An Overview of Recent Research in the Conversion of Glycerol into Biofuels, Fuel Additives and other Bio-Based Chemicals

 ,
, 
Abstract
:1. Introduction
2. Glycerol, Its Uniqueness and Availability
3. Biofuels from Glycerol
3.1. Hydrogen and Syngas
3.2. Ethanol
3.3. Methanol
4. Fuel Additives
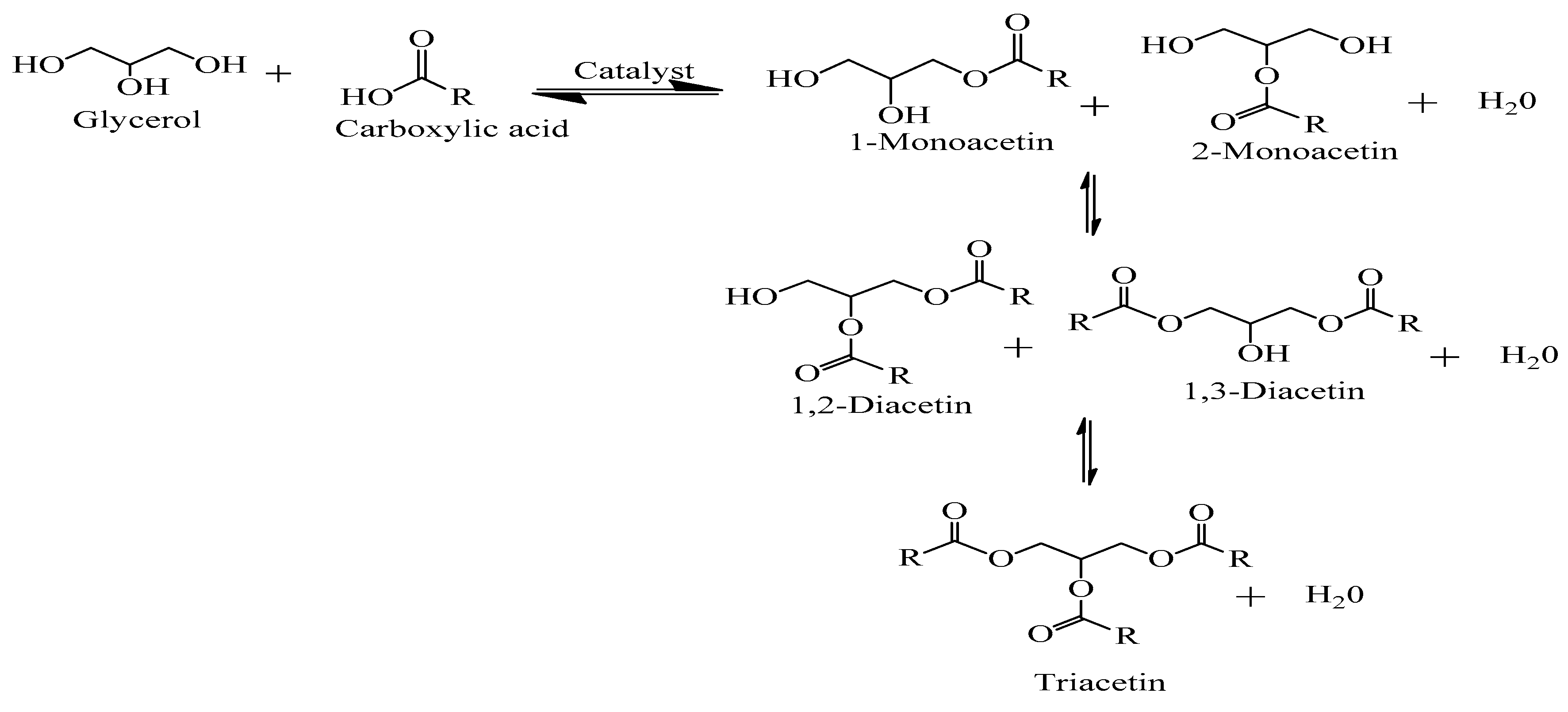
4.1. Acetin (Glycerol Esters)
4.2. Glycerol Ethers
4.3. Glycerol Formal
4.3.1. Solketal
4.3.2. Acetal
5. Other Precursor Bio-Based Chemicals from Glycerol
5.1. Acrolein
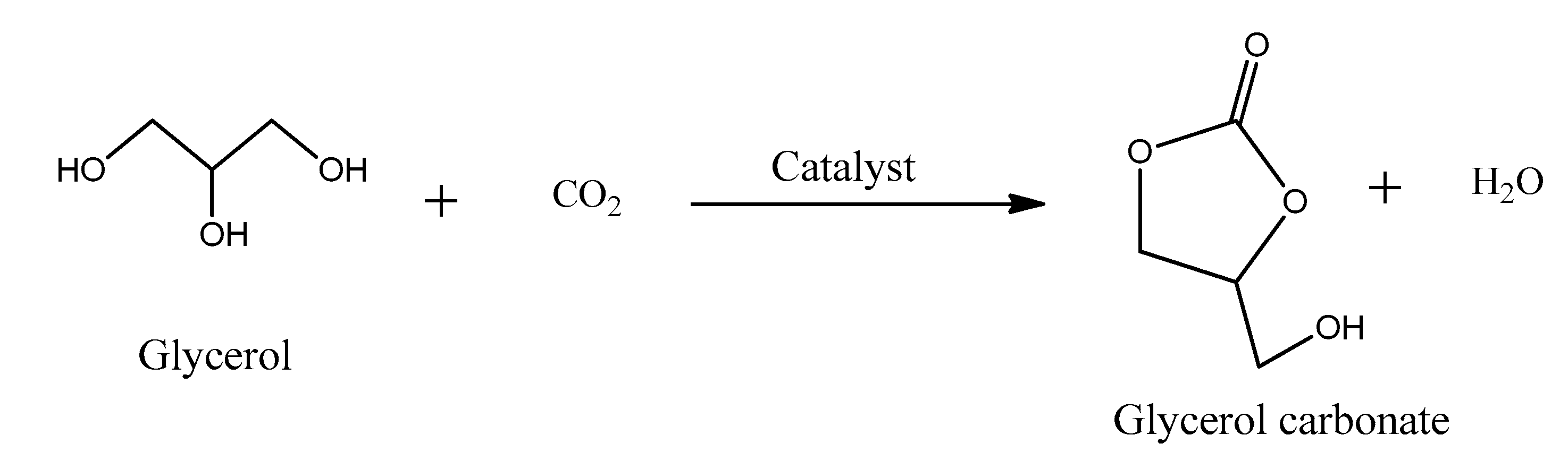
5.2. Glycerol Carbonate
5.3. 1,3–Propanediol
5.4. Polyglycerol (Oligomers)
5.5. Olefins
6. Prospects and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, H.; Su, L.; Shao, Y.; Zou, L. Biodiesel production catalyzed by cinder supported CaO/KF particle catalyst. Fuel 2012, 97, 651–657. [Google Scholar] [CrossRef]
- Akia, M.; Yazdani, F.; Motaee, E.; Han, D.; Arandiyan, H. A review on conversion of biomass to biofuel by nanocatalysts. Biofuel Res. J. 2014, 1, 16–25. [Google Scholar] [CrossRef]
- Da Silva, G.P.; Mack, M.; Contiero, J. Glycerol: A promising and abundant carbon source for industrial microbiology. Biotechnol. Adv. 2009, 27, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Markočič, E.; Kramberger, B.; van Bennekom, J.G.; Heeres, H.J.; Vos, J.; Knez, Ž. Glycerol reforming in supercritical water; a short review. Renew. Sustain. Energy Rev. 2013, 23, 40–48. [Google Scholar] [CrossRef]
- Yang, C.-Y.; Fang, Z.; Li, B.; Long, Y.-F. Review and prospects of Jatropha biodiesel industry in China. Renew. Sustain. Energy Rev. 2012, 16, 2178–2190. [Google Scholar] [CrossRef]
- Balat, M. Potential alternatives to edible oils for biodiesel production—A review of current work. Energy Convers. Manag. 2011, 52, 1479–1492. [Google Scholar] [CrossRef]
- Thanh, L.T.; Okitsu, K.; Boi, L.V.; Maeda, Y. Catalytic technologies for biodiesel fuel production and utilization of glycerol: A review. Catalysts 2012, 2, 191–222. [Google Scholar] [CrossRef]
- EU-Commission. Directive 2003/30/EC of the European Parliament and of the Council of 8 May 2003 on the promotion of the use of biofuels or other renewable fuels for transport. Off. J. Eur. Union 2003, 5. Available online: http://data.europa.eu/eli/dir/2003/30/oj (accessed on 22 September 2017).
- Knothe, G.; Razon, L.F. Biodiesel fuels. Prog. Energy Combust. Sci. 2017, 58, 36–59. [Google Scholar] [CrossRef]
- Fan, X.; Burton, R. Recent development of biodiesel feedstocks and the applications of glycerol: A review. Open Fuels Energy Sci. J. 2009, 2, 100–109. [Google Scholar] [CrossRef]
- Babajide, O. Sustaining biodiesel production via value-added applications of glycerol. J. Energy 2013, 2013. [Google Scholar] [CrossRef]
- Refaat, A. Different techniques for the production of biodiesel from waste vegetable oil. Int. J. Environ. Sci. Technol. 2010, 7, 183–213. [Google Scholar] [CrossRef]
- Banerjee, N.; Ramakrishnan, R.; Jash, T. Biodiesel production from used vegetable oil collected from shops selling fritters in Kolkata. Energy Procedia 2014, 54, 161–165. [Google Scholar] [CrossRef]
- Zhang, F.; Fang, Z.; Wang, Y.-T. Biodiesel production direct from high acid value oil with a novel magnetic carbonaceous acid. Appl. Energy 2015, 155, 637–647. [Google Scholar] [CrossRef]
- Neumann, K.; Werth, K.; Martín, A.; Górak, A. Biodiesel production from waste cooking oils through esterification: Catalyst screening, chemical equilibrium and reaction kinetics. Chem. Eng. Res. Des. 2016, 107, 52–62. [Google Scholar] [CrossRef]
- Dimitratos, N.; Lopez-Sanchez, J.A.; Hutchings, G.J. Green catalysis with alternative feedstocks. Top. Catal. 2009, 52, 258–268. [Google Scholar] [CrossRef]
- European Biodiesel Board. European Biodiesel Board Statistics on Biodiesel Production. Available online: http://www.ebb-eu.org/stats.php# (accessed on 22 September 2017).
- Stelmachowski, M. Utilization of glycerol, a by-product of the transestrification process of vegetable oils: A review. Ecol. Chem. Eng. 2011, 18, 9–30. [Google Scholar] [CrossRef]
- National Biodiesel Board, U.S. Biodiesel Production Statistics. Available online: http://biodiesel.org/production/production-statist (accessed on 22 September 2017).
- Ren21. Renewables 2016 Global Status Report. Available online: http://www.ren21.net/status-of-renewables/global-status-report/ (accessed on 23 September 2017).
- Comelli, R.A. Glycerol, the co-product of biodiesel: One key for the future bio-refinery. In Biodiesel-Quality, Emissions and By-Products; InTech: Rijeka, Crotia, 2011. [Google Scholar] [CrossRef]
- Union, E. Directive 2009/28/EC of the European Parliament and of the Council of 23 April 2009 on the promotion of the use of energy from renewable sources and amending and subsequently repealing Directives 2001/77/EC and 2003/30/EC. Off. J. Eur. Union 2009, 5. Available online: http://data.europa.eu/eli/dir/2009/28/oj (accessed on 22 September 2017).
- Kawentar, W.A.; Budiman, A. Synthesis of biodiesel from second-used cooking oil. Energy Procedia 2013, 32, 190–199. [Google Scholar] [CrossRef]
- Kumar, A.; Shukla, S.; Tierkey, J. A review of research and policy on using different biodiesel oils as fuel for CI engine. Energy Procedia 2016, 90, 292–304. [Google Scholar] [CrossRef]
- Kurnia, J.C.; Jangam, S.V.; Akhtar, S.; Sasmito, A.P.; Mujumdar, A.S. Advances in biofuel production from oil palm and palm oil processing wastes&58; A review. Biofuel Res. J. 2016, 3, 332–346. [Google Scholar] [CrossRef]
- Mofijur, M.; Atabani, A.; Masjuki, H.A.; Kalam, M.; Masum, B. A study on the effects of promising edible and non-edible biodiesel feedstocks on engine performance and emissions production: A comparative evaluation. Renew. Sustain. Energy Rev. 2013, 23, 391–404. [Google Scholar] [CrossRef]
- Aransiola, E.; Ojumu, T.; Oyekola, O.; Madzimbamuto, T.; Ikhu-Omoregbe, D. A review of current technology for biodiesel production: State of the art. Biomass Bioenergy 2014, 61, 276–297. [Google Scholar] [CrossRef]
- Galadima, A.; Muraza, O. Biodiesel production from algae by using heterogeneous catalysts: A critical review. Energy 2014, 78, 72–83. [Google Scholar] [CrossRef]
- Gaurav, A.; Ng, F.T.; Rempel, G.L. A new green process for biodiesel production from waste oils via catalytic distillation using a solid acid catalyst—Modeling, economic and environmental analysis. Green Energy Environ. 2016, 1, 62–74. [Google Scholar] [CrossRef]
- Demirbas, A. Importance of biodiesel as transportation fuel. Energy Policy 2007, 35, 4661–4670. [Google Scholar] [CrossRef]
- Singhabhandhu, A.; Tezuka, T. A perspective on incorporation of glycerin purification process in biodiesel plants using waste cooking oil as feedstock. Energy 2010, 35, 2493–2504. [Google Scholar] [CrossRef]
- Quispe, C.A.; Coronado, C.J.; Carvalho, J.A., Jr. Glycerol: Production, consumption, prices, characterization and new trends in combustion. Renew. Sustain. Energy Rev. 2013, 27, 475–493. [Google Scholar] [CrossRef]
- Yazdani, S.S.; Gonzalez, R. Anaerobic fermentation of glycerol: A path to economic viability for the biofuels industry. Curr. Opin. Biotechnol. 2007, 18, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Hanna, M.A.; Sun, R. Value-added uses for crude glycerol--a byproduct of biodiesel production. Biotechnol. Biofuels 2012, 5, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagheri, S.; Julkapli, N.M.; Yehye, W.A. Catalytic conversion of biodiesel derived raw glycerol to value added products. Renew. Sustain. Energy Rev. 2015, 41, 113–127. [Google Scholar] [CrossRef]
- Gupta, M.; Kumar, N. Scope and opportunities of using glycerol as an energy source. Renew. Sustain. Energy Rev. 2012, 16, 4551–4556. [Google Scholar] [CrossRef]
- Luo, X.; Ge, X.; Cui, S.; Li, Y. Value-added processing of crude glycerol into chemicals and polymers. Bioresour. Technol. 2016, 215, 144–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anitha, M.; Kamarudin, S.K.; Kofli, N.T. The potential of glycerol as a value-added commodity. Chem. Eng. J. 2016, 295, 119–130. [Google Scholar] [CrossRef]
- Gholami, Z.; Abdullah, A.Z.; Lee, K.-T. Dealing with the surplus of glycerol production from biodiesel industry through catalytic upgrading to polyglycerols and other value-added products. Renew. Sustain. Energy Rev. 2014, 39, 327–341. [Google Scholar] [CrossRef]
- Len, C.; Luque, R. Continuous flow transformations of glycerol to valuable products: An overview. Sustain. Chem. Process. 2014, 2, 1. [Google Scholar] [CrossRef]
- Christoph, R.; Schmidt, B.; Steinberner, U.; Dilla, W.; Karinen, R. Glycerol. Ullmann’s Encycl. Ind. Chem. 2006. [Google Scholar] [CrossRef]
- Kenar, J.A. Glycerol as a platform chemical: Sweet opportunities on the horizon? Lipid Technol. 2007, 19, 249–253. [Google Scholar] [CrossRef]
- Rahmat, N.; Abdullah, A.Z.; Mohamed, A.R. Recent progress on innovative and potential technologies for glycerol transformation into fuel additives: A critical review. Renew. Sustain. Energy Rev. 2010, 14, 987–1000. [Google Scholar] [CrossRef]
- Ayoub, M.; Abdullah, A.Z. Critical review on the current scenario and significance of crude glycerol resulting from biodiesel industry towards more sustainable renewable energy industry. Renew. Sustain. Energy Rev. 2012, 16, 2671–2686. [Google Scholar] [CrossRef]
- Hu, S.; Luo, X.; Wan, C.; Li, Y. Characterization of crude glycerol from biodiesel plants. J. Agric. Food Chem. 2012, 60, 5915–5921. [Google Scholar] [CrossRef] [PubMed]
- Din, N.; Idris, Z.; Kian, Y.S.; Hassan, H.A. Preparation of polyglycerol from palm-biodiesel crude glycerin. J. Oil Palm Res. 2013, 25, 289–297. [Google Scholar]
- Xiao, Y.; Xiao, G.; Varma, A. A universal procedure for crude glycerol purification from different feedstocks in biodiesel production: Experimental and simulation study. Ind. Eng. Chem. Res. 2013, 52, 14291–14296. [Google Scholar] [CrossRef]
- Hazimah, A.; Ooi, T.; Salmiah, A. Recovery of glycerol and diglycerol from glycerol pitch. J. Oil Palm Res. 2003, 15, 1–5. [Google Scholar]
- You, Y.-D.; Shie, J.-L.; Chang, C.-Y.; Huang, S.-H.; Pai, C.-Y.; Yu, Y.-H.; Chang, C.H. Economic cost analysis of biodiesel production: Case in soybean oil. Energy Fuels 2007, 22, 182–189. [Google Scholar] [CrossRef]
- Moita, R.; Freches, A.; Lemos, P. Crude glycerol as feedstock for polyhydroxyalkanoates production by mixed microbial cultures. Water Res. 2014, 58, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Koh, C.M.J.; Ji, L. Bioconversion of crude glycerol to glycolipids in Ustilago maydis. Bioresour. Technol. 2011, 102, 3927–3933. [Google Scholar] [CrossRef] [PubMed]
- Garlapati, V.K.; Shankar, U.; Budhiraja, A. Bioconversion technologies of crude glycerol to value added industrial products. Biotechnol. Rep. 2016, 9, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Demirbas, A.; Dincer, K. Sustainable green diesel: A futuristic view. Energy Sources Part A 2008, 30, 1233–1241. [Google Scholar] [CrossRef]
- Balat, H.; Kırtay, E. Hydrogen from biomass—Present scenario and future prospects. Int. J. Hydrog. Energy 2010, 35, 7416–7426. [Google Scholar] [CrossRef]
- Wei, Z.; Sun, J.; Li, Y.; Datye, A.K.; Wang, Y. Bimetallic catalysts for hydrogen generation. Chem. Soc. Rev. 2012, 41, 7994–8008. [Google Scholar] [CrossRef] [PubMed]
- Avasthi, K.S.; Reddy, R.N.; Patel, S. Challenges in the production of hydrogen from glycerol—A biodiesel byproduct via steam reforming process. Procedia Eng. 2013, 51, 423–429. [Google Scholar] [CrossRef]
- Hu, H.; Wood, T.K. An evolved Escherichia coli strain for producing hydrogen and ethanol from glycerol. Biochem. Biophys. Res. Commun. 2010, 391, 1033–1038. [Google Scholar] [CrossRef] [PubMed]
- Leoneti, A.B.; Aragão-Leoneti, V.; De Oliveira, S.V.W.B. Glycerol as a by-product of biodiesel production in Brazil: Alternatives for the use of unrefined glycerol. Renew. Energy 2012, 45, 138–145. [Google Scholar] [CrossRef]
- Lin, K.-H.; Chang, A.C.-C.; Lin, W.-H.; Chen, S.-H.; Chang, C.-Y.; Chang, H.-F. Autothermal steam reforming of glycerol for hydrogen production over packed-bed and Pd/Ag alloy membrane reactors. Int. J. Hydrog. Energy 2013, 38, 12946–12952. [Google Scholar] [CrossRef]
- Schwengber, C.A.; Alves, H.J.; Schaffner, R.A.; da Silva, F.A.; Sequinel, R.; Bach, V.R.; Ferracin, R.J. Overview of glycerol reforming for hydrogen production. Renew. Sustain. Energy Rev. 2016, 58, 259–266. [Google Scholar] [CrossRef]
- Dou, B.; Rickett, G.L.; Dupont, V.; Williams, P.T.; Chen, H.; Ding, Y.; Ghadiri, M. Steam reforming of crude glycerol with in situ CO2 sorption. Bioresour. Technol. 2010, 101, 2436–2442. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-C. Catalytic valorization of glycerol to hydrogen and syngas. Int. J. Hydrog. Energy 2013, 38, 2678–2700. [Google Scholar] [CrossRef]
- Fan, X.; Burton, R.; Zhou, Y. Glycerol (byproduct of biodiesel production) as a source for fuels and chemicals mini review. Open Fuels Energy Sci. J. 2010, 3. [Google Scholar] [CrossRef]
- Tan, H.; Aziz, A.A.; Aroua, M. Glycerol production and its applications as a raw material: A review. Renew. Sustain. Energy Rev. 2013, 27, 118–127. [Google Scholar] [CrossRef]
- Onwudili, J.A.; Williams, P.T. Hydrothermal reforming of bio-diesel plant waste: Products distribution and characterization. Fuel 2010, 89, 501–509. [Google Scholar] [CrossRef]
- Tapah, B.; Santos, R.; Leeke, G. Processing of glycerol under sub and supercritical water conditions. Renew. Energy 2014, 62, 353–361. [Google Scholar] [CrossRef]
- Adhikari, S.; Fernando, S.D.; Haryanto, A. Hydrogen production from glycerol: An update. Energy Convers. Manag. 2009, 50, 2600–2604. [Google Scholar] [CrossRef]
- Demsash, H.D.; Mohan, R. Steam reforming of glycerol to hydrogen over ceria promoted nickel–alumina catalysts. Int. J. Hydrog. Energy 2016, 41, 22732–22742. [Google Scholar] [CrossRef]
- Liu, B.; Greeley, J. Decomposition pathways of glycerol via C–H, O–H, and C–C bond scission on Pt (111): A density functional theory study. J. Phys. Chem. C 2011, 115, 19702–19709. [Google Scholar] [CrossRef]
- Manfro, R.L.; Ribeiro, N.F.; Souza, M.M. Production of hydrogen from steam reforming of glycerol using nickel catalysts supported on Al2O3, CeO2 and ZrO2. Catal. Sustain. Energy 2013, 1, 60–70. [Google Scholar] [CrossRef]
- Sanchez, E.A.; Comelli, R.A. Hydrogen production by glycerol steam-reforming over nickel and nickel-cobalt impregnated on alumina. Int. J. Hydrog. Energy 2014, 39, 8650–8655. [Google Scholar] [CrossRef]
- Zamzuri, N.H.; Mat, R.; Amin, N.A.S.; Talebian-Kiakalaieh, A. Hydrogen production from catalytic steam reforming of glycerol over various supported nickel catalysts. Int. J. Hydrog. Energy 2017, 42, 9087–9098. [Google Scholar] [CrossRef]
- Bepari, S.; Pradhan, N.C.; Dalai, A.K. Selective production of hydrogen by steam reforming of glycerol over Ni/Fly ash catalyst. Catal. Today 2017, 291, 36–46. [Google Scholar] [CrossRef]
- Özgür, D.Ö.; Uysal, B.Z. Hydrogen production by aqueous phase catalytic reforming of glycerine. Biomass Bioenergy 2011, 35, 822–826. [Google Scholar] [CrossRef]
- Menezes, A.O.; Rodrigues, M.T.; Zimmaro, A.; Borges, L.E.; Fraga, M.A. Production of renewable hydrogen from aqueous-phase reforming of glycerol over Pt catalysts supported on different oxides. Renew. Energy 2011, 36, 595–599. [Google Scholar] [CrossRef]
- Seretis, A.; Tsiakaras, P. Aqueous phase reforming (APR) of glycerol over platinum supported on Al2O3 catalyst. Renew. Energy 2016, 85, 1116–1126. [Google Scholar] [CrossRef]
- Remón, J.; Jarauta-Córdoba, C.; García, L.; Arauzo, J. Analysis and optimisation of H2 production from crude glycerol by steam reforming using a novel two step process. Fuel Process. Technol. 2016, 145, 130–147. [Google Scholar] [CrossRef]
- Yoon, S.J.; Yun, Y.M.; Seo, M.W.; Kim, Y.K.; Ra, H.W.; Lee, J.-G. Hydrogen and syngas production from glycerol through microwave plasma gasification. Int. J. Hydrog. Energy 2013, 38, 14559–14567. [Google Scholar] [CrossRef]
- Skoulou, V.K.; Zabaniotou, A.A. Co-gasification of crude glycerol with lignocellulosic biomass for enhanced syngas production. J. Anal. Appl. Pyrolysis 2013, 99, 110–116. [Google Scholar] [CrossRef]
- Guo, S.; Guo, L.; Cao, C.; Yin, J.; Lu, Y.; Zhang, X. Hydrogen production from glycerol by supercritical water gasification in a continuous flow tubular reactor. Int. J. Hydrog. Energy 2012, 37, 5559–5568. [Google Scholar] [CrossRef]
- Dianningrum, L.W.; Choi, H.; Kim, Y.; Jung, K.-D.; Susanti, R.F.; Kim, J.; Sang, B.-I. Hydrothermal gasification of pure and crude glycerol in supercritical water: A comparative study. Int. J. Hydrog. Energy 2014, 39, 1262–1273. [Google Scholar] [CrossRef]
- Haryanto, A.; Fernando, S.D.; Pordesimo, L.O.; Adhikari, S. Upgrading of syngas derived from biomass gasification: A thermodynamic analysis. Biomass Bioenergy 2009, 33, 882–889. [Google Scholar] [CrossRef]
- Sad, M.; Duarte, H.; Vignatti, C.; Padró, C.; Apesteguía, C. Steam reforming of glycerol: Hydrogen production optimization. Int. J. Hydrog. Energy 2015, 40, 6097–6106. [Google Scholar] [CrossRef]
- Wang, W. Thermodynamic analysis of glycerol partial oxidation for hydrogen production. Fuel Process. Technol. 2010, 91, 1401–1408. [Google Scholar] [CrossRef]
- Liu, S.-K.; Lin, Y.-C. Autothermal partial oxidation of glycerol to syngas over Pt-, LaMnO3−, and Pt/LaMnO3-coated monoliths. Ind. Eng. Chem. Res. 2012, 51, 16278–16287. [Google Scholar] [CrossRef]
- Rennard, D.C.; Kruger, J.S.; Michael, B.C.; Schmidt, L.D. Long-time behavior of the catalytic partial oxidation of glycerol in an autothermal reactor. Ind. Eng. Chem. Res. 2010, 49, 8424–8432. [Google Scholar] [CrossRef]
- Maru, B.T.; Constanti, M.; Stchigel, A.M.; Medina, F.; Sueiras, J.E. Biohydrogen production by dark fermentation of glycerol using Enterobacter and Citrobacter sp. Biotechnol. Prog. 2013, 29, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, N.; Ngadi, M.O.; Simpson, B.K.; Kassama, L.S. Biosynthesis of ethanol and hydrogen by glycerol fermentation using Escherichia coli. Adv. Chem. Eng. Sci. 2011, 1, 83. [Google Scholar] [CrossRef]
- Mangayil, R.; Karp, M.; Santala, V. Bioconversion of crude glycerol from biodiesel production to hydrogen. Int. J. Hydrog. Energy 2012, 37, 12198–12204. [Google Scholar] [CrossRef]
- Lo, Y.-C.; Chen, X.-J.; Huang, C.-Y.; Yuan, Y.-J.; Chang, J.-S. Dark fermentative hydrogen production with crude glycerol from biodiesel industry using indigenous hydrogen-producing bacteria. Int. J. Hydrog. Energy 2013, 38, 15815–15822. [Google Scholar] [CrossRef]
- Maru, B.T.; Bielen, A.; Kengen, S.; Constantí, M.; Medina, F. Biohydrogen production from glycerol using Thermotoga spp. Energy Procedia 2012, 29, 300–307. [Google Scholar] [CrossRef]
- Nomanbhay, S.; Hussain, R.; Rahman, M.M.; Palanisamy, K. Review paper integration of biodiesel and bioethanol processes: Convertion of low cost waste glycerol to bioethanol. Adv. Nat. Appl. Sci. 2012, 6, 802–818. [Google Scholar]
- Amaral, P.F.F.; Ferreira, T.F.; Fontes, G.C.; Coelho, M.A.Z. Glycerol valorization: New biotechnological routes. Food Bioprod. Process. 2009, 87, 179–186. [Google Scholar] [CrossRef]
- Posada, J.; Cardona, C. Design and analysis of fuel ethanol production from raw glycerol. Energy 2010, 35, 5286–5293. [Google Scholar] [CrossRef]
- Loaces, I.; Rodríguez, C.; Amarelle, V.; Fabiano, E.; Noya, F. Improved glycerol to ethanol conversion by E. coli using a metagenomic fragment isolated from an anaerobic reactor. J. Ind. Microbiol. Biotechnol. 2016, 43, 1405–1416. [Google Scholar] [CrossRef] [PubMed]
- Suhaimi, S.N.; Phang, L.-Y.; Maeda, T.; Abd-Aziz, S.; Wakisaka, M.; Shirai, Y.; Hassan, M.A. Bioconversion of glycerol for bioethanol production using isolated Escherichia coli SS1. Braz. J. Microbiol. 2012, 43, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Oh, B.-R.; Seo, J.-W.; Heo, S.-Y.; Hong, W.-K.; Luo, L.H.; Joe, M.-H.; Park, D.-H.; Kim, C.H. Efficient production of ethanol from crude glycerol by a Klebsiella pneumoniae mutant strain. Bioresour. Technol. 2011, 102, 3918–3922. [Google Scholar] [CrossRef] [PubMed]
- Metsoviti, M.; Paraskevaidi, K.; Koutinas, A.; Zeng, A.-P.; Papanikolaou, S. Production of 1,3-propanediol, 2,3-butanediol and ethanol by a newly isolated Klebsiella oxytoca strain growing on biodiesel-derived glycerol based media. Process. Biochem. 2012, 47, 1872–1882. [Google Scholar] [CrossRef]
- Metsoviti, M.; Paramithiotis, S.; Drosinos, E.H.; Galiotou-Panayotou, M.; Nychas, G.J.E.; Zeng, A.P.; Papanikolaou, S. Screening of bacterial strains capable of converting biodiesel-derived raw glycerol into 1,3-propanediol, 2,3-butanediol and ethanol. Eng. Life Sci. 2012, 12, 57–68. [Google Scholar] [CrossRef]
- Jitrwung, R.; Yargeau, V. Biohydrogen and bioethanol production from biodiesel-based glycerol by Enterobacter aerogenes in a continuous stir tank reactor. Int. J. Mol. Sci. 2015, 16, 10650–10664. [Google Scholar] [CrossRef] [PubMed]
- Thapa, L.P.; Lee, S.J.; Yang, X.; Lee, J.H.; Choi, H.S.; Park, C.; Kim, S.W. Improved bioethanol production from metabolic engineering of Enterobacter aerogenes ATCC 29007. Process. Biochem. 2015, 50, 2051–2060. [Google Scholar] [CrossRef]
- Kata, I.; Semkiv, M.V.; Ruchala, J.; Dmytruk, K.V.; Sibirny, A.A. Overexpression of the genes PDC1 and ADH1 activates glycerol conversion to ethanol in the thermotolerant yeast Ogataea (Hansenula) polymorpha. Yeast 2016, 33, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Varrone, C.; Fiocchetti, F.; Giussani, B.; Izzo, A.; Marone, A.; Massini, G.; Patriarca, C.; Rosa, S.; Signorini, A.; Wang, A. Bio-Conversion of Biodiesel-Derived Glycerol into Hydrogen and Ethanol: Beyond Second-Generation Biofuels. In Proceedings of the 20th European Biomass Conference & Exhibition, Milan, Italy, 18–22 June 2012; pp. 713–716, ISBN 978-88-89407-54-7. [Google Scholar]
- Sankaranarayanan, S.; Srinivasan, K. Carbon dioxide—A potential raw material for the production of fuel, fuel additives and bio-derived chemicals. Indian J. Chem. A 2012, 51, 1252–1262. [Google Scholar]
- Haider, M.H.; Dummer, N.F.; Knight, D.W.; Jenkins, R.L.; Howard, M.; Moulijn, J.; Taylor, S.H.; Hutchings, G.J. Efficient green methanol synthesis from glycerol. Nat. Chem. 2015, 7, 1028. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.G.; Shi, X.; Domene, C.; Leung, A.K.; Green, W.H. Methanol formation from the treatment of glycerol in supercritical water and with ethylsulfide. J. Supercrit. Fluids 2016, 117, 80–88. [Google Scholar] [CrossRef]
- García, J.I.; García-Marín, H.; Pires, E. Glycerol based solvents: Synthesis, properties and applications. Green Chem. 2014, 16, 1007–1033. [Google Scholar] [CrossRef]
- Cornejo, A.; Barrio, I.; Campoy, M.; Lázaro, J.; Navarrete, B. Oxygenated fuel additives from glycerol valorization. Main production pathways and effects on fuel properties and engine performance: A critical review. Renew. Sustain. Energy Rev. 2017, 79, 1400–1413. [Google Scholar] [CrossRef]
- Farinha, J.; Caiado, M.; Castanheiro, J. Valorisation of glycerol into biofuel additives over heterogeneous catalysts. In Materials and Processes for Energy: Communicating Current Research and Technological Developments; Méndez-Vilas, A.S., Ed.; Formatex Research Center Country: Badajoz, Spain, 2013; pp. 422–429. ISBN 978-84-939843-7-3. Available online: http://hdl.handle.net/10174/10317 (accessed on 29 May 2017).
- Usai, E.; Gualdi, E.; Solinas, V.; Battistel, E. Simultaneous enzymatic synthesis of FAME and triacetyl glycerol from triglycerides and methyl acetate. Bioresour. Technol. 2010, 101, 7707–7712. [Google Scholar] [CrossRef] [PubMed]
- Morales, G.; Paniagua, M.; Melero, J.A.; Vicente, G.; Ochoa, C. Sulfonic acid-functionalized catalysts for the valorization of glycerol via transesterification with methyl acetate. Ind. Eng. Chem. Res. 2011, 50, 5898–5906. [Google Scholar] [CrossRef]
- Khayoon, M.; Hameed, B. Synthesis of hybrid SBA-15 functionalized with molybdophosphoric acid as efficient catalyst for glycerol esterification to fuel additives. Appl. Catal. A Gen. 2012, 433, 152–161. [Google Scholar] [CrossRef]
- Liao, X.; Zhu, Y.; Wang, S.-G.; Li, Y. Producing triacetylglycerol with glycerol by two steps: Esterification and acetylation. Fuel Process. Technol. 2009, 90, 988–993. [Google Scholar] [CrossRef]
- Gonçalves, V.L.; Pinto, B.P.; Silva, J.C.; Mota, C.J. Acetylation of glycerol catalyzed by different solid acids. Catal. Today 2008, 133, 673–677. [Google Scholar] [CrossRef]
- Silva, L.N.; Gonçalves, V.L.; Mota, C.J. Catalytic acetylation of glycerol with acetic anhydride. Catal. Commun. 2010, 11, 1036–1039. [Google Scholar] [CrossRef]
- Zhou, L.; Al-Zaini, E.; Adesina, A.A. Catalytic characteristics and parameters optimization of the glycerol acetylation over solid acid catalysts. Fuel 2013, 103, 617–625. [Google Scholar] [CrossRef]
- Khayoon, M.; Triwahyono, S.; Hameed, B.; Jalil, A. Improved production of fuel oxygenates via glycerol acetylation with acetic acid. Chem. Eng. J. 2014, 243, 473–484. [Google Scholar] [CrossRef]
- Chandrakala, U.; Prasad, R.B.; Prabhavathi Devi, B.L. Glycerol valorization as biofuel additives by employing a carbon-based solid acid catalyst derived from glycerol. Ind. Eng. Chem. Res. 2014, 53, 16164–16169. [Google Scholar] [CrossRef]
- Sun, J.; Tong, X.; Yu, L.; Wan, J. An efficient and sustainable production of triacetin from the acetylation of glycerol using magnetic solid acid catalysts under mild conditions. Catal. Today 2016, 264, 115–122. [Google Scholar] [CrossRef]
- Zhu, S.; Zhu, Y.; Gao, X.; Mo, T.; Zhu, Y.; Li, Y. Production of bioadditives from glycerol esterification over zirconia supported heteropolyacids. Bioresour. Technol. 2013, 130, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Arellano, C.; De, S.; Luque, R. Selective glycerol transformations to high value-added products catalysed by aluminosilicate-supported iron oxide nanoparticles. Catal. Sci. Technol. 2014, 4, 4242–4249. [Google Scholar] [CrossRef]
- Dalla Costa, B.; Decolatti, H.; Legnoverde, M.; Querini, C. Influence of acidic properties of different solid acid catalysts for glycerol acetylation. Catal. Today 2017, 289, 222–230. [Google Scholar] [CrossRef]
- Reddy, P.S.; Sudarsanam, P.; Raju, G.; Reddy, B.M. Synthesis of bio-additives: Acetylation of glycerol over zirconia-based solid acid catalysts. Catal. Commun. 2010, 11, 1224–1228. [Google Scholar] [CrossRef]
- Reddy, P.S.; Sudarsanam, P.; Raju, G.; Reddy, B.M. Selective acetylation of glycerol over CeO2–M and SO42−/CeO2–M (M = ZrO2 and Al2O3) catalysts for synthesis of bioadditives. J. Ind. Eng. Chem. 2012, 18, 648–654. [Google Scholar] [CrossRef]
- Zhang, Z.; Huang, H.; Ma, X.; Li, G.; Wang, Y.; Sun, G.; Teng, Y.; Yan, R.; Zhang, N.; Li, A. Production of diacylglycerols by esterification of oleic acid with glycerol catalyzed by diatomite loaded SO42−/TiO2. J. Ind. Eng. Chem. 2017, 53, 307–316. [Google Scholar] [CrossRef]
- Sandesh, S.; Kristachar, P.K.R.; Manjunathan, P.; Halgeri, A.; Shanbhag, G.V. Synthesis of biodiesel and acetins by transesterification reactions using novel CaSn (OH) 6 heterogeneous base catalyst. Appl. Catal. A Gen. 2016, 523, 1–11. [Google Scholar] [CrossRef]
- Okoye, P.; Abdullah, A.; Hameed, B. Synthesis of oxygenated fuel additives via glycerol esterification with acetic acid over bio-derived carbon catalyst. Fuel 2017, 209, 538–544. [Google Scholar] [CrossRef]
- Gonçalves, C.E.; Laier, L.O.; Cardoso, A.L.; da Silva, M.J. Bioadditive synthesis from H3PW12O40-catalyzed glycerol esterification with HOAc under mild reaction conditions. Fuel Process. Technol. 2012, 102, 46–52. [Google Scholar] [CrossRef]
- García, J.I.; García-Marín, H.; Mayoral, J.A.; Pérez, P. Green solvents from glycerol. Synthesis and physico-chemical properties of alkyl glycerol ethers. Green Chem. 2010, 12, 426–434. [Google Scholar] [CrossRef]
- Gonçalves, M.; Souza, V.C.; Galhardo, T.S.; Mantovani, M.; Figueiredo, F.V.C.; Mandelli, D.; Carvalho, W.A. Glycerol conversion catalyzed by carbons prepared from agroindustrial wastes. Ind. Eng. Chem. Res. 2013, 52, 2832–2839. [Google Scholar] [CrossRef]
- Ayoub, M.; Khayoon, M.; Abdullah, A.Z. Synthesis of oxygenated fuel additives via the solventless etherification of glycerol. Bioresour. Technol. 2012, 112, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Xia, S.; Chen, P.; Hou, Z.; Zheng, X. Etherification of biodiesel-based glycerol with bioethanol over tungstophosphoric acid to synthesize glyceryl ethers. Energy Fuels 2011, 25, 3186–3191. [Google Scholar] [CrossRef]
- Viswanadham, N.; Saxena, S.K. Etherification of glycerol for improved production of oxygenates. Fuel 2013, 103, 980–986. [Google Scholar] [CrossRef]
- Veiga, P.M.; Gomes, A.C.; Veloso, C.O.; Henriques, C.A. Acid zeolites for glycerol etherification with ethyl alcohol: Catalytic activity and catalyst properties. Appl. Catal. A Gen. 2017, 548, 2–15. [Google Scholar] [CrossRef]
- Pinto, B.P.; de Lyra, J.T.; Nascimento, J.A.; Mota, C.J. Ethers of glycerol and ethanol as bioadditives for biodiesel. Fuel 2016, 168, 76–80. [Google Scholar] [CrossRef]
- Estevez, R.; Lopez, M.; Jiménez-Sanchidrián, C.; Luna, D.; Romero-Salguero, F.; Bautista, F. Etherification of glycerol with tert-butyl alcohol over sulfonated hybrid silicas. Appl. Catal. A Gen. 2016, 526, 155–163. [Google Scholar] [CrossRef]
- Agirre, I.; Garcia, I.; Requies, J.; Barrio, V.; Güemez, M.; Cambra, J.; Arias, P. Glycerol acetals, kinetic study of the reaction between glycerol and formaldehyde. Biomass Bioenergy 2011, 35, 3636–3642. [Google Scholar] [CrossRef]
- Mota, C.J.; da Silva, C.X.; Rosenbach, N., Jr.; Costa, J.; da Silva, F.V. Glycerin derivatives as fuel additives: The addition of glycerol/acetone ketal (solketal) in gasolines. Energy Fuels 2010, 24, 2733–2736. [Google Scholar] [CrossRef]
- Maksimov, A.; Nekhaev, A.; Ramazanov, D.; Arinicheva, Y.A.; Dzyubenko, A.; Khadzhiev, S. Preparation of high-octane oxygenate fuel components from plant-derived polyols. Pet. Chem. 2011, 51, 61–69. [Google Scholar] [CrossRef]
- Suriyaprapadilok, N.; Kitiyanan, B. Synthesis of solketal from glycerol and its reaction with benzyl alcohol. Energy Procedia 2011, 9, 63–69. [Google Scholar] [CrossRef]
- Vicente, G.; Melero, J.A.; Morales, G.; Paniagua, M.; Martín, E. Acetalisation of bio-glycerol with acetone to produce solketal over sulfonic mesostructured silicas. Green Chem. 2010, 12, 899–907. [Google Scholar] [CrossRef]
- De Torres, M.; Jimenez-Oses, G.; Mayoral, J.A.; Pires, E.; de los Santos, M. Glycerol ketals: Synthesis and profits in biodiesel blends. Fuel 2012, 94, 614–616. [Google Scholar] [CrossRef]
- Nanda, M.R.; Yuan, Z.; Qin, W.; Ghaziaskar, H.S.; Poirier, M.-A.; Xu, C.C. A new continuous-flow process for catalytic conversion of glycerol to oxygenated fuel additive: Catalyst screening. Appl. Energy 2014, 123, 75–81. [Google Scholar] [CrossRef]
- Nanda, M.R.; Yuan, Z.; Qin, W.; Ghaziaskar, H.S.; Poirier, M.-A.; Xu, C.C. Catalytic conversion of glycerol to oxygenated fuel additive in a continuous flow reactor: Process optimization. Fuel 2014, 128, 113–119. [Google Scholar] [CrossRef]
- Nanda, M.R.; Yuan, Z.; Qin, W.; Ghaziaskar, H.S.; Poirier, M.-A.; Xu, C.C. Thermodynamic and kinetic studies of a catalytic process to convert glycerol into solketal as an oxygenated fuel additive. Fuel 2014, 117, 470–477. [Google Scholar] [CrossRef]
- Gadamsetti, S.; Rajan, N.P.; Rao, G.S.; Chary, K.V. Acetalization of glycerol with acetone to bio fuel additives over supported molybdenum phosphate catalysts. J. Mol. Catal. A Chem. 2015, 410, 49–57. [Google Scholar] [CrossRef]
- Jamil, F.; Saxena, S.K.; Ala’a, H.; Baawain, M.; Al-Abri, M.; Viswanadham, N.; Kumar, G.; Abu-Jrai, A.M. Valorization of waste “date seeds” bio-glycerol for synthesizing oxidative green fuel additive. J. Clean. Prod. 2017, 165, 1090–1096. [Google Scholar] [CrossRef]
- Manjunathan, P.; Maradur, S.P.; Halgeri, A.; Shanbhag, G.V. Room temperature synthesis of solketal from acetalization of glycerol with acetone: Effect of crystallite size and the role of acidity of beta zeolite. J. Mol. Catal. A Chem. 2015, 396, 47–54. [Google Scholar] [CrossRef]
- Khayoon, M.; Hameed, B. Solventless acetalization of glycerol with acetone to fuel oxygenates over Ni–Zr supported on mesoporous activated carbon catalyst. Appl. Catal. A Gen. 2013, 464, 191–199. [Google Scholar] [CrossRef]
- De Carvalho, D.C.; Oliveira, A.C.; Ferreira, O.P.; Josué Filho, M.; Tehuacanero-Cuapa, S.; Oliveira, A.C. Titanate nanotubes as acid catalysts for acetalization of glycerol with acetone: Influence of the synthesis time and the role of structure on the catalytic performance. Chem. Eng. J. 2017, 313, 1454–1467. [Google Scholar] [CrossRef]
- Rodrigues, R.; Mandelli, D.; Gonçalves, N.S.; Pescarmona, P.P.; Carvalho, W.A. Acetalization of acetone with glycerol catalyzed by niobium-aluminum mixed oxides synthesized by a sol–gel process. J. Mol. Catal. A Chem. 2016, 422, 122–130. [Google Scholar] [CrossRef]
- Kapkowski, M.; Ambrożkiewicz, W.; Siudyga, T.; Sitko, R.; Szade, J.; Klimontko, J.; Balin, K.; Lelątko, J.; Polanski, J. Nano silica and molybdenum supported Re, Rh, Ru or Ir nanoparticles for selective solvent-free glycerol conversion to cyclic acetals with propanone and butanone under mild conditions. Appl. Catal. B Environ. 2017, 202, 335–345. [Google Scholar] [CrossRef]
- Umbarkar, S.B.; Kotbagi, T.V.; Biradar, A.V.; Pasricha, R.; Chanale, J.; Dongare, M.K.; Mamede, A.-S.; Lancelot, C.; Payen, E. Acetalization of glycerol using mesoporous MoO3/SiO2 solid acid catalyst. J. Mol. Catal. A Chem. 2009, 310, 150–158. [Google Scholar] [CrossRef]
- Sudarsanam, P.; Mallesham, B.; Prasad, A.N.; Reddy, P.S.; Reddy, B.M. Synthesis of bio–additive fuels from acetalization of glycerol with benzaldehyde over molybdenum promoted green solid acid catalysts. Fuel Process. Technol. 2013, 106, 539–545. [Google Scholar] [CrossRef]
- Chen, L.; Nohair, B.; Kaliaguine, S. Glycerol acetalization with formaldehyde using water-tolerant solid acids. Appl. Catal. A Gen. 2016, 509, 143–152. [Google Scholar] [CrossRef]
- Possato, L.G.; Diniz, R.N.; Garetto, T.; Pulcinelli, S.H.; Santilli, C.V.; Martins, L. A comparative study of glycerol dehydration catalyzed by micro/mesoporous MFI zeolites. J. Catal. 2013, 300, 102–112. [Google Scholar] [CrossRef]
- Rajan, N.P.; Rao, G.S.; Pavankumar, V.; Chary, K.V. Vapour phase dehydration of glycerol over VPO catalyst supported on zirconium phosphate. Catal. Sci. Technol. 2014, 4, 81–92. [Google Scholar] [CrossRef]
- Akizuki, M.; Oshima, Y. Kinetics of glycerol dehydration with WO3/TiO2 in supercritical water. Ind. Eng. Chem. Res. 2012, 51, 12253–12257. [Google Scholar] [CrossRef]
- Gu, Y.; Liu, S.; Li, C.; Cui, Q. Selective conversion of glycerol to acrolein over supported nickel sulfate catalysts. J. Catal. 2013, 301, 93–102. [Google Scholar] [CrossRef]
- Yadav, G.D.; Sharma, R.V.; Katole, S.O. Selective dehydration of glycerol to acrolein: Development of efficient and robust solid acid catalyst MUICaT-5. Ind. Eng. Chem. Res. 2013, 52, 10133–10144. [Google Scholar] [CrossRef]
- Alhanash, A.; Kozhevnikova, E.F.; Kozhevnikov, I.V. Gas-phase dehydration of glycerol to acrolein catalysed by caesium heteropoly salt. Appl. Catal. A Gen. 2010, 378, 11–18. [Google Scholar] [CrossRef]
- Ma, T.; Ding, J.; Shao, R.; Xu, W.; Yun, Z. Dehydration of glycerol to acrolein over Wells–Dawson and Keggin type phosphotungstic acids supported on MCM-41 catalysts. Chem. Eng. J. 2017, 316, 797–806. [Google Scholar] [CrossRef]
- Dar, B.A.; Dadhwal, S.; Singh, G.; Garg, P.; Sharma, P.; Singh, B. Vapour phase conversion of glycerol to acrolein over supported copper. Arab. J. Sci. Eng. 2013, 38, 37–40. [Google Scholar] [CrossRef]
- Jia, C.-J.; Liu, Y.; Schmidt, W.; Lu, A.-H.; Schüth, F. Small-sized HZSM-5 zeolite as highly active catalyst for gas phase dehydration of glycerol to acrolein. J. Catal. 2010, 269, 71–79. [Google Scholar] [CrossRef]
- Possato, L.G.; Chaves, T.F.; Cassinelli, W.H.; Pulcinelli, S.H.; Santilli, C.V.; Martins, L. The multiple benefits of glycerol conversion to acrolein and acrylic acid catalyzed by vanadium oxides supported on micro-mesoporous MFI zeolites. Catal. Today 2017, 289, 20–28. [Google Scholar] [CrossRef]
- Zhao, H.; Zhou, C.H.; Wu, L.M.; Lou, J.Y.; Li, N.; Yang, H.M.; Tong, D.S.; Yu, W.H. Catalytic dehydration of glycerol to acrolein over sulfuric acid-activated montmorillonite catalysts. Appl. Clay Sci. 2013, 74, 154–162. [Google Scholar] [CrossRef]
- Ochoa-Gómez, J.R.; Gómez-Jiménez-Aberasturi, O.; Maestro-Madurga, B.; Pesquera-Rodríguez, A.; Ramírez-López, C.; Lorenzo-Ibarreta, L.; Torrecilla-Soria, J.; Villarán-Velasco, M.C. Synthesis of glycerol carbonate from glycerol and dimethyl carbonate by transesterification: Catalyst screening and reaction optimization. Appl. Catal. A Gen. 2009, 366, 315–324. [Google Scholar] [CrossRef]
- Climent, M.J.; Corma, A.; De Frutos, P.; Iborra, S.; Noy, M.; Velty, A.; Concepción, P. Chemicals from biomass: Synthesis of glycerol carbonate by transesterification and carbonylation with urea with hydrotalcite catalysts. The role of acid–base pairs. J. Catal. 2010, 269, 140–149. [Google Scholar] [CrossRef]
- Casiello, M.; Monopoli, A.; Cotugno, P.; Milella, A.; Dell’Anna, M.M.; Ciminale, F.; Nacci, A. Copper (II) chloride-catalyzed oxidative carbonylation of glycerol to glycerol carbonate. J. Mol. Catal. A Chem. 2014, 381, 99–106. [Google Scholar] [CrossRef]
- Zhang, J.; He, D. Surface properties of Cu/La2O3 and its catalytic performance in the synthesis of glycerol carbonate and monoacetin from glycerol and carbon dioxide. J. Colloid Interface Sci. 2014, 419, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Zuhaimi, N.A.S.; Indran, V.P.; Deraman, M.A.; Mudrikah, N.F.; Maniam, G.P.; Taufiq-Yap, Y.H.; Rahim, M.H.A. Reusable gypsum based catalyst for synthesis of glycerol carbonate from glycerol and urea. Appl. Catal. A Gen. 2015, 502, 312–319. [Google Scholar] [CrossRef]
- Wu, Y.; Song, X.; Cai, F.; Xiao, G. Synthesis of glycerol carbonate from glycerol and diethyl carbonate over Ce-NiO catalyst: The role of multiphase Ni. J. Alloys Compd. 2017, 720, 360–368. [Google Scholar] [CrossRef]
- Ozorio, L.P.; Pianzolli, R.; da Cruz Machado, L.; Miranda, J.L.; Turci, C.C.; Guerra, A.C.; Souza-Aguiar, E.F.; Mota, C.J. Metal-impregnated zeolite Y as efficient catalyst for the direct carbonation of glycerol with CO2. Appl. Catal. A Gen. 2015, 504, 187–191. [Google Scholar] [CrossRef]
- Indran, V.P.; Zuhaimi, N.A.S.; Deraman, M.A.; Maniam, G.P.; Yusoff, M.M.; Hin, T.-Y.Y.; Rahim, M.H.A. An accelerated route of glycerol carbonate formation from glycerol using waste boiler ash as catalyst. RSC Adv. 2014, 4, 25257–25267. [Google Scholar] [CrossRef]
- Naik, P.U.; Petitjean, L.; Refes, K.; Picquet, M.; Plasseraud, L. Imidazolium-2-Carboxylate as an Efficient, Expeditious and Eco-Friendly Organocatalyst for Glycerol Carbonate Synthesis. Adv. Synth. Catal. 2009, 351, 1753–1756. [Google Scholar] [CrossRef]
- Ishak, Z.I.; Sairi, N.A.; Alias, Y.; Aroua, M.K.T.; Yusoff, R. Production of glycerol carbonate from glycerol with aid of ionic liquid as catalyst. Chem. Eng. J. 2016, 297, 128–138. [Google Scholar] [CrossRef]
- Saxena, R.; Anand, P.; Saran, S.; Isar, J. Microbial production of 1,3-propanediol: Recent developments and emerging opportunities. Biotechnol. Adv. 2009, 27, 895–913. [Google Scholar] [CrossRef] [PubMed]
- Ten Dam, J.; Hanefeld, U. Renewable chemicals: Dehydroxylation of glycerol and polyols. ChemSusChem 2011, 4, 1017–1034. [Google Scholar] [CrossRef] [PubMed]
- Zeng, A.-P.; Sabra, W. Microbial production of diols as platform chemicals: Recent progresses. Curr. Opin. Biotechnol. 2011, 22, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.-Z.; Song, M.-J.; Chen, C.-L. Aqueous-phase deoxygenation of glycerol to 1,3-propanediol over Pt/WO3/ZrO2 catalysts in a fixed-bed reactor. Green Chem. 2010, 12, 1466–1472. [Google Scholar] [CrossRef]
- Gong, L.; Lu, Y.; Ding, Y.; Lin, R.; Li, J.; Dong, W.; Wang, T.; Chen, W. Selective hydrogenolysis of glycerol to 1,3-propanediol over a Pt/WO3/TiO2/SiO2 catalyst in aqueous media. Appl. Catal. A Gen. 2010, 390, 119–126. [Google Scholar] [CrossRef]
- García-Fernández, S.; Gandarias, I.; Requies, J.; Soulimani, F.; Arias, P.L.; Weckhuysen, B.M. The role of tungsten oxide in the selective hydrogenolysis of glycerol to 1,3-propanediol over Pt/WOx/Al2O3. Appl. Catal. B Environ. 2017, 204, 260–272. [Google Scholar] [CrossRef]
- García-Fernández, S.; Gandarias, I.; Requies, J.; Güemez, M.; Bennici, S.; Auroux, A.; Arias, P. New approaches to the Pt/WOx/Al2O3 catalytic system behavior for the selective glycerol hydrogenolysis to 1,3-propanediol. J. Catal. 2015, 323, 65–75. [Google Scholar] [CrossRef]
- Longjie, L.; Zhang, Y.; Aiqin, W.; Zhang, T. Mesoporous WO3 supported Pt catalyst for hydrogenolysis of glycerol to 1,3-propanediol. Chin. J. Catal. 2012, 33, 1257–1261. [Google Scholar] [CrossRef]
- Zhu, S.; Gao, X.; Zhu, Y.; Zhu, Y.; Xiang, X.; Hu, C.; Li, Y. Alkaline metals modified Pt–H4SiW12O40/ZrO2 catalysts for the selective hydrogenolysis of glycerol to 1,3-propanediol. Appl. Catal. B Environ. 2013, 140, 60–67. [Google Scholar] [CrossRef]
- Zhu, S.; Qiu, Y.; Zhu, Y.; Hao, S.; Zheng, H.; Li, Y. Hydrogenolysis of glycerol to 1,3-propanediol over bifunctional catalysts containing Pt and heteropolyacids. Catal. Today 2013, 212, 120–126. [Google Scholar] [CrossRef]
- Zhu, S.; Gao, X.; Zhu, Y.; Cui, J.; Zheng, H.; Li, Y. SiO2 promoted Pt/WOx/ZrO2 catalysts for the selective hydrogenolysis of glycerol to 1,3-propanediol. Appl. Catal. B Environ. 2014, 158, 391–399. [Google Scholar] [CrossRef]
- Deng, C.; Duan, X.; Zhou, J.; Chen, D.; Zhou, X.; Yuan, W. Size effects of Pt-Re bimetallic catalysts for glycerol hydrogenolysis. Catal. Today 2014, 234, 208–214. [Google Scholar] [CrossRef]
- Zhou, W.; Zhao, Y.; Wang, S.; Ma, X. The effect of metal properties on the reaction routes of glycerol hydrogenolysis over platinum and ruthenium catalysts. Catal. Today 2017, 298, 2–8. [Google Scholar] [CrossRef]
- Kant, A.; He, Y.; Jawad, A.; Li, X.; Rezaei, F.; Smith, J.D.; Rownaghi, A.A. Hydrogenolysis of glycerol over Ni, Cu, Zn, and Zr supported on H-β. Chem. Eng. J. 2017, 317, 1–8. [Google Scholar] [CrossRef]
- Sivaiah, M.; Robles-Manuel, S.; Valange, S.; Barrault, J. Recent developments in acid and base-catalyzed etherification of glycerol to polyglycerols. Catal. Today 2012, 198, 305–313. [Google Scholar] [CrossRef]
- Beltramini, J.N.; Zhou, C.-H.C. Catalytic conversion of glycerol to valuable commodity chemicals. In Thermochemical Conversion of Biomass to Liquid Fuels and Chemicals; Crocker, M.S., Ed.; RSC Publishing: London, UK, 2010; pp. 435–467. ISBN 978-1-84973-035-8. [Google Scholar] [CrossRef]
- Martin, A.; Checinski, M.P.; Richter, M. Tuning of diglycerol yield and isomer distribution in oligomerization of glycerol supported by DFT-calculations. Catal. Commun. 2012, 25, 130–135. [Google Scholar] [CrossRef]
- Ruppert, A.M.; Meeldijk, J.D.; Kuipers, B.W.; Erné, B.H.; Weckhuysen, B.M. Glycerol Etherification over Highly Active CaO-Based Materials: New Mechanistic Aspects and Related Colloidal Particle Formation. Chem.-A Eur. J. 2008, 14, 2016–2024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Sancho, C.; Moreno-Tost, R.; Mérida-Robles, J.M.; Santamaría-González, J.; Jiménez-López, A.; Torres, P.M. Etherification of glycerol to polyglycerols over MgAl mixed oxides. Catal. Today 2011, 167, 84–90. [Google Scholar] [CrossRef]
- Anuar, M.R.; Abdullah, A.Z.; Othman, M.R. Etherification of glycerol to polyglycerols over hydrotalcite catalyst prepared using a combustion method. Catal. Commun. 2013, 32, 67–70. [Google Scholar] [CrossRef]
- Ayoub, M.; Abdullah, A.Z. Diglycerol synthesis via solvent-free selective glycerol etherification process over lithium-modified clay catalyst. Chem. Eng. J. 2013, 225, 784–789. [Google Scholar] [CrossRef]
- Gholami, Z.; Abdullah, A.Z.; Lee, K.-T. Heterogeneously catalyzed etherification of glycerol to diglycerol over calcium-lanthanum oxide supported on MCM-41: A heterogenous basic catalyst. Appl. Catal. A Gen. 2014, 479, 76–86. [Google Scholar] [CrossRef]
- Gholami, Z.; Abdullah, A.Z.; Lee, K.-T. Glycerol etherification to polyglycerols using Ca1+xAl1−xLaxO3 composite catalysts in a solventless medium. J. Taiwan Inst. Chem. Eng. 2013, 44, 117–122. [Google Scholar] [CrossRef]
- Guerrero-Urbaneja, P.; García-Sancho, C.; Moreno-Tost, R.; Mérida-Robles, J.; Santamaría-González, J.; Jiménez-López, A.; Maireles-Torres, P. Glycerol valorization by etherification to polyglycerols by using metal oxides derived from MgFe hydrotalcites. Appl. Catal. A Gen. 2014, 470, 199–207. [Google Scholar] [CrossRef]
- Ayoub, M.; Abdullah, A.Z.; Ahmad, M.; Sultana, S. Performance of lithium modified zeolite Y catalyst in solvent-free conversion of glycerol to polyglycerols. J. Taibah Univ. Sci. 2014, 8, 231–235. [Google Scholar] [CrossRef] [Green Version]
- Bookong, P.; Ruchirawat, S.; Boonyarattanakalin, S. Optimization of microwave-assisted etherification of glycerol to polyglycerols by sodium carbonate as catalyst. Chem. Eng. J. 2015, 275, 253–261. [Google Scholar] [CrossRef]
- Pérez-Barrado, E.; Pujol, M.C.; Aguiló, M.; Llorca, J.; Cesteros, Y.; Díaz, F.; Pallarès, J.; Marsal, L.F.; Salagre, P. Influence of acid–base properties of calcined MgAl and CaAl layered double hydroxides on the catalytic glycerol etherification to short-chain polyglycerols. Chem. Eng. J. 2015, 264, 547–556. [Google Scholar] [CrossRef] [Green Version]
- Karam, A.; Sayoud, N.; Vigier, K.D.O.; Lai, J.; Liebens, A.; Oldani, C.; Jérôme, F. Heterogeneously-acid catalyzed oligomerization of glycerol over recyclable superacid Aquivion® PFSA. J. Mol. Catal. A Chem. 2016, 422, 84–88. [Google Scholar] [CrossRef]
- Galy, N.; Nguyen, R.; Blach, P.; Sambou, S.; Luart, D.; Len, C. Glycerol oligomerization in continuous flow reactor. J. Ind. Eng. Chem. 2017, 51, 312–318. [Google Scholar] [CrossRef]
- Barros, F.; Moreno-Tost, R.; Cecilia, J.; Ledesma-Muñoz, A.; de Oliveira, L.; Luna, F.; Vieira, R. Glycerol oligomers production by etherification using calcined eggshell as catalyst. Mol. Catal. 2017, 433, 282–290. [Google Scholar] [CrossRef]
- Zakaria, Z.; Mohamad, N.; Amin, N. Catalysts Screening for Catalytic Conversion of Glycerol to Olefins. J. Appl. Sci. 2010, 10, 1166–1170. [Google Scholar] [CrossRef]
- Li, X.; Shen, B.; Guo, Q.; Gao, J. Effects of large pore zeolite additions in the catalytic pyrolysis catalyst on the light olefins production. Catal. Today 2007, 125, 270–277. [Google Scholar] [CrossRef]
- Masih, M.; Algahtani, I.; De Mello, L. Price dynamics of crude oil and the regional ethylene markets. Energy Econ. 2010, 32, 1435–1444. [Google Scholar] [CrossRef]
- Zakaria, Z.Y.; Linnekoski, J.; Amin, N. Catalyst screening for conversion of glycerol to light olefins. Chem. Eng. J. 2012, 207, 803–813. [Google Scholar] [CrossRef]
- Blass, S.D.; Hermann, R.J.; Persson, N.E.; Bhan, A.; Schmidt, L.D. Conversion of glycerol to light olefins and gasoline precursors. Appl. Catal. A Gen. 2014, 475, 10–15. [Google Scholar] [CrossRef]
- Zakaria, Z.Y.; Amin, N.A.S.; Linnekoski, J. Thermodynamic analysis of glycerol conversion to olefins. Energy Procedia 2014, 61, 2489–2492. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Medical, Pharmaceutical and Personal Care Industries | Food Industry | Automotive and Chemical Industries | Other |
|---|---|---|---|
| Cough syrups, expectorant and elixirs | Humidifier/Humectant | Antifreeze | Denitrification |
| Mild laxative | Flavor | hydraulic and lubricating agents | Enzymatic reagents |
| Allergen treatment | Solvent | Tannins prevention | Cryoprotectant |
| Plasticizer | Emulsifier | Alcohol free alternatives | Animal feed |
| Lubrication | Antioxidant | Preserving agent | |
| Flavor | Lubricant | Explosives | |
| Additive | Sweetener | Synthesis of resins and many compound | |
| Toothpaste | Preservation | Polymers and fuels | |
| Mouth washers | Thickening agent | ||
| Skin and hair care | |||
| Moisturizer | |||
| Softener | |||
| Soap |
| S/N | Properties | Values Reported by Different Authors | ||
|---|---|---|---|---|
| [44] | [36] | [41] | ||
| 1 | Form and color | Liquid and colorless | na | na |
| 2 | Formula weight | 92.09 | 92.09 | 92.09 |
| 3 | Specific gravity/Density (20 °C) | 1.26050/4 | 1.261 g/cm3 | 1.261 g/cm3 |
| 4 | Melting point | 17.9 °C | 18.17 °C | 18.0 °C |
| 5 | Boiling point | 290 °C | 290 °C | 290 °C |
| 6 | Solubility in 100 parts | |||
| Water | Infinity | na | na | |
| Alcohol | Infinity | na | na | |
| 7 | Viscosity of liquid glycerol at 100% purity | 10 cP | 1499 cP at 20 °C | 1.410 Pa·s |
| 8 | Difusivity in (DL × 105 sqcm/s) | |||
| i-Amyl alcohol | 0.12 | na | na | |
| Ethanol | 0.56 | na | na | |
| Water | 0.94 | na | 1.33 × 10–11 m2/s | |
| 9 | Vapour pressure | na | 0.0025 mmHg at 50 °C | <0.0001 kPa at 20 °C |
| na | 0.195 mmHg at 100 °C | 0.03 kPa at 100 °C | ||
| na | 4.3 mmHg at 150 °C | 0.67 kPa at 152 °C | ||
| na | 46 mmHg at 200 °C | 6.67 kPa at 204 °C | ||
| 10 | Refractive index | na | 1.474 | 1.474 |
| 11 | Surface tension (20 °C) | na | 63.4 dyne/cm | 63.4 mN/m |
| 12 | Compressibility (28.5 °C) | na | 2.1 × 10 MPa | 2.1 × 10−4 MPa |
| 13 | Heat of vaporization | na | 21,060 cal/mole at 55 °C | 88.2 KJ/mol |
| na | 18,170 cal/mole at 195 °C | 76.1 KJ/mol | ||
| 14 | Heat of formation | na | 159.6 Kcal/gm mole | −669 KJ/mol |
| 15 | Heat of combustion | na | 1662 KJ/mole = 18.05 MJ/kg | −1662 KJ/mol |
| 16 | Heat fusion (18 °C) | 47.49 cal/g | 18.3 KJ/mole | 18.3 KJ/mol |
| 17 | Heat of solution | na | na | −5.8 KJ/mol |
| 18 | Thermal conductivity | na | 0.29 w/°K | 0.29 W/m/K |
| 19 | Flash point | na | 177 °C | 177 °C |
| 20 | Fire point | na | 204 °C | 204 °C |
| 21 | Relative dielectric constant (25 °C) | na | na | 42.48 |
| 22 | Autoignition temperature on glass | na | na | 429 °C |
| 23 | Calorific value | na | na | 18 KJ/g |
| 24 | Specific electrical conductivity (20 °C) | na | na | 0.1 µS/cm |
| S/N | Parameters | Crude Glycerol | Purified Glycerol | Refined/Commercial Glycerol |
|---|---|---|---|---|
| 1 | Glycerol content (%) | 60–80 | 99.1–99.8 | 99.2–99.98 |
| 2 | Moisture content (%) | 1.5–6.5 | 0.11–0.8 | 0.14–0.29 |
| 3 | Ash (%) | 1.5–2.5 | 0.054 | <0.002 |
| 4 | Soap (%) | 3–5 | 0.1–0.16 | na |
| 5 | Acidity (meq/100 g) | 0.7–1.3 | 0.10–0.16 | 0.04–0.07 |
| 6 | Chloride (ppm) | BDL | 1.0 | 0.6–9.5 |
| 7 | Color (APHA) | Dark | 34–45 | 1.8–10.3 |
| Feedstock | Element | Reference | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| C (wt%) | H (wt%) | N (wt%) | O (wt%) | Na (ppm) | K (ppm) | P (ppm) | Ca (ppm) | Mg (ppm) | Fe (ppm) | ||
| na | 52.77 | 11.08 | <0.0001 | 36.15 | na | na | na | na | na | na | [49] |
| SB oil (1) | 24.3 | na | 0.3 | na | 11,769 | 118.8 | 38.7 | BDL | BDL | 31.6 | [45] |
| SB oil (2) | 44.3 | na | 0.6 | na | 19,361 | 140.5 | 101.3 | 8.4 | 3.7 | 34.2 | |
| SB oil (3) | 46.8 | na | 0.7 | na | 19,108 | 1057 | 24.7 | 4 | BDL | 52.8 | |
| WV oil | 54.2 | na | 0.9 | na | 16,263 | 486.2 | 233.8 | 20.5 | 14.2 | 92.1 | |
| Feedstock | Glycerol Content (%) | Methanol Content (%) | Moisture Content (%) | Ash Content (%) | Soap Content (%) | pH | Appearance | Reference |
|---|---|---|---|---|---|---|---|---|
| Commercial pure glycerol | 99.5 | na | 0.5 | 0 | 0 | 6.7 | Colorless liquid | [46] |
| Crude palm oil | 88.56 | na | 3.41 | 7.12 | 0.91 | 6.68 | Dark brown liquid | |
| RBDP oil (1) | 85.2 | na | 8.11 | 5.26 | 1.43 | 7.73 | Dark brown semi solid | |
| RBDP oil (2) | 93.4 | na | 1.43 | 3.43 | 1.74 | 12.13 | Dark brown semi solid | |
| Mixture of vegetable oils | 71.7 | 25.7 | na | na | na | na | na | [50] |
| SB oil (1) | 63 | 6.2 | 28.7 | 2.7 | BDL | 6.9 | na | [45] |
| SB oil (2) | 22.9 | 10.9 | 18.2 | 3.0 | 26.2 | 9.7 | na | |
| SB oil (3) | 33.3 | 12.6 | 6.5 | 2.8 | 26.1 | 9.5 | na | |
| WV oil | 27.8 | 8.6 | 4.1 | 2.7 | 20.5 | 9.4 | na | |
| Waste cooking oil | 83.4 | BDL | 11.6 | 2.7 | 1.3 | na | na | [51] |
| Catalyst (A) | Operation Parameters | Performance | TOF | Reference |
|---|---|---|---|---|
| 7.5 wt% Ni/Fly ash (10.4) | T = 550 °C, WGMR = 12:1, Cat = 1 g, ST = 8.4 kg cat h/kmol G | GC = 98.6%, Y = 5.8 mol/mol G, S = na | nd | [73] |
| 10 wt% Ni/Al2O3 (123.4) | T = 650 °C, t = 5 h, Cat = 0.3 g, WGMR = 6:1 | GC = 80%, Y = na, S = 75.1% | nd | [72] |
| 5% Pt/Al2O3 (na) | T = 240 °C, t = 4 h, Cat = 1 g | GC = 84%, Y = 14.1%, S = na | 2.1 | [76] |
| Ni-Co/Al-Mg (na) | T = 680 °C, t = 2 h, Cat = 3 g | GC = 95%, Y = 67 vol%, S = 91% | 58 | [77] |
| 1.44 wt% Pt/MgO (221) | T = 225 °C, P = 2.3 MPa. t = 3 h, Cat = 100 mg | GC = na, Y = 71.9%, S = na | * 24 | [75] |
| 1 wt% Pt/Al2O3 (na) | T = 230 °C, P = na. t = na, Cat = 2 g, LHSV = 0.73 h−1 | GC = na, Y = 67 mol%, S = na | nd | [74] |
| Biocatalyst | Operating Parameters | Performance | Reference |
|---|---|---|---|
| Ogataeo polymorpha | T = 37 °C, pH = na, rpm = 140, t = na | C = 0.80 ± 0.100 g/L, Y = 0.077 ± 0.036 g/g, P = 0.007 ± 0.001 g/L/h | [102] |
| Ogataeo polymorpha/PDC1 | C = 2.91 ± 0.240, Y = 0.270 ± 0.159 g/g, P = 0.022 ± 0.002 g/L/h | ||
| Ogataeo polymorpha/PDC1/ADH1 | C = 4.29 ± 0.040, Y = 0.357 ± 0.073 g/g, P = 0.036 ± 0.000 g/L/h | ||
| Enterobacter aerogenes ATCC 29007 | T = 34 °C, pH = 7.5, t = 78 h | C = 13.09 g/L, Y = 0.156 g/g, P = 0.161 g/L/h | [101] |
| Enterobacter aerogenes SUMI1014 | C = 34.54 g/L, Y = 0.428 g/g, P = 0.439 g/L/h | ||
| Enterobacter aerogenes SUMI2008 | C = 38.32 g/L, Y = 0.479 g/g, P = 0.491 g/L/h | ||
| Enterobacter aerogenes ATCC 35029 | T = 34 °C, pH = 6.4, rpm = 500, t = 20–24 h | C = na, Y = 0.75 mol/mol, P = na | [100] |
| Escherichia coli SS1 | T = 37 °C, pH = na, rpm = 120, t = 24 h | C = 9.23 ± 0.6 g/L, Y = 1.00 mol/mol, P = na | [96] |
| Escherichia coli BL 21 | C = 9.50 ± 0.67 g/L, Y = 0.991 mol/mol, P = na | ||
| Escherichia coli BW25113 | C = 5.33 ± 1.00 g/L, Y = 0.592 mol/mol, P = na | ||
| Enterobacter aerogenes HU101 | C = 3.98 ± 0.27 g/L, Y = 0.594 mol/mol, P = na | ||
| Mixed culture | T = 37.5 °C, pH = 7.9, t = na | C = 6.75 g/L, Y = na, P = na | [103] |
| Optimization | C = 26 g/L, Y = na, P = na | ||
| Escherichia coli MG1655 | T = 37 °C, pH = na, t = 110 h | C = 4.00 g/L, Y = 0.40 g/g, P = na | [88] |
| Catalyst (A) | Operation Parameters | Performance (%) | TOF | Reference |
|---|---|---|---|---|
| Diatomite loaded 0.3%SO42−/TiO2 (na) | T = 210 °C, P = 0.002 MPa, Cat = 0.1% wt Oleic acid, t = 6 h, 200 rpm, Crude Glycerol/Oleic acid (1:2) | GC = na, MA = na, DA = 59.1 ± 0.6, TA = na | * 2955 | [125] |
| 0.3%SO42−/TiO2 (na) | GC = na, MA = na, DA = 41.4 ± 1.0, TA = na | * 2070 | ||
| 0.3%SO42−/ZrO2-Al2O3 (na) | GC = na, MA = na, DA = 39.9 ± 0.7, TA = na | * 1995 | ||
| Sulphonated glycerol-based carbon catalyst (na) | T = 110 °C, Cat = 2 wt%CG, t = 3 h, Crude Glycerol/Acetic Acid (1:3) | GC = 99, MA = 12, DA+TA = 88 | 165 | [127] |
| Pr-SO3HSBA-15 (366) | T = 120 °C, P = atm. press., Cat = 4 wt% G, t = 2.5 h, Glycerol/Acetic Acid (1:6) | GC = 96, MA = 13, DA+TA = 87 | ** 16.25 S−1 | [122] |
| CaSn(OH)6 (3.95) | T = 30 °C, Cat = 7 wt% G, t = 2 h, Glycerol/Methyl Acetate (1:10) | GC = 78.2, MA = 67.3, DA = 32.6, TA = na | nd | [126] |
| MgSn(OH)6 (18.1) | GC = 65.4, MA = 66.1, DA = 33.8, TA = na | |||
| ZnSn(OH)6 (7.0) | GC = 40, MA = 62.5, DA = 37.5, TA = na | |||
| SrSn(OH)6 (5.0) | GC = 25.2, MA = 70.1, DA = 30.2, TA = na | |||
| Ca(OH)2 (12.0) | GC = 52.1, MA = 87.1, DA = 12.9, TA = na | |||
| CaO (62.0) | GC = 47.2, MA = 90.2, DA = 9.6, TA = na | |||
| MgO (44.4) | GC = 25.2, MA = 99.8, DA = na, TA = na | |||
| Fe-Sn-Ti (SO42−) (16.96) | T = 80 °C, Cat = 0.05 g, t = 0.5 h, Glycerol/Acetic Anhydride (1:6) | GC = 58, MA = 83, DA = 17, TA = 0 | 348 | [119] |
| Fe-Sn-Ti (SO42−)-400 (18.88) | GC = 100, MA = 0, DA = 1, TA = 99 | 600 | ||
| Fe-Sn-Ti (SO42−)-500 (16.55) | GC = 99, MA = 0, DA = 3, TA = 97 | 594 | ||
| Fe-Sn-Ti (SO42−)-600 (14.91) | GC = 99, MA = 0, DA = 4, TA = 96 | 594 | ||
| Fe-Sn-Ti (SO42−)-700 (11.65) | GC = 68, MA = 82, DA = 17, TA = 1 | 408 | ||
| Amberlyst-15 (na) | GC = 99, MA = 0, DA = 1, TA = 99 | 594 | ||
| Blank | GC = 49, MA = 48, DA = 46, TA = 6 | - | ||
| 3 wt% Al-SBA-15 (747) | T = 120 °C, Cat = 50 mg/mmol, t = 8 h, 1000 rpm, Glycerol/Levulnic Acid (1:4) | GC = 78, MA = 94, DA = 5, TA = 1 | 2 | [121] |
| 0.63 wt% Fe/Al-SBA-15 (688) | GC = >99, MA = 0, DA = 71, TA = 28 | 2 | ||
| 0.63 wt% Fe/Al-SBA-15 (688) | T = 140 °C, Cat = 50 mg/mmol, t = 8 h, 1000 rpm, Glycerol/Levulnic Acid (1:4) | GC = >99, MA = 0, DA = 80, TA = 20 | 2 | |
| 0.63 wt% Fe/Al-SBA-15 | T = 100 °C, Cat = 50 mg/mmol, t = 8 h, 1000 rpm, Glycerol/Levulnic Acid (1:4) | GC = -, MA = -, DA = -, TA = - | nd | |
| 1% Y/SBA-3 (na) | T = 110 °C, Cat = 0.2 g, t = 3 h, Glycerol/Acetic Acid (1:4) | GC = 65, MA = 95, DA = 5, TA = 0 | 54 | [117] |
| 2% Y/SBA-3 (na) | GC = 68, MA = 82, DA = 11, TA = 7 | 56 | ||
| 2.5% Y/SBA-3 (na) | GC = 82, MA = 54, DA = 26, TA = 20 | 68 | ||
| 3% Y/SBA-3 (1568) | GC = 100, MA = 11, DA = 34, TA = 55 | 83 | ||
| 3.5% Y/SBA-3 (1622) | GC = 94, MA = 24, DA = 34, TA = 55 | 78 | ||
| Blank | GC = 20, MA = 71, DA = 28, TA = 1 | - | ||
| Glycerol Based Carbon Catalyst (na) | T = 115 °C, Cat = 0.46 g, t = 1 h, Glycerol/Acetic Acid (1:4) | GC = 100, MA = 22, DA = 67, TA = 11 | 217 | [118] |
| T = 115 °C, Cat = 0.46 g, t = 1 h, Glycerol/Acetic Acid (1:6) | GC = 100, MA = 15.8, DA = 72.2, TA = 12 | 217 | ||
| T = 115 °C, Cat = 0.46 g, t = 1 h, Glycerol/Acetic Acid (1:8) | GC = 100, MA = 12.3, DA = 73.2, TA = 14.5 | 217 | ||
| T = 115 °C, Cat = 0.46 g, t = 1 h, Glycerol/Acetic Acid (1:10) | GC = 100, MA = 8.4, DA = 71.8, TA = 19.8 | 217 | ||
| Amberlyst-15 (1. 6wt) (37.6) | T = 110 °C, Cat = 82.86 mmolH+/L, t = 4.5 h, Glycerol/Acetic Acid (1:9) | GC = 97, MA = -, DA = 47.7, TA = 44.5 | nd | [116] |
| Amberlyst-15 (3.2 wt) (37.6) | GC = 93.5, MA = -, DA = 43.2, TA = 38.3 | |||
| HZSM-5 (368) | GC = 85.6, MA = -, DA = 25.7, TA = 7.7 | |||
| HUSY (552) | GC = 78.4, MA = -, DA = 20.6, TA = 5.6 | |||
| Blank | GC = 73.2, MA = -, DA = 13.8, TA = 1.5 | |||
| ZrO2 (59.7) | T = 120 °C, Cat = 0.3 g, t = 4 h, 250 rpm, Glycerol/Acetic Acid (1:10) | GC = 72.4, MA = 48.3, DA = 45.4, TA = 6.3 | nd | [120] |
| 20 wt% HSiW/ZrO2 (48.7) | GC = 100, MA = 6.4, DA = 61.3, TA = 32.3 | |||
| 20 wt% HPW/ZrO2 (52.5) | GC = 99.2, MA = 7.3, DA = 62.8, TA = 29.9 | |||
| 20 wt% HPMo/ZrO2 (49.3) | GC = 98.2, MA = 12.0, DA = 62.0, A = 26.0 | |||
| 0.01 mmol H3PW12O40 | T = 60 °C, P = 0.101 MPa, Cat = 0.03 mmol, t = 8 h, Glycerol/Acetic Acid (1:3) | GC = 96, MA = 66, DA = 34, TA = 0 | ** 5.3 min−1 | [128] |
| 0.01 mmol H3PW12O40-calcined 200 °C | GC = 93, MA = 65, DA = 35, TA = 0 | ** 4.28 min−1 | ||
| 0.03 mmol PTSA | GC = 85, MA = 86, DA = 8, TA = 0 | ** 3.1 min−1 | ||
| 0.015 mmol H2SO4 | GC = 98, MA = 54, DA = 27, TA = Trace | ** 4.1 min−1 | ||
| CeO2-Al2O3 (101) | T = 120 °C, Cat = 5 wt% G, t = 1 h, Glycerol/Acetic Acid (1:6) | GC = 59.4, MA = 88.1, DA = 11.6, TA = 0.3 | 119 | [124] |
| CeO2-ZrO2 (49) | GC = 68.1, MA = 75.2, DA = 22.7, TA = 2.1 | 136 | ||
| SO42−/CeO2-Al2O3 (136) | GC = 79.9, MA = 58.9, DA = 35.5, TA = 5.6 | 160 | ||
| SO42−/CeO2-ZrO2 (92) | GC = ≈100, MA = 25.8, DA = 57.7, TA = 16.5 | 200 |
| Catalyst (A) | Operation Parameters | Performance (%) | TOF | Reference |
|---|---|---|---|---|
| USY-650-L-2 (741) | T = 200 °C, Cat = 3.5 wt% G, t = 6 h, Glycerol/Ethyl alcohol (1:9) | GC = 67, ME = 73, DE = 25, TE = 2 | nd | [134] |
| H-Beta (623) | GC = 81, ME = 72, DE = 25, TE = 2 | |||
| HZSM-5 (337) | GC = 60, ME = 90, DE = 10, TE = 0 | |||
| USY-650-L-2 (741) | T = 90 °C, Cat = 7.6 wt% G, t = 4 h, Glycerol/tert-butyl alcohol (1:4) | GC = 74, ME = 79, DE = 21, TE = 0 | ||
| H-Beta (623) | GC = 75, ME = 74, DE = 26, TE = 0 | |||
| HZSM-5 (337) | GC = 15, ME = na, DE = na, TE = na | |||
| Blank | T = 180 °C, Cat = none, t = 8 h, Glycerol/Ethanol (1:6) | GC = 0, ME = 0, DE = 0, TE = 0 | - | [135] |
| K-10 (260) | T = 180 °C, Cat = 2.1 g, t = 8 h, Glycerol/Ethanol (1:3) | GC = 70, ME = 79, DE = 14, TE = 7 | 5 | |
| K-10 (260) | T = 180 °C, Cat = 2.1 g, t = 4 h, Glycerol/Ethanol (1:6) | GC = 59, ME = 72, DE = 18, TE = 10 | 8 | |
| HZSM-5 (408) | T = 180 °C, Cat = 1.9 g, t = 8 h, Glycerol/Ethanol (1:3) | GC = 61, ME = 94, DE = 4, TE = 2 | 4 | |
| HZSM-5 (408) | T = 180 °C, Cat = 1.9 g, t = 4 h, Glycerol/Ethanol (1:6) | GC = 40, ME = 95, DE = 3, TE = 2 | 5 | |
| H-Beta (564) | T = 180 °C, Cat = 1.1 g, t = 8 h, Glycerol/Ethanol (1:3) | GC = 92, ME = 71, DE = 17, TE = 12 | 6 | |
| H-Beta (564) | T = 180 °C, Cat = 1.1 g, t = 4 h, Glycerol/Ethanol (1:6) | GC = 89, ME = 75, DE = 14, TE = 10 | 12 | |
| Amberlyst-15 (53) | T = 180 °C, Cat = 0.32 g, t = 8 h, Glycerol/Ethanol (1:3) | GC = 96, ME = 65, DE = 19, TE = 16 | 6 | |
| Amberlyst-15 (53) | T = 180 °C, Cat = 0.32 g, t = 4 h, Glycerol/Ethanol (1:6) | GC = 95, ME = 79, DE = 12, TE = 9 | 12 | |
| S90TS10O (538) | T = 75 °C, Cat = 5 wt% G, t = 17 h, Glycerol/tert-butyl alcohol (1:4) | GC = 27, ME = 92, DE + TE = 2.0 | nd | [136] |
| S50TS50O (18) | GC = 74, ME = 76, DE + TE = 18.0 | |||
| BS90TS10O (601) | GC = 6, ME = 97, DE + TE = 0.2 | |||
| BS50TS50O (48) | GC = 32, ME = 87, DE + TE = 4.0 | |||
| Amberlyst-15 (39) | GC = 51, ME = 65, DE + TE = 18.0 | |||
| S50TS50O (18) | T = 75 °C, Cat = 5 wt% G, t = 24 h, Glycerol/tert-butyl alcohol (1:4) | GC = 98, ME = 71, DE + TE = 28.0 | ||
| Amberlyst-15 (530 | T = 90 °C, Cat = 0.38 g, t = 4 h, Glycerol/tert-butyl alcohol (1:4) | GC = 97.8, ME = 94.2, DE = 5.8, TE = na | 35 | [133] |
| Amberlyst-35 (50) | GC = 97.7, ME = 94.8, DE = 5.2, TE = na | 17 | ||
| Zeolite-BEA (450) | GC = 96.7, ME = 81.8, DE = 18.2, TE = na | 34 | ||
| Mordenite (400) | GC = 66.1, ME = 99.4, DE = 10.6, TE = na | 24 | ||
| Ultra-Stable Y (USY) (614) | GC = 100, ME = 100, DE = -, TE = na | 36 | ||
| N-BEA (565) | GC = 98.4, ME = 75.6, DE = 21.8, TE = 2.6 | 35 | ||
| Sulfonated sugar cane bagasse (<10) | T = 120 °C, Cat = 5 wt% G, t = 4 h, Glycerol/tert-butyl alcohol (1:4) | GC = 81.8, ME = 60.5, DE + TE = 21.8 | nd | [130] |
| Sulfonated coconut husk (<10) | GC = 61.5, ME = <40, DE + TE = 21.1 | |||
| Sulfonated coffee grounds (<10) | GC = 61.5, ME = <40, DE + TE = 14 | |||
| LiOH (na) | T = 240 °C, P = atm. press., Cat = 2 wt% G, t = 8 h, Glycerol, no solvent used | GC = 100, ME = na, DE = <40, TE = na | nd | [131] |
| NaOH (na) | GC = 100, ME = na, DE = <40, TE = na | |||
| KOH (na) | GC = 100, ME = na, DE = <20, TE = na | |||
| Na2CO3 (na) | GC = 100, ME = na, DE = <20, TE = na | |||
| Blank | T = 160 °C, P = 3.0 MPa N2, Cat = 1 g, t = 20 h, Glycerol/Ethanol (1:6) | GC = 0, ME = na, DE = -, TE = na | - | [132] |
| HZSM5-20 (na) | GC = 7.3, ME = 80, DE = 7.9, TE = 12.1 | 2 | ||
| HZSM5-100 (na) | GC = 1.1, ME = 100, DE = 0, TE = 0 | 3 | ||
| H-β (na) | GC = 5.7, ME = 100, DE = 0, TE = 0 | 2 | ||
| H3PO40W12 (na) | GC = 68.9, ME = 79, DE = 14.8, TE = 6.2 | 19 | ||
| H2SO4 (na) | GC = 70, ME = 78.6, DE = 14.9, TE = 6.5 | 19 | ||
| FeCl3 (na) | GC = 19.2, E = 72.4, DE = 16.3, E = 11.3 | 5 | ||
| AlCl3 (na) | GC = 10.2, ME = 79.6, DE = 9.4, TE = 11 | 3 | ||
| H3PO40W12 (na) | T = 160 °C, Cat = 3.3 g, t = 20 h, Glycerol/Ethanol (1:6) | GC = 97.1, ME = 61.9, DE = 28.1, =10.0 | 26 |
| Catalyst (A) | Operation Parameters | Performance (%) | TOF | Reference |
|---|---|---|---|---|
| β-Zeolite (467.4) | T = 60 °C, Cat = 10 wt% G, t = 4 h, Bioglycerol/Acetone (1:6) | GC = 70.13, SK = 64.65, AT = 5.48 | nd | [147] |
| HCl-treated β-Zeolite (597.15) | GC = 86.32, SK = 83.71, AT = 2.61 | |||
| HNO3-treated β-Zeolite (607.22) | GC = 94.26, SK = 94.21, AT = 0.05 | |||
| Oxalic acid-treated β-Zeolite (527.09) | GC = 92.45, SK = 89.60, AT = 2.85 | |||
| Blank | T = 55 °C, Cat = 50 mg, t = 1.5 h, Glycerol/butanone (1:10), under N2 flow | GC = 0.3, SK = 100, AT = 0, OT = 0 | - | [152] |
| SiO2 (na) | GC = 0.6, SK = 100, AT = na, OT = 0 | ** 0 | ||
| Mo (na) | GC = 0.3, SK = 100, AT = na, OT = 0 | ** 0 | ||
| 1% Re/SiO2 (na) | GC = 92.6, SK = 94.6, AT = na, T = 5.4 | ** 124 | ||
| 1% Ru/Mo (na) | GC = 67.1, SK = 93.4, AT = na, OT = 6.6 | ** 67.4 | ||
| Titanate nanotubes (HTNT-72) (182) | T = 50 °C, Cat = 130 mg, t = 6 h, 150 rpm, Glycerol/Acetone (1:1) under N2 flow | GC = 45.5, SK = 83, AT = 15, OT = 2 | nd | [150] |
| T = 50 °C, Cat = 130 mg, t = 6 h, 150 rpm, Glycerol/Acetone (1:4) under N2 flow | GC = 65.5, SK = 35, AT = 0, OT = 65 | |||
| T = 50 °C, Cat = 130 mg, t = 6 h, 150 rpm, Glycerol/Acetone (1:8) under N2 flow | GC = 85.4, SK = 10, AT = 0, OT = 90 | |||
| 1:1 Nb-Al (182) | T = Room temp., Cat = 2.7 wt% G, t = 6 h, Glycerol/Acetone (1:4), 600 rpm | GC = 36, SK = 80, AT = 20 | 24 | [151] |
| 1:0.05 Nb-Al (<10) | GC = 73, SK = 93, AT = 7 | 48 | ||
| 1:1 Nb-Al (182) | T = 50 °C, Cat = 2.7 wt% G, t = 6 h, Glycerol/Acetone (1:4), 600 rpm | GC = 62, SK = 94, AT = 6 | 41 | |
| 1:0.05 Nb-Al (<10) | GC = 84, SK = 98, AT = 2 | 56 | ||
| 1:1 Nb-Al (182) | T = 80 °C, Cat = 2.7 wt% G, t = 6 h, Glycerol/Acetone (1:4), 600 rpm | GC = 78, SK = 97, AT = 3 | 52 | |
| 1:0.05 Nb-Al (<10) | GC = 77, SK = 97, AT = 3 | 51 | ||
| 10 wt% MoPO/SBA-15 (573) | T = room temp., P = atm. press., Cat = 50 mg, t = 2 h, Glycerol/Acetone (1:3) | GC = 75, SK = 98, AT = na, OT = na | 75 | [146] |
| 40 wt% MoPO/SBA-15 (164) | GC = 100, SK = 98, AT = na, OT = na | 100 | ||
| 100 wt% MoPO/SBA-15 (na) | GC = 64, SK = 97, AT = na, OT = na | 64 | ||
| Amberlyst-15 (na) | T = room temp. (28 °C), Cat = 5 wt% G, t = 1 h, Glycerol/Acetone (1:2) | GC = 73, SK = 91, AT = - | 197 | [148] |
| K-10 Clay (na) | GC = 27.3, SK = 89, AT = - | 74 | ||
| CsHPW (na) | GC = 67.3, SK = 99, AT = - | 182 | ||
| MoO3/SiO2 (na) | GC = 46.8, SK = 90, AT = - | 126 | ||
| HZSM-5 Zeolite (na) | GC = 19.5, SK = 88, AT = - | 53 | ||
| H-mordenite Zeolite (na) | GC = 8, SK = 85.5, AT = - | 22 | ||
| H-Y Zeolite (na) | GC = 74.2, SK = 98.2, AT = - | 200 | ||
| H-Beta-1 Zeolite (na) | GC = 86, SK = 98.5, AT = - | 232 | ||
| Amberlyst-35 Dry (35) | T = 40 °C, P = 4.1 MPa, WHSV = 4/h, Glycerol/Acetone (1:2) | GC = 71 ± 2, SK = 70 ± 1, AT = na, OT = na | 3547 | [143] |
| Amberlyst-35 Wet (35) | GC = 71 ± 3, SK = 71 ± 3, AT = na, OT = na | 3547 | ||
| Zeolite (480) | GC = 73 ± 3, SK = 72 ± 2, AT = na, OT = na | 3646 | ||
| Montmorillonite (264) | GC = 60 ± 4, SK = 60 ± 1, AT = na, OT = na | 2997 | ||
| Polymax (na) | GC = 51 ± 3, SK = 50 ± 1, AT = na, OT = na | 2548 | ||
| Zirconium Sulphate (na) | GC = 66 ± 1, SK = 65 ± 3, AT = na, OT = na | 3297 | ||
| Amberlyst-35 Dry (35) | T = 40 °C, P = 4.1 MPa, WHSV = 4/h, Glycerol/Acetone (1:6) | GC = 88 ± 3, SK = 86 ± 3, AT = na, OT = na | 4396 | |
| Amberlyst-35 Wet (35) | GC = 89 ± 3, SK = 88 ± 4, AT = na, OT = na | 4446 | ||
| Zeolite (480) | GC = 85 ± 2, SK = 84 ± 2, AT = na, OT = na | 4246 | ||
| Montmorillonite (264) | GC = 69 ± 1, SK = 68 ± 1, AT = na, OT = na | 3447 | ||
| Polymax (na) | GC = 61 ± 2, SK = 60 ± 2, AT = na, OT = na | 3047 | ||
| Zirconium Sulphate (na) | GC = 79 ± 2, SK = 77 ± 2, AT = na, OT = na | 3946 | ||
| ZrO2 (42) | T = 100 °C, Cat = 5 wt%, t = 30 min, Glycerol/Benzaldehyde (1:1) | GC = 58, SK = 47, AT = 53 | 252 | [154] |
| TiO2-ZrO2 (30) | GC = 64, SK = 47, AT = 53 | 278 | ||
| 10 wt% MoOx/ZrO2 (94) | GC = 70, SK = 47, AT = 53 | 304 | ||
| 10 wt% MoOx/TiO2-ZrO2 (7) | GC = 74, SK = 49, AT = 51 | 321 | ||
| Blank | T = 45 °C, Cat = Nil, t = 3 h, Glycerol/Acetone (1:8), 530 rpm, under N2 flow | GC = 28, SK = 75, AT = 6, OT = 25 | - | [149] |
| Activated Carbon (AC) (780) | T = 45 °C, Cat = 0.2g, t = 3 h, Glycerol/Acetone (1:8), 530 rpm, under N2 flow | GC = 33, SK = 81, AT = 0, OT = 19 | 30 | |
| 1% Ni/AC (na) | GC = 65, SK = 91, AT = 3, OT = 6 | 59 | ||
| 3% Ni/AC (na) | GC = 83, SK = 62, AT = 26, OT = 12 | 75 | ||
| 5% Ni/AC (582) | GC = 98, SK = 86, AT = 10, OT = 4 | 89 | ||
| 1% Zr/AC (799) | GC = 54, SK = 77, AT = 23, OT = 0 | 49 | ||
| 3% Zr/AC (na) | GC = 57, SK = 68, AT = 32, OT = 0 | 51 | ||
| 5% Zr/AC (na) | GC = 67, SK = 63, AT = 37, OT = 0 | 61 | ||
| 5% Ni-1% Zr/AC (612) | GC = 100, SK = 74, AT = 26, OT = 0 | 90 | ||
| 1% Ni-5% Zr/AC (74.9) | GC = 74, SK = 61, AT = 39, OT = 0 | 67 |
| Catalyst (A) | Operation Parameters | Performance (%) | TOF | Reference |
|---|---|---|---|---|
| H3PW12O40/MCM-41 (615) | T = 320 °C, P = atm. press., t = 1–5 h, Cat = 0.5 g, 10 wt% glycerol solution, FFR = 3 mL/h, under 10 mL/min N2 flow | GC = >95, S = 43.8 | ** 103.7 | [162] |
| H6P2W18O62/MCM-41 (601) | GC = >93, S = 51.3 | ** 184.2 | ||
| ZSM-5 (na) | T = 300 °C, P = atm. press., t = 1 h, Cat = 100 mg, 10 wt% glycerol solution, FFR = 0.05 mL/min, under 30 mL/min N2 flow | GC = 90, S = ≈33 | 900 | [165] |
| ZSM-5 (NaOH-treated) (na) | GC = 89, S = ≈24 | 890 | ||
| ZSM-5 (H2C2O4-treated) (na) | GC = 100, S = ≈29 | 1000 | ||
| ZSM-5 (HCl-treated) (na) | GC = 100, S = ≈35 | 1000 | ||
| 10 wt% V2O5/ZSM-5 (na) | GC = 100, S = ≈26 | 1000 | ||
| 10 wt% V2O5/ZSM-5 (NaOH-treated) (na) | GC = 89, S = ≈35 | 890 | ||
| 10 wt% V2O5/ZSM-5 (H2C2O4-treated) (na) | GC = 93, S = ≈33 | 930 | ||
| 10 wt% V2O5/ZSM-5 (HCl-treated) (na) | GC = 100, S = ≈30 | 1000 | ||
| 17NiSO4-350 | T = 340 °C, P = atm. press., Cat = 1 g, GHSV = 873/h, 20 wt% glycerol solution, FFR = 0.13 mL/min, t = >10 h | GC = >90, S = >70 | nd | [159] |
| 29NiSO4-350 | GC = >85, S = >70 | |||
| 17NiSO4-550 | GC = <85, S = <60 | |||
| 29NiSO4-550 | GC = >89.2, S = >62.1 | |||
| 20% w/w DTP/K-10 (na) | T = 225 °C, P = atm. press., Cat = 1 g, WHSV = 10.74/h, 20 wt% glycerol solution, FFR = 10.2 mL/h, t = 4 h, under 1.5 L/min N2 flow | GC = 89, S = 50 | 46 | [160] |
| 20% w/w DTP/HMS (299.6) | GC = 94, S = 80 | 47 | ||
| 20% w/w DTP/OMS (na) | GC = 62, S = 45 | 31 | ||
| Sulfuric acid Treated-Zr/HMS (na) | T = 275 °C, P = atm. press., Cat = 1 g, WHSV = 10.74/h, 20 wt% glycerol solution, FFR = 10.2 mL/h, under 1.5 L/min N2 flow, t = 4 h | GC = 60, S = 28 | 3 | |
| Persulfated Zr-Al/HMS (na) | GC = 68, S = 38 | 34 | ||
| Chlorosulfonic acid-treated-Zr/HMS | GC = 71, S = 29 | 36 | ||
| W-Zr-Al/HMS (143.9) | GC = 86, S = 60 | 43 | ||
| 10 wt% CuO/CeO2(na) | T = 320 °C, P = atm. press., Cat = 0.5 g, 30 mL/min H2 flow, t = 5 h, 30 wt% glycerol solution | GC = 12.21, S = 14.42 | 15 | [163] |
| 10 wt% CuO/CeO2-ZrO2 (na) | GC = 69.96, S = 56.92 | 35 | ||
| 10 wt% CuO/CeO2-SiO2 (na) | GC = 44.68, S = 43.14 | 22 | ||
| SO3H-Activated Montmorillonite (46.9) | T = 320 °C, P = atm. press., Cat = 0.5 g, 10 wt% glycerol solution, FFR = 0.3 mL/min, under 10 mL/min N2 flow, LHSV = 18.5/h. | GC = 54.2, S = 83 | nd | [166] |
| Catalyst (A) | Operation Parameters | Performance (%) | TOF | Reference |
|---|---|---|---|---|
| NiO-400 (56.07) | T = 80 °C, P = na, t = 6 h, Cat = 5 wt% G, Glycerol/DEC (≈1:3) | GC = 17.3, S = 100 | 6 | [172] |
| 0.2CeNiO-400 (30.49) | GC = 76.95, S = 100 | 28 | ||
| 0.6CeNiO-400 (41.10) | GC = ≈75, S = 100 | 27 | ||
| 1.0CeNiO-400 (71.80) | GC = 66.51, S = 100 | 24 | ||
| Methylammonium nitrate | T = 120 °C, P = na, t = 2 h, Cat = 0.5 mol%, Glycerol/DEC (≈1:2) | GC = <10, Y = <10 | nd | [176] |
| Ethylammonium nitrate | GC = <10.Y = <10 | |||
| 2-hydrozyethylammonium formate | GC = 24.08, Y = 24.00 | |||
| 1-ethyl-3-methylimidazolium dimethyl phosphate | GC = 22.20, Y = 22 | |||
| 1-butyl-3-methylimidazolium dicyanamide | GC = 45, Y = 45 | |||
| 1-butyl-3-methylimidazolium chloride | GC = <10, Y = <10 | |||
| 1-butyl-3-methylimidazolium tetrafluoroborate | GC = <5, Y = <5 | |||
| 1-ethyl-3-methylimidazolium acetate | GC = 93.5, Y = 88.7 | |||
| Gypsum (CaSO4.2H2O) (10.8) | T = 150 °C, P = na, t = 4 h, Cat = 0.25 g, 340 rpm, Glycerol/Urea (1:1.5) | GC = 92.5, S = 64.8, Y = 59.9 | ** 89.7 | [171] |
| Gypsum-150 (2.6) | GC = 92.8, S = 90.1, Y = 83.6 | ** 110.5 | ||
| Gypsum-800 (1.4) | GC = 89.1, S = 82.8, Y = 73.8 | ** 125.4 | ||
| Gypsum-30% H2O2 | GC = 91.0, S = 43.8, Y = 39.8 | ** 59.6 | ||
| Blank | GC = 78.7, S = 32.8, Y = 25.8 | |||
| Waste boiler ash-110 (8.05) | T = 150 °C, P = na, t = 4 h, Cat = 0.25 g, 340 rpm, Glycerol/Urea (1:1.5) | GC = 91.1, S = 83.5, Y = 76.2 | ** 136.7 | [174] |
| Waste boiler ash-700 (na) | GC = 94.1, S = 88.6, Y = 83.4 | ** 141.2 | ||
| Waste boiler ash-900 (2.00) | GC = 93.6, S = 90.1, Y = 84.3 | ** 140.4 | ||
| Waste boiler ash-1100 (na) | GC = 89.8, S = 85.6, Y = 77.0 | ** 134.7 | ||
| Blank | GC = 78.7, S = 32.8, Y = 25.8 | - | ||
| La2O3 (21) | T = 150 °C, P = 7 MPa, t = 12 h, Cat = 0.23 g, Glycerol/CH3CN/CO2 | GC = 0.8, S = 6.3, Y = 0.05 | 0.1 | [170] |
| 0.7% Cu/La2O3 (21) | GC = 19.3, S = 47.7, Y = 9.2 | 4 | ||
| 2.3% Cu/La2O3 (20) | GC = 33.4, S = 45.4, Y = 15.2 | 6 | ||
| 4.6% Cu/La2O3 (20) | GC = 30.6, S = 44.2, Y = 13.5 | 6 | ||
| 13.4% Cu/La2O3 (na) | GC = 1.7, S = 35.3, Y = 0.6 | 0.3 | ||
| Blank | GC = 0.4, S = 5.0, Y = 0.02 | - | ||
| NaY-Zeolite (760) | T = 180 °C, P = 10 MPa, t = 3 h, Cat = 0.5 g, Glycerol/CO2 | GC = na, Y = 0.0, S = na | 0 | [173] |
| 5.9 wt% AgY-Zeolite (289) | GC = na, Y = 5.6, S = na | 10 | ||
| 6.0 wt% ZnY-Zeolite (407) | GC = na, Y = 5.8, S = na | 11 | ||
| 10.3 wt% SnY-Zeolite (442) | GC = na, Y = 5.1, S = na | 9 | ||
| AgNO3(na) | GC = na, Y = 3.5, S = na | 6 | ||
| Zn (NO3)2 (na) | GC = na, Y = 2.7, S = na | 5 | ||
| SnCl2 (na) | GC = na, Y = 2.6, S = na | 5 | ||
| CuCl2 (na)) | T = 130 °C, P = 4 MPa, t = 4 h, Cat = 0.6 mmol, Glycerol/DMA/CO:O2 (5:1), Pyridine = 0.3 mmol | GC = 93, S = 91 | nd | [169] |
| CuBr2 (na)) | GC = 51, S = 89 | |||
| CuI2 (na)) | GC = 69, S = 94 | |||
| CuSO4 (na)) | GC = 75, S = 95 | |||
| Cu (OAc)2 (na)) | GC = 10, S = 40 | |||
| Cu (OTf)2 (na)) | GC = 72, S = 60 | |||
| Cu (ClO4)2 (na)) | GC = 41, S = 40 |
| Catalyst (A) | Operation Parameters | Performance (%) | TOF | Reference |
|---|---|---|---|---|
| H-beta (484) | T = 200 °C, P = 4.1 MPa H2, t = 10 h, Cat = 0.5 g, 600 rpm, 50 wt% glycerol solution | GC = 85, S = 0.2, Y = 0.2 | 85 | [190] |
| 5 wt% Zr/H-beta (445) | GC = 70, S = 8, Y = 5.6 | 7 | ||
| 10 wt% Zr/H-beta (434) | GC = 69, S = 7, Y = 4.8 | 69 | ||
| 5 wt% Ni-Zr/H-beta (407) | GC = 77, S = 14, Y = 10.8 | 77 | ||
| 5 wt% Cu-Zr/H-beta (417) | GC = 70, S = 9, Y= 6.3 | 7 | ||
| 5 wt% Zn-Zr/H-beta (420) | GC = 68, S = 9.3, Y = 6.3 | 68 | ||
| 2 wt% Pt/ZrO2 (34.2) | T = 180 °C, P = 8 MPa, t = 18 h, Cat = 1 g, 1000 rpm, 10 wt% glycerol solution (in 40 mL H2O) | GC = 13.7, S = 4.8 | 0.3 | [189] |
| 2 wt% Pt/ZrO2-WO3(76.3) | GC = 10.4, S = 30.6 | 0.3 | ||
| 2 wt% Ru/ZrO2 (34.4) | GC = 100, S = 0.3 | 2.4 | ||
| 2 wt% Ru/ZrO2-WO3(81.5) | GC = 12.0, S = 9.5 | 0.3 | ||
| 9 wt% Pt-8 wt% WO3/Al2O3 (95) | T = 220 °C, P = 4.5 MPa H2, t = 16 h, Cat = 0.35 g, 550 rpm under H2 atm. 5 wt% glycerol solution | GC = 80.3, S = 38.5 | 7 | [182] |
| 1 wt% Pt-1 wt% WO3/Al2O3 (128) | GC = 25.6, S = 0 | 2 | [183] | |
| 1 wt% Pt-9 wt% WO3/Al2O3 (104) | GC = 16.7, S = 16.4 | 2 | ||
| 4 wt% Pt-8 wt% WO3/Al2O3 (na) | GC = 42,1, S = 27.8 | 4 | ||
| 9 wt% Pt-8 wt% WO3/Al2O3 (81) | GC = 60.3, S = 31.2 | 6 | ||
| 9 wt% Pt-8 wt% WO3/Al2O3 (81) | T = 200 °C, P = 4.5 MPa H2, t = 24 h, Cat = 0.35 g, 550 rpm, 5 wt% glycerol solution | GC = 53.1, S = 51.9 | 3 | |
| 2 wt% Pt-15 wt% WO3/ZrO2 (35.1) | T = 180 °C, P = 5 MPa, Cat = 3 g, WHSV = 1.0 g/g cat/h, 10 wt% glycerol solution | GC = 30, S = 44.3 | nd | [187] |
| 2 wt% Pt-15 wt% WO3/ZrO2-2.5 wt% SiO2 (101.2) | GC = 41.5, S = 46.3 | |||
| 2 wt% Pt-15 wt% WO3/ZrO2-5 wt% SiO2 (113.3) | GC = 54.3, S = 52 | |||
| 2 wt% Pt-15 wt% WO3/ZrO2-7.5 wt% SiO2 (121.2) | GC = 40.1, S = 46.7 | |||
| 2 wt% Pt-15 wt% WO3/ZrO2-10 wt% SiO2 (152.4) | GC = 32.1, S = 47.1 | |||
| 1 wt% Pt-20 wt% HSiWOX/ZrO2 (46.9) | T = 180 °C, P = 5 MPa, Cat = 2 g, WHSV = 0.09/h, 10 wt% glycerol solution | GC = 26.7, S = 38.9 | nd | [185] |
| 1 wt% Pt–LiSiWOX/ZrO2 (50.5) | GC = 43.5, S = 53.6 | |||
| 1 wt% Pt–KSiWOX/ZrO2 (51.8) | GC = 24.0, S = 36.8 | |||
| 1 wt% Pt–RbSiWOX/ZrO2 (53.3) | GC = 16.6, S = 31.6 | |||
| 1 wt% Pt–CsSiWOX/ZrO2 (53.8) | GC = 41.2, S = 40.2 | |||
| 2 wt% Pt/ZrO2 (61.0) | T = 180 °C, P = 5 MPa, Cat = 2 g, WHSV = 0.09/h, 10 wt% glycerol solution | GC = 18.7, S = 3.9 | nd | [186] |
| 2 wt% Pt-15 wt% HSiW/ZrO2 (54.8) | GC = 24.1, S = 48.1 | |||
| 2 wt% Pt-15 wt% HPW/ZrO2 (57.0) | GC = 25.5, S = 32.9 | |||
| 2 wt% Pt-15 wt% HPMo/ZrO2 (55.3) | GC = 27.1, S = 7.8 | |||
| Pt/mesoporous-WO3 (22) | T = 180 °C, P = 5.5 MPa H2, t = 12 h, Cat = 1 g, 10 wt% glycerol solution | GC = 18.0, S = 39.3 | 1.5 | [184] |
| Pt/commercial-WO3 (9.0) | GC = 4.5, S = 29.9 | 0.3 |
| Catalyst (A) | Operation Parameters | Performance (%) | TOF | Reference |
|---|---|---|---|---|
| Calcined Eggshell (29) | T = 245 °C, Cat = 2 wt% G, t = 6 h, under N2 flow | GC = 80, DG = 40, TG = 2.5, Oth = 57.5 | 72 | [206] |
| T = 220 °C, Cat = 2 wt% G, t = 24 h, under N2 flow | GC = 85, DG = 35, TG = 8, Oth = 57 | 19 | ||
| K2CO3 (na) | T = 250 °C, Cat = 18 g, 4.69 mol glycerol (in 50 mL H2O) | G = 59.9 ± 2.7, DG = 18.8 ± 0.2, CDG = 1.5 ± 0.4, TG = 8.4 ± 1.2, CTG = 1.0 ± 0.2, TtG = 4.4 ± 1.0, PG = 3.1 ± 0.1, HG = 1.0 ± 0.5 | nd | [205] |
| Aquivion PW 98 (na) | T = 150 °C, Cat = 0.45 mmol, t = 6 h | GC = 82, YDG = 21, YTG = 17, Y>TtG = 33, SO = 87 | [204] | |
| Amberlyst 70 (na) | GC = 76, YDG = 31, YTG = 15, Y>TtG = 16, SO = 81 | |||
| Nafion NR 50 (na) | GC = 78, YDG = 14, YTG = 21, Y>TtG = 24, SO = 75 | |||
| Aquivion PW 98 (na) | T = 150 °C, Cat = 0.45 mmol, t = 7 h | GC = 95, YDG = 13, YTG = 15, Y>TtG = 47, SO = 79 | ||
| Na2CO3 (na) | T = 270 °C, P = atm. press., Cat = 1 wt% G, t = 1 h | GC = 85.26, Y(DG+TG+tTtG) = 59.13, SPG = 11.44, SCDG = 7.18 | nd | [202] |
| T = 270 °C, P = atm. press., Cat = 1 wt% G, t = 2 h | GC = 96.03, Y(DG+TG+tTtG) = 66.83, SPG = 12.50, SCDG = 12.94 | |||
| T = 270 °C, P = atm. press., Cat = 1 wt% G, t = 3 h | GC = 96.12, Y(DG+TG+tTtG) = 47.33, SPG = 37.53, SCDG = 8.33 | |||
| T = 270 °C, P = atm. press., Cat = 3 wt% G, t = 1 h | GC = 92.83, Y(DG+TG+tTtG) = 70.13, SPG = 9.06, SCDG = 6.50 | |||
| T = 270 °C, P = atm. press., Cat = 3 wt% G, t = 2 h | GC = 96.20, Y(DG+TG+tTtG) = 53.30, SPG = 13.33, SCDG = 12.63 | |||
| T = 270 °C, P = atm. press., Cat = 3 wt% G, t = 3 h | GC = 87.18, Y(DG+TG+tTtG) = 57.77, SPG = 0.00, SCDG = 35.60 | |||
| MgAl-LDH1-723K (237) | T = 235 °C, Cat = 2 wt% G, t = 24 h, under N2 flow | GC = 96, SDG = 9, STG = 3, SOth = 88, YO = 12 | 22 | [203] |
| MgAl-LDH2-723K (188) | GC = 24, SDG = 89, STG = 11, SOth = 0, YO = 24 | 5 | ||
| CaAl-LDH1-723K (15) | GC = 75, SDG = 36, STG = 6, SOth = 58, YO = 32 | 17 | ||
| CaAl-LDH4-723K (3) | GC = 60, SDG = 44, STG = 11, SOth = 45%, Y = 33 | 14 | ||
| CaAl-LDH4-923K (5) | GC = 59, SD = 38, STG = 13, SOth = 49, YO = 35 | 13 | ||
| CaAl-LDH4-1073K (3) | GC = 84, SDG = 29, STG = 20, SOth = 51%, YO = 59 | 19 | ||
| Blank | T = 250 °C, Cat = 2 wt% G, t = 8 h | GC = 2, SDG = 3, YDG = 0, STG = 0, YTG = 0, SOth = 97, Y = 2. | - | [198] |
| MCM-41 (938) | GC = 2, SDG = 4, YDG = 0, STG = 0, YTG = 0, SOth = 96, Y = 2. | 1 | ||
| CaO (14) | GC = 72, SDG = 27, YDG = 20, STG = 10, YTG = 7, SOth = 63, Y = 46. | 49 | ||
| La2O3 (na) | GC = 54, SDG = 32, YDG = 17, STG = 11, YTG = 6, SOth = 57, Y = 30. | 37 | ||
| Ca1.8La0.8O2 (20) | GC = 76, SDG = 30, YDG = 23, STG = 15, YTG = 12, SOth = 55, Y = 42. | 52 | ||
| 30% Ca/MCM-41 (na) | GC = 69, SDG = 33, YDG = 22, STG = 24, YTG = 17, SOth = 43, Y = 30. | 47 | ||
| 30% La/MCM-41 (na) | GC = 74, SDG = 25, YDG = 19, STG = 18, YTG = 14, SOth = 56, Y = 42. | 50 | ||
| 30% Ca1.6La0.6/MCM-41 (128) | GC = 85, SDG = 22, YDG = 19, STG = 14, YTG = 12, SOth = 64, Y = 55. | 57 | ||
| LiOH (na) | T = 240 °C, P = atm. press. Cat = 2 wt% G, t = 8 h, under N2 flow | GC = 99, SDG = 18, STG = 21, STtG = 13, SPG = 52, Y = 51.5, Oth S = 48. | 67 | [201] |
| Zeolite-Y (690) | GC = 15, SDG = 2, STG = 7, STtG = 4, SPG = 13, Y = 2, SOth = 67. | 10 | ||
| Li-ZeY (531) | GC = 98, SDG = 21, STG = 32, STtG = 19, SPG = 72, Y = 70.5, SOth = 28. | 67 |
| Catalyst (A) | Operation Parameters | Performance (%) | TOF | Reference |
|---|---|---|---|---|
| HZSM-5 (≈400) | T = 600 °C, P = 0.1 MPa, WHSV = 105/h, Cat = na | Ethene S = 7.94 Y = 3.99, Propene S = 0.35 Y = 0.18, Butene S = 0.00 Y = 0.00, Methane S = 4.75, Y = 2.38. | nd | [210] |
| 30%Al/ZSM-5 (≈350) | Ethene S = 6.45 Y = 5.26, Propene S = 0.07 Y = 0.06, Butene S = 0.00 Y = 0.00, Methane S = 9.63, Y = 7.85. | |||
| 30%Ca/ZSM-5 (>215) | Ethene S = 3.95 Y = 1.93, Propene S = 0.83 Y = 0.41, Butene S = 0.00 Y = 0.00, Methane S = 5.95, Y = 2.90. | |||
| 30%Cr/ZSM-5 (>230) | Ethene S = 15.20 Y = 14.82, Propene S = 2.60 Y = 2.53, Butene S = 0.96 Y = 0.93, Methane S = 8.16, Y = 7.96. | |||
| 30%Cu/ZSM-5 (≈260) | Ethene S = 18.62 Y = 14.97, Propene S = 1.06 Y = 0.86, Butene S = 0.56 Y = 0.45, Methane S = 5.60, Y = 4.50. | |||
| 30%Li/ZSM-5 (≈230) | Ethene S = 2.61 Y = 0.90, Propene S = 1.76 Y = 0.61, Butene S = 0.96 Y = 0.33, Methane S = 6.04, Y = 2.10. | |||
| 30%Mg/ZSM-5 (≈250) | Ethene S = 6.142 Y = 4.19, Propene S = 0.271 Y = 0.19, Butene S = 0.00 Y = 0.00, Methane S = 5.457, Y = 3.72. | |||
| 30%Ni-ZSM-5 (≈270) | Ethene S = 3.27 Y = 3.20, Propene S = 0.5 Y = 0.5, Butene S = 0.00 Y = 0.00, Methane S = 7.21, Y = 7.05. | |||
| HZSM-5 | T = 700 °C, P = 0.1 MPa, LSV = 20–80 s, Cat = 0.2 g | GC = 11.62, Olefin S = 1.20 | nd | [207] |
| 10 wt% Cr-ZSM-5 (na) | GC = 10.85, Olefin S = 0.63 | |||
| 10 wt% Al-ZSM-5 (na) | GC = 10.44, Olefin S = 1.09 | |||
| 10 wt% Ca-ZSM-5 (na) | GC = 15.02, Olefin S = 1.34 | |||
| 10 wt% CuZSM-5 (na) | GC = 17.72, Olefin S = 3.55 | |||
| 10 wt% Ni-ZSM-5 (na) | GC = 16.07, Olefin S = 1.84 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nda-Umar, U.I.; Ramli, I.; Taufiq-Yap, Y.H.; Muhamad, E.N. An Overview of Recent Research in the Conversion of Glycerol into Biofuels, Fuel Additives and other Bio-Based Chemicals. Catalysts 2019, 9, 15. https://doi.org/10.3390/catal9010015
Nda-Umar UI, Ramli I, Taufiq-Yap YH, Muhamad EN. An Overview of Recent Research in the Conversion of Glycerol into Biofuels, Fuel Additives and other Bio-Based Chemicals. Catalysts. 2019; 9(1):15. https://doi.org/10.3390/catal9010015
Chicago/Turabian StyleNda-Umar, Usman Idris, Irmawati Ramli, Yun Hin Taufiq-Yap, and Ernee Noryana Muhamad. 2019. "An Overview of Recent Research in the Conversion of Glycerol into Biofuels, Fuel Additives and other Bio-Based Chemicals" Catalysts 9, no. 1: 15. https://doi.org/10.3390/catal9010015
APA StyleNda-Umar, U. I., Ramli, I., Taufiq-Yap, Y. H., & Muhamad, E. N. (2019). An Overview of Recent Research in the Conversion of Glycerol into Biofuels, Fuel Additives and other Bio-Based Chemicals. Catalysts, 9(1), 15. https://doi.org/10.3390/catal9010015