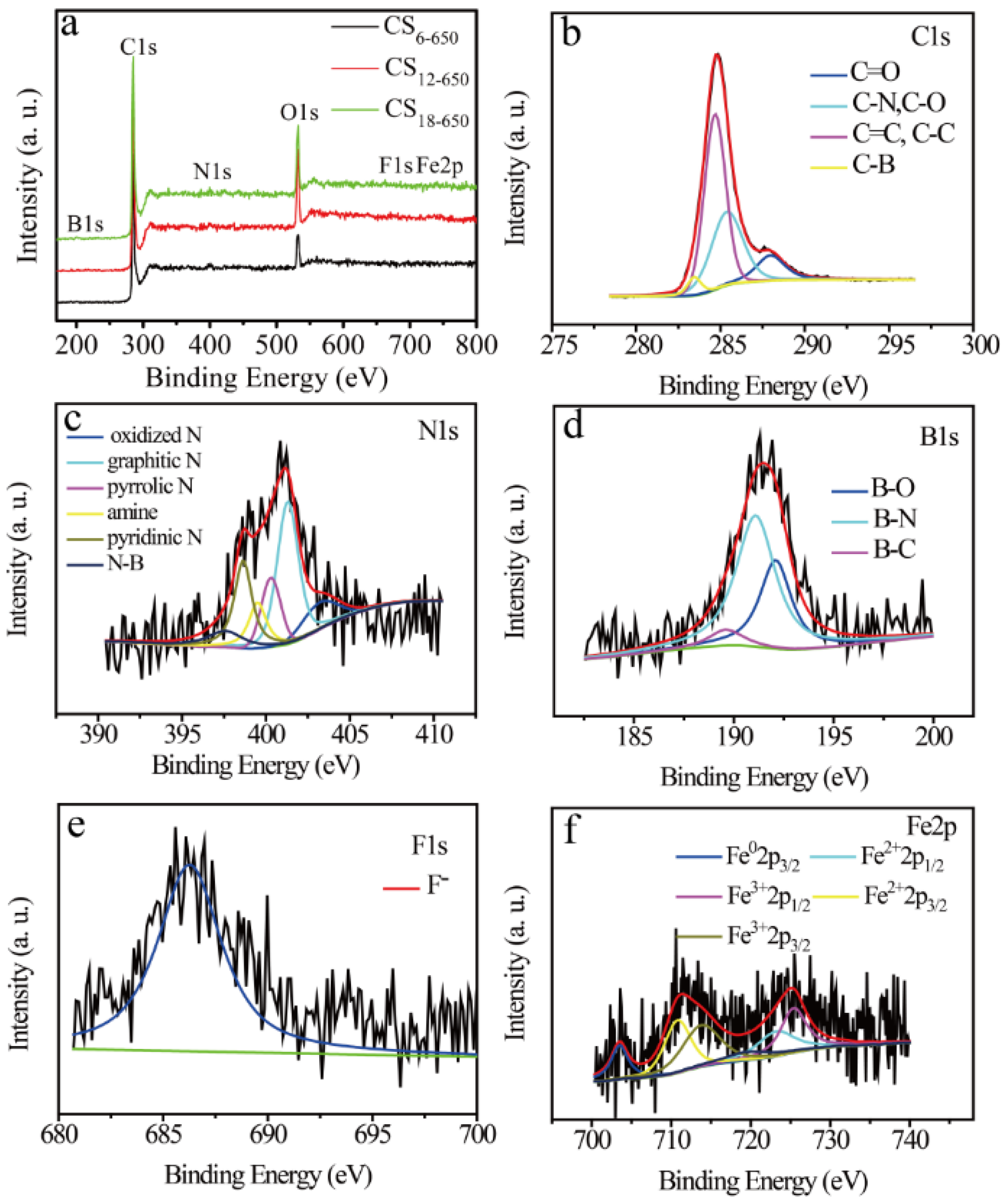
2.2. Composition and Structure Characterization
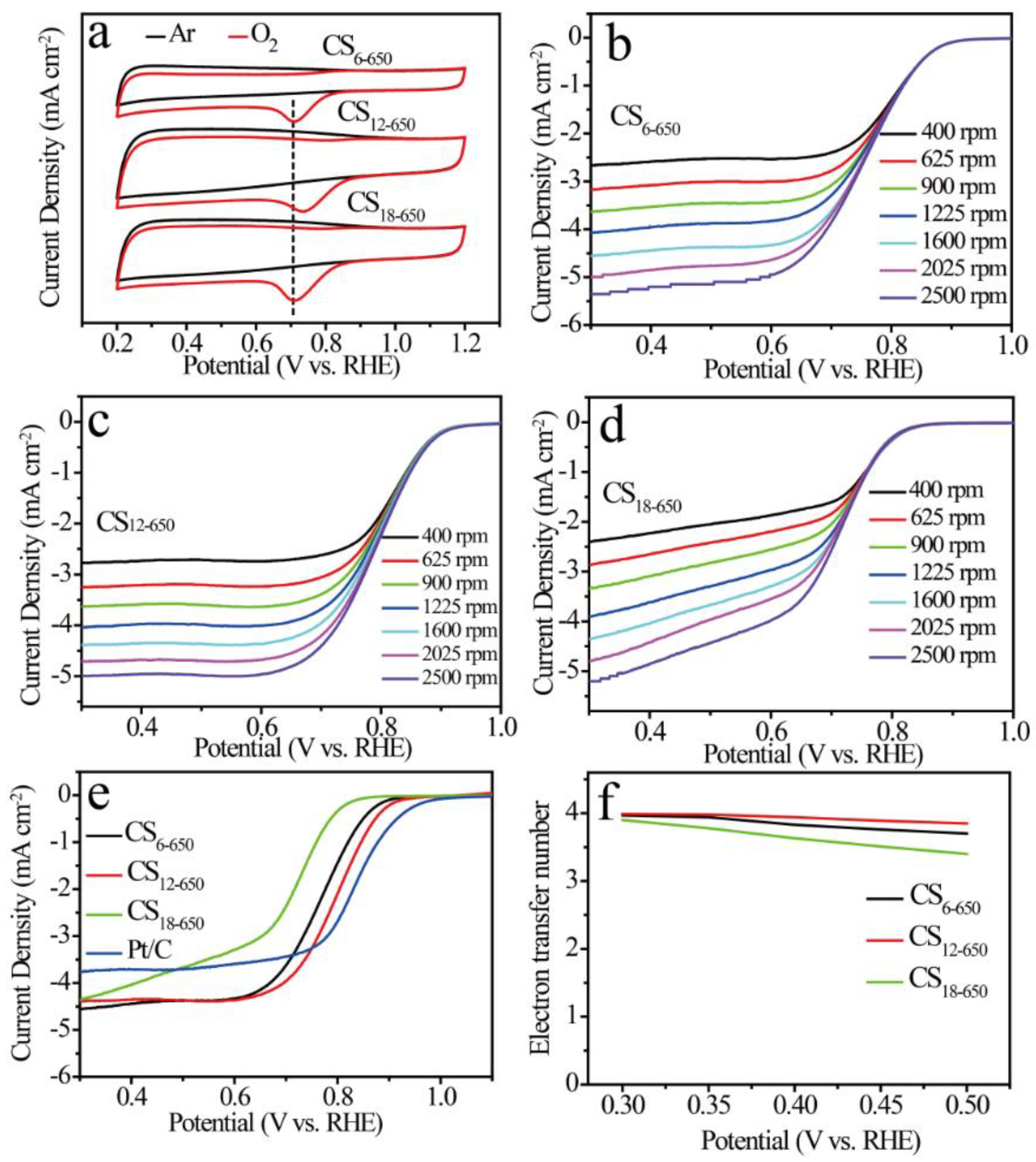
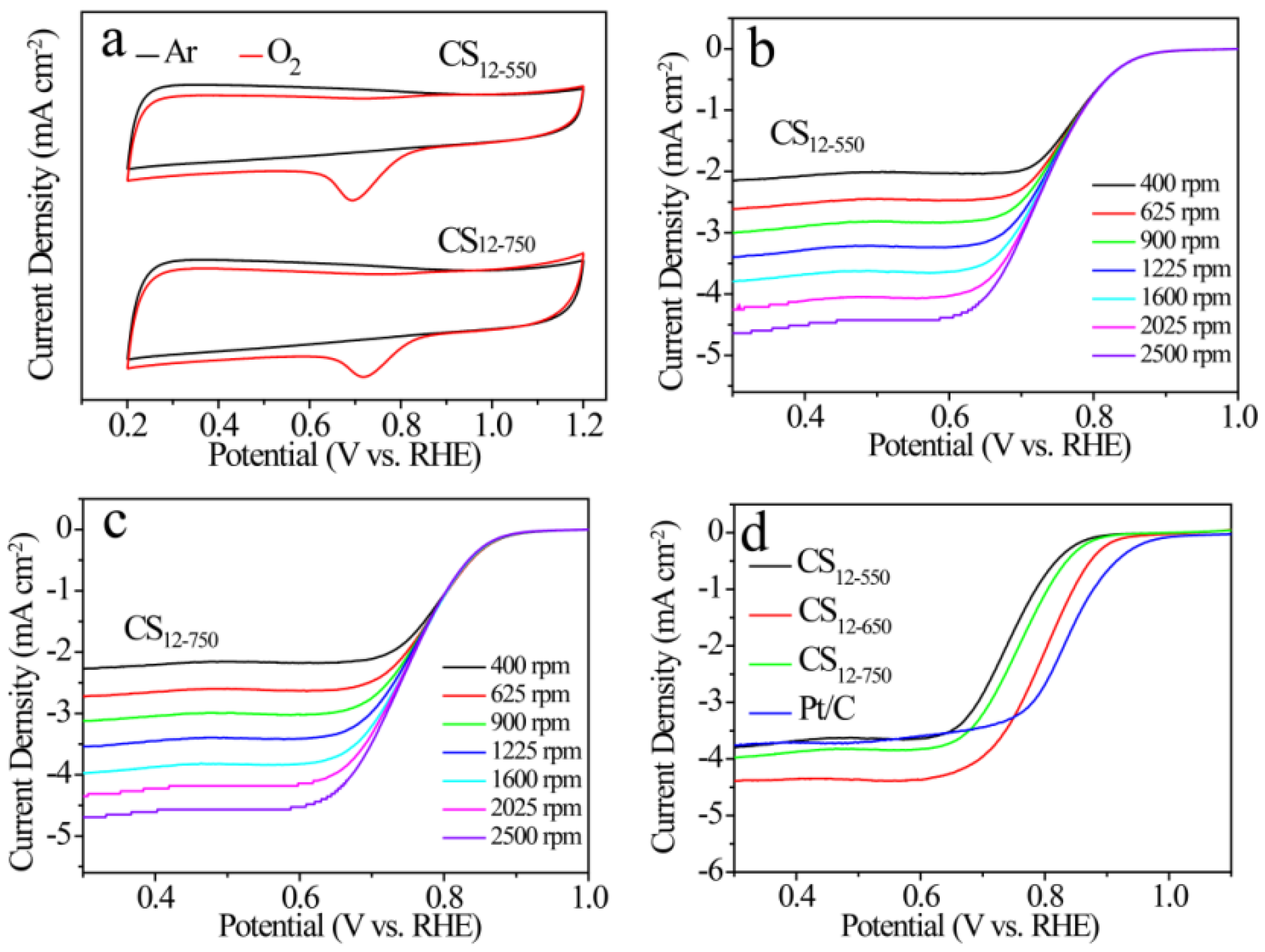
2.3. ORR Performances
3. Materials and Methods
3.1. Materials
3.2. Catalyst Preparation
3.3. Characterization
3.4. Electrochemical Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Shao, M.H.; Chang, Q.W.; Dodelet, J.P.; Chenitz, R. Recent Advances in Electrocatalysts for Oxygen Reduction Reaction. Chem. Rev. 2016, 116, 3594–3657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, A.; Siahrostami, S.; Patel, A.; Nørskov, J.K. Understanding Catalytic Activity Trends in the Oxygen Reduction Reaction. Chem. Rev. 2018, 118, 2302–2312. [Google Scholar] [CrossRef] [PubMed]
- Mano, N.; Poulpiquet, A.D. O2 Reduction in Enzymatic Biofuel Cells. Chem. Rev. 2018, 118, 2392–2468. [Google Scholar] [CrossRef] [PubMed]
- Kakati, N.; Maiti, J.; Lee, S.; Jee, S.H.; Viswanathan, B.; Yoon, Y.S. Anode catalysts for direct methanol fuel cells in acidic media: Do we have any alternative for Pt or Pt-Ru? Chem. Rev. 2014, 114, 12397–12429. [Google Scholar] [CrossRef] [PubMed]
- Pegis, M.L.; Wise, C.F.; Martin, D.J.; Mayer, J.M. Oxygen Reduction by Homogeneous Molecular Catalysts and Electrocatalysts. Chem. Rev. 2018, 118, 2340–2391. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.F.; Zhu, C.Z.; Song, J.H.; Engelhard, M.H.; Xia, H.B.; Du, D.; Lin, Y.H. Kinetically Controlled Synthesis of Pt-Based One-Dimensional Hierarchically Porous Nanostructures with Large Mesopores as Highly Efficient ORR Catalysts. ACS Appl. Mater. Interfaces 2016, 8, 35213–35218. [Google Scholar] [CrossRef]
- Unwin, P.R.; Guell, A.G.; Zhang, G.H. Nanoscale Electrochemistry of sp (2) Carbon Materials: From Graphite and Graphene to Carbon Nanotubes. Acc. Chem. Res. 2016, 49, 2041–2048. [Google Scholar] [CrossRef]
- Rao, C.V.; Viswanathan, B. ORR Activity and Direct Ethanol Fuel Cell Performance of Carbon-Supported Pt-M (M = Fe, Co, and Cr) Alloys Prepared by Polyol Reduction Method. Phys. Chem. C. 2009, 113, 18907–18913. [Google Scholar]
- Kim, J.H.; Sa, Y.J.; Jeong, H.Y.; Joo, S.H. Roles of Fe-Nx and Fe-Fe3C@C Species in Fe-N/C Electrocatalysts for Oxygen Reduction Reaction. ACS Appl. Mater. Interfaces 2017, 9, 9567–9575. [Google Scholar] [CrossRef]
- Zhang, Q.; Mamtani, K.; Jain, D.; Ozkan, U.; Asthagiri, A. CO Poisoning Effects on FeNC and CNx ORR Catalysts: A Combined Experimental-Computational Study. J. Phys. Chem. C 2016, 120, 15173–15184. [Google Scholar] [CrossRef]
- Loget, G.; Zigah, D.; Bouffier, L.; Sojic, N.; Kuhn, A. Bipolar electrochemistry: From materials science to motion and beyond. Acc. Chem. Res. 2013, 4, 2513–2523. [Google Scholar] [CrossRef] [PubMed]
- Ambrosi, A.; Chua, C.K.; Bonanni, A.; Pumera, M. Electrochemistry of graphene and related materials. Chem. Rev. 2014, 114, 7150–7188. [Google Scholar] [CrossRef]
- Ghasemi, M.; Daud, W.R.W.; Hassan, S.H.A.; Ismail, O.; Rahimnejad, M.; Jahim, J.M. Nano-structured carbon as electrode material in microbial fuel cells: A comprehensive review. J. Alloy Compd. 2013, 580, 245–255. [Google Scholar] [CrossRef]
- Lin, F.; Liu, Y.J.; Yu, X.Q.; Cheng, L.; Singer, A.; Shpyrko, O.G.; Huolin, L.; Xin, L.H.; Tamura, N.; Tian, C.X.; et al. Synchrotron X-ray Analytical Techniques for Studying Materials Electrochemistry in Rechargeable Batteries. Chem. Rev. 2017, 117, 13123–13186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, X.; Wei, Q.L.; Zhan, X.X.; Zhang, G.X.; Shu, H.; Sun, S.H. The New Graphene Family Materials: Synthesis and Applications in Oxygen Reduction Reaction. Catalysts 2017, 7, 1. [Google Scholar] [CrossRef]
- Brouzgou, A.; Song, S.Q.; Liang, Z.X.; Tsiakaras, P. Non-Precious Electrocatalysts for Oxygen Reduction Reaction in Alkaline Media: Latest Achievements on Novel Carbon Materials. Catalysts 2016, 6, 159. [Google Scholar] [CrossRef]
- Wu, Z.X.; Song, M.; Wang, J.; Liu, X. Recent Progress in Nitrogen-Doped Metal-Free Electrocatalysts for Oxygen Reduction Reaction. Catalysts 2018, 8, 196. [Google Scholar] [CrossRef]
- Paraknowitsch, J.P.; Thomasa, A. Doping carbons beyond nitrogen: An overview of advanced heteroatom doped carbons with boron, sulphur and phosphorus for energy applications. Energy Environ. Sci. 2013, 6, 2839–2855. [Google Scholar] [CrossRef]
- Bag, S.; Roy, K.; Gopinath, C.; Raj, C.R. Facile single-step synthesis of nitrogen-doped reduced graphene oxide-Mn(3)O(4) hybrid functional material for the electrocatalytic reduction of oxygen. ACS Appl. Mater. Interfaces 2014, 6, 2692–2699. [Google Scholar] [CrossRef] [PubMed]
- Xiong, D.B.; Li, X.F.; Fan, L.L.; Bai, Z.M. Three-Dimensional Heteroatom-Doped Nanocarbon for Metal-Free Oxygen Reduction Electrocatalysis: A Review. Catalysts 2018, 8, 301. [Google Scholar] [CrossRef]
- Liu, J.; Ji, Y.G.; Qiao, B.; Zhao, F.Q.; Gao, H.X.; Chen, P.; An, Z.W.; Chen, X.B.; Chen, Y. N, S Co-Doped Carbon Nanofibers Derived from Bacterial Cellulose/Poly (Methylene Blue) Hybrids: Efficient Electrocatalyst for Oxygen Reduction Reaction. Catalysts 2018, 8, 269. [Google Scholar] [CrossRef]
- Kim, D.W.; Li, O.L.; Saito, N. Enhancement of ORR catalytic activity by multiple heteroatom-doped carbon materials. Phys. Chem. Chem. Phys. 2015, 17, 407–413. [Google Scholar] [CrossRef]
- Chang, Y.; Yuan, C.H.; Li, Y.T.; Liu, C.; Wu, T.; Zeng, B.R.; Xu, Y.T.; Dai, L.Z. Controllable fabrication of a N and B co-doped carbon shell on the surface of TiO2 as a support for boosting the electrochemical performances. J. Mater. Chem. A 2017, 5, 1672–1678. [Google Scholar] [CrossRef]
- Chang, Y.; Yuan, C.H.; Liu, C.; Mao, J.; Li, Y.T.; Wu, H.Y.; Wu, Y.Z.; Xu, Y.T.; Dai, L.Z. B, N co-doped carbon from cross-linking induced self-organization of boronate polymer for supercapacitor and oxygen reduction reaction. J. Power Sources 2017, 365, 354–361. [Google Scholar] [CrossRef]
- Bezerra, C.W.B.; Zhang, L.; Lee, K.C.; Liu, H.S.; Marques, A.L.B.; Marques, E.P.; Wang, H.J.; Zhang, J.J. A review of Fe-N/C and Co-N/C catalysts for the oxygen reduction reaction. Electrochim. Acta 2008, 53, 4937–4951. [Google Scholar] [CrossRef]
- Liua, Y.Y.; Yue, X.P.; Li, K.X.; Qiao, J.L.; Wilkinsone, D.; Zhang, J.J. PEM fuel cell electrocatalysts based on transition metal macrocyclic compounds. Coord. Chem. Rev. 2016, 315, 153–177. [Google Scholar] [CrossRef]
- Guo, J.N.; Cheng, Y.H.; Xiang, Z.H. Confined-Space-Assisted Preparation of Fe3O4 Nanoparticle Modified Fe-N-C Catalysts Derived from a Covalent Organic Polymer for Oxygen Reduction. ACS Sustain. Chem. Eng. 2017, 5, 7871–7877. [Google Scholar] [CrossRef]
- Liu, J.; Li, E.L.; Ruan, M.B.; Song, P.; Xu, W.L. Recent Progress on Fe/N/C Electrocatalysts for the Oxygen Reduction Reaction in Fuel Cells. Catalysts 2015, 5, 1167–1192. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.J.; Gu, L.; Li, L.; Zhang, Y.; Zhang, X.; Zhang, L.J.; Wang, J.Q.; Hu, J.S.; Wei, Z.D.; Wan, L.J. Understanding the High Activity of Fe-N-C Electrocatalysts in Oxygen Reduction: Fe/Fe3C Nanoparticles Boost the Activity of Fe-N(x). J. Am. Chem. Soc. 2016, 138, 3570–3578. [Google Scholar] [CrossRef]
- Li, L.Y.; Yuan, C.H.; Dai, L.Z. Thermoresponsive Polymeric Nanoparticles: Nucleation from Cooperative Polymerization Driven by Dative Bonds. Macromolecules 2014, 47, 5869–5876. [Google Scholar] [CrossRef]
- Li, L.Y.; Yuan, C.Y.; Zhou, D.M.; Ribbe, D.E.; Kittilstved, P.R.; Thayumanavan, S. Utilizing Reversible Interactions in Polymeric Nanoparticles to Generate Hollow Metal-Organic Nanoparticles. Angew. Chem. Int. Ed. 2015, 127, 13183–13187. [Google Scholar] [CrossRef]
- Ejima, H.; Richardson, J.J.; Liang, K.; Best, J.P.; Koeverden, M.P.; Such, G.H.; Cui, J.W.; Such, G.H.; Cui, J.W.; Caruso, F. One-step assembly of coordination complexes for versatile film and particle engineering. Science 2013, 341, 154–157. [Google Scholar] [CrossRef]
- Rahim, M.A.; Bjornmalm, M.; Suma, T.; Faria, M.; Ju, Y.; Kempe, K.; Mgllner, M.; Ejima, H.; Stickland, A.D.; Caruso, F. Metal-Phenolic Supramolecular Gelation. Angew. Chem. Int. Ed. 2016, 55, 13803–13807. [Google Scholar] [CrossRef]
- Barrett, D.G.; Fullenkamp, D.E.; He, L.H.; Holten, N.; Lee, K.C. pH-Based Regulation of Hydrogel Mechanical Properties Through Mussel-Inspired Chemistry and Processing. Adv. Funct. Mater. 2013, 23, 1111–1119. [Google Scholar] [CrossRef]
- He, J.R.; Luo, L.; Chen, Y.F.; Manthiram, A. Yolk-Shelled C@Fe3O4 Nanoboxes as Effcient Sulfur Hosts for High-Performance Lithium-Sulfur Batteries. Adv. Mater. 2017, 29, 1702707. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.X.; Wang, Y.H.; Dong, L.; Chen, X.; Xin, G.X.; Zhang, Y.; Zang, J.B. One-step synthesis of shell/core structural boron and nitrogen co-doped graphitic carbon/nanodiamond as efficient electrocatalyst for the oxygen reduction reaction in alkaline media. Electrochim. Acta 2016, 194, 161–167. [Google Scholar] [CrossRef]
- Sun, X.J.; Zhang, Y.W.; Song, P.S.; Pan, J.; Zhuang, L.; Xu, W.L.; Xing, W. Fluorine-Doped Carbon Blacks: Highly Efficient Metal-Free Electrocatalysts for Oxygen Reduction Reaction. ACS Catal. 2013, 3, 1726–1729. [Google Scholar] [CrossRef]
- Ali-Löytty, H.; Louie, M.W.; Singh, M.R.; Li, L.; Casalongue, H.G.; Ogasawara, H.; Crumlin, E.J.; Liu, Z.; Bell, A.T.; Nilsson, A.; et al. Ambient-Pressure XPS Study of a Ni-Fe Electrocatalyst for the Oxygen Evolution Reaction. J. Phys. Chem. C 2016, 120, 2247–2253. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.Y.; Iyyamperumal, E.; Roy, A.; Xue, Y.H.; Yu, D.S.; Dai, L.M. Vertically aligned BCN nanotubes as efficient metal-free electrocatalysts for the oxygen reduction reaction: A synergetic effect by co-doping with boron and nitrogen. Angew. Chem. Int. Ed. 2011, 50, 11756–11760. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | SBET a [m2 g−1] | Smicro b [m2 g−1] | Smeso + macro c [m2 g−1] | Vtotal d [cm3 g−1] |
|---|---|---|---|---|
| CS6-650 | 361.62 | 149.54 | 212.08 | 0.34 |
| CS12-550 | 394.38 | 182.54 | 211.84 | 0.34 |
| CS12-650 | 439.47 | 180.79 | 258.68 | 0.40 |
| CS12-750 | 445.20 | 178.97 | 266.23 | 0.35 |
| CS18-650 | 451.13 | 261.13 | 190.00 | 0.36 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.; Li, Y.; Mao, J.; Wu, H.; Wu, T.; Li, Y.; Zeng, B.; Xu, Y.; Yuan, C.; Dai, L. Metallosupramolecular Polymer Precursor Design for Multi-Element Co-Doped Carbon Shells with Improved Oxygen Reduction Reaction Catalytic Activity. Catalysts 2019, 9, 102. https://doi.org/10.3390/catal9010102
Wu Y, Li Y, Mao J, Wu H, Wu T, Li Y, Zeng B, Xu Y, Yuan C, Dai L. Metallosupramolecular Polymer Precursor Design for Multi-Element Co-Doped Carbon Shells with Improved Oxygen Reduction Reaction Catalytic Activity. Catalysts. 2019; 9(1):102. https://doi.org/10.3390/catal9010102
Chicago/Turabian StyleWu, Yuzhe, Yuntong Li, Jie Mao, Haiyang Wu, Tong Wu, Yaying Li, Birong Zeng, Yiting Xu, Conghui Yuan, and Lizong Dai. 2019. "Metallosupramolecular Polymer Precursor Design for Multi-Element Co-Doped Carbon Shells with Improved Oxygen Reduction Reaction Catalytic Activity" Catalysts 9, no. 1: 102. https://doi.org/10.3390/catal9010102