Abstract
A one-step catalytic process was used to catalyze the hydrodeoxygenation of 5-methyl-3-heptanone (C8 ketone) to a mixture of 5-methyl-3-heptene, 5-methyl-2-heptene (C8 alkenes), and 3-methyl heptane (C8 alkane). High conversion of C8 ketone to the desired products was achieved over a single bed of a supported catalyst (bifunctional heterogeneous catalyst) consisting of one transition metal (copper (Cu) or platinum (Pt)) loaded on alumina (Al2O3) under mild operating conditions (reaction temperatures were varied between 180 °C to 260 °C, and the pressure was 1 atm). The C8 ketone was hydrogenated to 5-methyl-3-heptanol (C8 alcohol) over metal sites, followed by dehydration of the latter on acid sites on the support to obtain a mixture of C8 alkenes. These C8 alkenes can be further hydrogenated on metal sites to make a C8 alkane. The results showed that the main products over copper loaded on alumina (20 wt% Cu–Al2O3) were a mixture of C8 alkenes and C8 alkane in different amounts depending on the operating conditions (the highest selectivity for C8 alkenes (~82%) was obtained at 220 °C and a H2/C8 ketone molar ratio of 2). However, over platinum supported on alumina (1 wt% Pt–Al2O3), the major product was a C8 alkane with a selectivity up to 97% and a conversion of 99.9% at different temperatures and all H2/C8 ketone ratios.
1. Introduction
Recently, significant effort has been devoted to the development of alternative pathways of producing biofuel and chemicals from biomass as a sustainable source due to declining petroleum resources and rising oil prices. Synthesizing fuel-range hydrocarbons from biomass is of particular interest because it offers the potential for a drop-in replacement of petroleum-based fuels.
This work envisions that fuel-range hydrocarbons can be produced with a C8 ketone as the key intermediate. This C8 ketone is produced by dehydrating 2,3 butanediol (2,3 BDO), which can be produced in high yields from biomass-derived sugars [1,2,3,4,5,6,7,8], to methyl ethyl ketone (MEK) [9,10,11,12,13,14,15,16] followed by aldol condensation and hydrogenation reactions [17]. Then, the C8 ketone can be converted to C8 alkenes and C8 alkanes, which can be used as gasoline blended components. This work extends previous work in our laboratory to convert 2,3 BDO and MEK to light alkenes (butene) [18,19,20], an intermediate to higher alkenes, and MEK to chemicals such as C8 ketone [21].
Hydrodeoxygenation of C8 ketones to C8 alkenes and C8 alkanes requires removing an oxygen atom from the ketone, for example, by the hydrogenation of the C=O bond via metal sites followed by dehydration of the produced alcohol by acidic sites. C=O hydrogenation catalysts also have high hydrogenation activity of C=C bonds, causing the produced C=C bonds (alkene) to be further hydrogenated to alkanes. An exception is Cu, which is relatively inactive for the hydrogenolysis of C–C bonds [22].
A variety of metals including Pt, Ru, Rh, Ni, and Cu catalyze the hydrogenation of carbonyl groups [23]. Cu [23,24,25,26] and Pt [27,28,29,30] based catalysts have been extensively studied for the hydrogenation of carbonyl groups. For example, Fan et al. [23] investigated Cu–γAl2O3 and Ni–γAl2O3 to hydrogenate 2,2,6,6-tetramethylpiperidin-4-one to 2,2,6,6 tetramethylpiperidin-4-ol at 140 °C. The results demonstrated that Cu–γAl2O3 had much better activity and selectivity than Ni–γAl2O3. They also studied the effect of introducing Cr into Cu–γAl2O3 and found that Cu–Cr/γAl2O3 displayed better catalytic performance over Cu–γAl2O3. Vannice et al. [29] investigated Pt supported on various supports (TiO2, SiO2, and Al2O3) for the hydrogenation of benzaldehyde to benzyl alcohol. The most active catalyst among all Pt catalysts was Pt–TiO2, giving a selectivity for benzyl alcohol of 100% and a conversion up to at least 80%.
Al2O3 is a traditional choice for alcohol dehydration and has been well known for centuries [31]. Kostestkyy et al. [32] studied the dehydration of simple alcohols such as 1-propanol, 2-propanol, and 2-methyl-2-propanol on TiO2, ZrO2, and γAl2O3 oxide catalysts and reported that γAl2O3 was more active in catalyzing the dehydration reactions than either TiO2 or ZrO2.
It is well known that the hydrodeoxygenation of ketones to alkanes is a significant reaction in the production of biofuels [33]. However, hydrodeoxygenation reactions are seldom used in industrial chemistry, and the development of successful catalytic systems has only been explored in recent years.
Ma et al. [34] investigated the hydrodeoxygenation of hypnone to phenylethane over different catalysts (Ni–γAl2O3, Cu–γAl2O3, and Cu–Cr/γAl2O3) at 200 °C. They found that the catalytic performance of Cu–γAl2O3 was better than that of Ni–γAl2O3 and its activity can be improved by adding Cr to it.
Alotaibi et al. [35] investigated a number of supported metal catalysts consisting of Pt, Pd, Ru, and Cu supported on silica and activated carbon for the hydrogenation of methyl isobutyl ketone (MIBK) in the temperature range between 100 to 400 °C. The results displayed that these catalysts were active for the hydrogenation of MIBK to 4-methyl-2-pentanol (MPol) at 100–200 °C. The best results were obtained using 10% Pt–C, 10% Pd–C, 5% Ru–C, and copper chromite, with selectivities for MPol ranging from 93 to 100% and conversions of MIBK of 92–96% at 100 °C. The selectivity to MPol reduced with increasing temperature because of the formation of 2-methyl pentane (MP) in addition to the hydrogenolysis of the C–C bonds, giving C1–C5 hydrocarbons. The highest selectivities of MP were 87% and 94% over 10% Pt–C and 10% Pd–C catalysts, respectively, with 52% and 84% conversions at a temperature as high as 300 °C. In addition, they studied the hydrodeoxygenation of MIBK over metal catalysts (Pt, Pd, Ru, or Cu) loaded on Cs2.5H0.5PW12O40 (CsPW) at 100 °C under a H2 flow. They stated that amongst the bifunctional catalysts studied, 0.5% Pt–CsPW was the best, yielding 99% MP at 100 °C with 100% MP selectivity and 99% MIBK conversion. Alharbi et al. [36] applied the Pt/CsPW system to other ketones such as acetone, 2-butanone, 3-pentanone, 2-hexanone, cyclohexanone, 2-octanone, and diisobutyl ketone and the results showed that the selectivity of the alkanes produced from different ketones were ≥93%, except for propane resulting from acetone, where its selectivity was 87%.
Yang et al. [37] studied the hydrodeoxygenation of 2-cyclopentylidene cyclopentanone, produced via the self-condensation of cyclopentanone [38,39,40], to bi-cyclopentane (bicyclopentyl) in a flow system at 230 °C. Higher yields of bicyclopentyl (≥90%) were obtained using Pd/SiO2 and Ni/SiO2 while catalysts with more acidic supports like Pd/SiO2–Al2O3, Pd/H–zeolites (H-beta, H-ZSM-5), and Pd/zirconium phosphate gave about an ~80% yield at 230 °C.
The present study involved using a one-step catalytic process to produce 5-methyl-3-heptene, 5-methyl-2-heptene (C8 alkenes), and 3-methyl-heptane (C8 alkane) from biomass-derived 5-methyl-3-heptanone (C8 ketone). The reaction uses a heterogenous catalyst comprised of a transition metal (Cu or Pt) loaded on Al2O3, chosen based on the above literature. This catalyst functions as a bifunctional catalyst with both hydrogenation and dehydration activity. This work shows that this approach can be quite selective to C8 alkenes and C8 alkane in the presence of hydrogen in a single stage.
2. Results and Discussions
2.1. Characterization of Catalysts
2.1.1. X-Ray Diffraction (XRD)
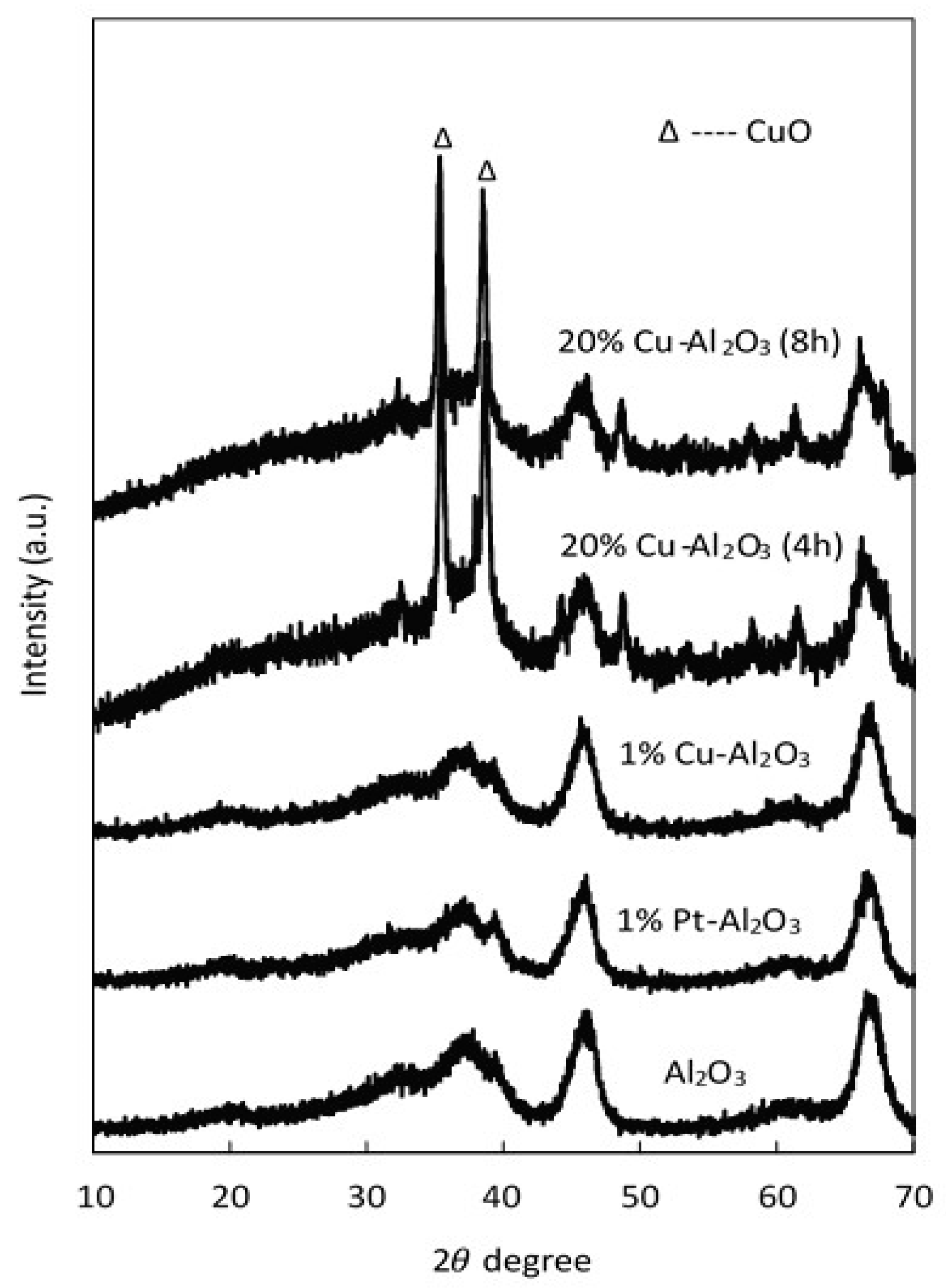
The XRD patterns of pure Al2O3, 1% Cu–Al2O3, 1% Pt–Al2O3, and catalysts with a high loading of Cu (20 wt% Cu on Al2O3) with different calcination times (4 h and 8 h) are shown in Figure 1.
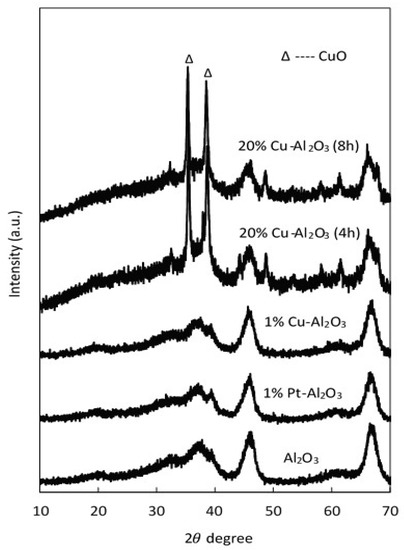
Figure 1.
XRD pattern for pure Al2O3 and different supported catalysts.
As seen in this figure, all characteristic peaks of the parent support (Al2O3) were observed in the supported metal catalysts. These peaks were maintained when metals were introduced; however, the intensity of the main peaks slightly decreased at high Cu loadings. Two peaks characteristic of CuO (35.5° and 38.6°) were observed on the Cu catalysts with 20 wt% Cu, confirming that at least some of the copper was present as CuO clusters. However, there were no distinguishable diffraction peaks representing crystalline CuO peaks at low Cu loadings (1% Cu–Al2O3), most likely because the copper particles were smaller than 40 Å in size, the detection limit of XRD [41,42]. Additionally, for the 1% Pt-Al2O3, no obvious diffraction peaks were observed, suggesting that the metal was well dispersed on the support. The crystallite sizes of CuO for 20% Cu–Al2O3 were determined using the Scherrer formula:
where K is an empirical constant (assumed to be 0.9, the value for spherical particles); λ is the x-ray source wavelength (0.15406 nm); B is the full width at half-maximum (FWHM); and θ is the Bragg angle. Crystallite sizes are reported in Table 1.
d = Kλ/Β cos θ

Table 1.
Physical properties as well as acidity and basicity values for the different supported catalysts.
2.1.2. Transmission Electron Microscopy (TEM)
The TEM images in Figure 2a,b mention the presence of a number of Pt and Cu nanoparticles (dark zones), and the measurements of the average distinct metal particle size were estimated to be about 7 and 8 nm in diameter, respectively. However, the pore structure could not be clearly observed in the TEM images either as rows or as ordered pore openings. The TEM image of the 20% Cu supported on Al2O3 with different calcined time (Figure 2c,d) suggests Cu particle sizes in the range of 18 nm, close to the Cu particle sizes calculated from the XRD.

Figure 2.
TEM images of (a) 1% Pt–Al2O3, (b) 1% Cu–Al2O3, (c) 20% Cu–Al2O3 (4 h), and (d) 20% Cu–Al2O3 (8 h).
2.1.3. N2 Adsorption
The surface area of all catalysts, according to the BET measurements, is summarized in Table 1. As seen in this table, the surface area decreased as the loading of Cu on Al2O3 increased. This is consistent with literature reports that show that surface area decreases as metal loading on a support increases when the catalyst is synthesized by the impregnation method [43].
2.1.4. Temperature Programmed Desorption (NH3–TPD) and (CO2–TPD)
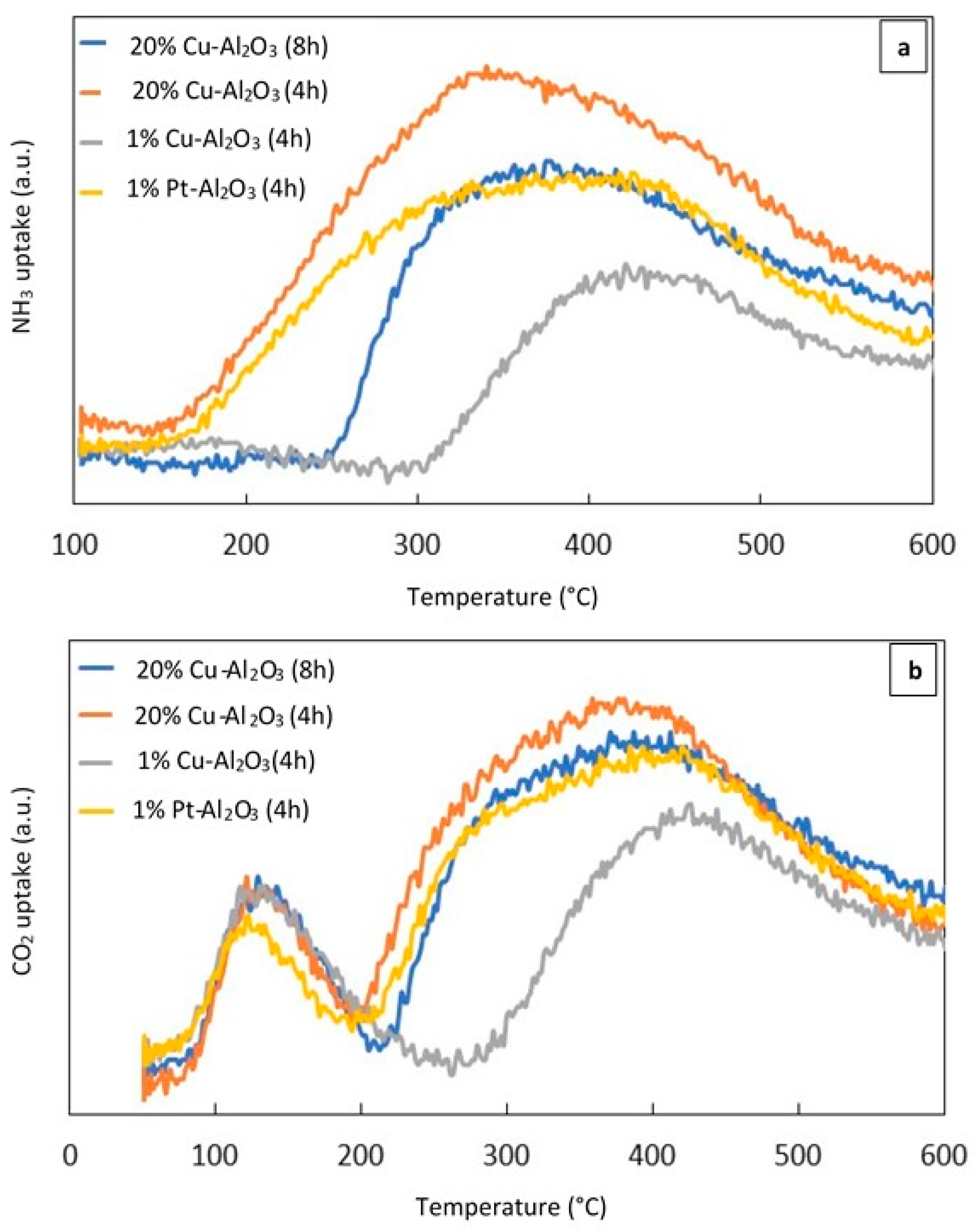
NH3–TPD and CO2–TPD experiments were performed to investigate the acid–base properties of the reduced catalysts. Figure 3a shows the NH3–TPD profile of the different reduced metal supported catalysts calcined for 4 h and 8 h, while Figure 3b displays the CO2–TPD profile for the same catalysts.
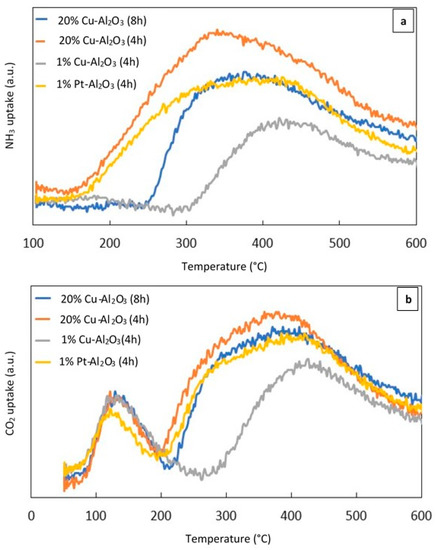
Figure 3.
(a) (NH3–TPD) profiles of the different catalysts, (b) (CO2–TPD) profiles of the different catalysts.
As can be seen from Figure 3a, broad NH3 desorption peaks over 20% Cu–Al2O3 (4 h) and 1% Pt–Al2O3 extended from 160 °C to 600 °C. However over 20% Cu–Al2O3 (8 h), the peak was narrower and less intense than that measured for 20% Cu–Al2O3 (4 h). For these three mentioned catalysts, the maximum NH3 desorption temperatures were centered around 360 °C while the peak for 1% Cu–Al2O3 shifted to a higher temperature and centered around 425 °C. The increase in the NH3 desorption temperature suggested that the strength of the acidic sites in 1% Cu-Al2O3 was higher than in the other. In addition, it had the lowest concentration of acidic sites while the highest concentration was noted for 20% Cu–Al2O3 (4 h) (see Table 1).
Figure 3b shows the CO2–TPD profiles of the different catalysts. Two obvious peaks could be observed for all catalysts, suggesting the presence of different sites of varying base strength. 20% Cu–Al2O3 (4 h and 8 h) and 1% Pt–Al2O3 had very similar maximum CO2 desorption temperatures (near 130 °C and 370 °C), although the first peak for 1% Pt–Al2O3 shifted slightly to a lower temperature (around 124 °C). For 1% Cu–Al2O3, the first peak was proportionally larger than the lower-temperature peak for the three other catalysts, while the second one was lower than both of them and shifted to a higher temperature (around 420 °C), suggesting that the number of weakly basic sites was higher for this catalyst. However, the total amount of base sites was still the lower than the other catalysts as shown in Table 1.
2.2. Catalytic Conversion of C8 Ketone to C8 Alkenes and C8 Alkane in a Fixed Bed Reactor
2.2.1. Effect of Different Supported Catalysts on the Reaction of C8 Ketone
Experiments were conducted to study the effect of using different metal (Cu and Pt) supported Al2O3 on the conversion of C8 ketone and the distribution of major products. The catalytic results are summarized in Table 2.
Table 2.
Catalytic activity in the conversion of C8 ketones to C8 alkenes and C8 alkane over several catalysts.
The results in Table 2 indicate that Pt was much more active than Cu. For the same weight loading, conversion over 1% Pt–Al2O3 was approximately double of that for 1% Cu–Al2O3. The other major difference for these two catalysts was that 1% Pt–Al2O3 had a nearly 98% selectivity for the C8 alkane, while 1% Cu–Al2O3 primarily produced C8 alkenes, with lesser amounts of C8 alkane and other products. When the amount of Cu loaded on the catalyst was increased to 20 wt%, an equivalent conversion was reached to that of 1% Pt–Al2O3, about 99.9%. This can be attributed to the increase in metal sites (see Table 1 and Figure 3). However, the selectivity difference between the Cu and Pt catalysts was still maintained: the selectivity for C8 alkenes was 45% for the Cu catalyst. These results suggest that Pt was more active than Cu, not only for the initial hydrogenation of the C8 ketone, but also for the hydrogenation of C8 alkenes to C8 alkane. These results are in accordance with the fact that copper catalysts selectively hydrogenate C=O bonds and are relatively inactive for the hydrogenolysis of C–C bonds [22], while platinum catalysts are very active in hydrogenating both C=O and C=C bonds [27,35].
It was also interesting to note the large amount of other products (24%) produced over 1% Cu–Al2O3 compared to the other catalysts. This was likely to be the result of the interplay between the chemistries on the two types of catalytic sites: metal and acidic sites. For the low loading of Cu, the hydrogenation of the C8 ketone was sufficiently slow that reactions over the acidic support could proceed, leading to products other than C8 alkenes and C8 alkane. When the metal chemistry was more pronounced, in the case of using Pt or increased Cu loadings, then hydrogenation of the C8 ketone took place fast enough that other reaction pathways did not play a significant role, and the selectivities for other products dropped.
2.2.2. Effect of Reaction Temperature
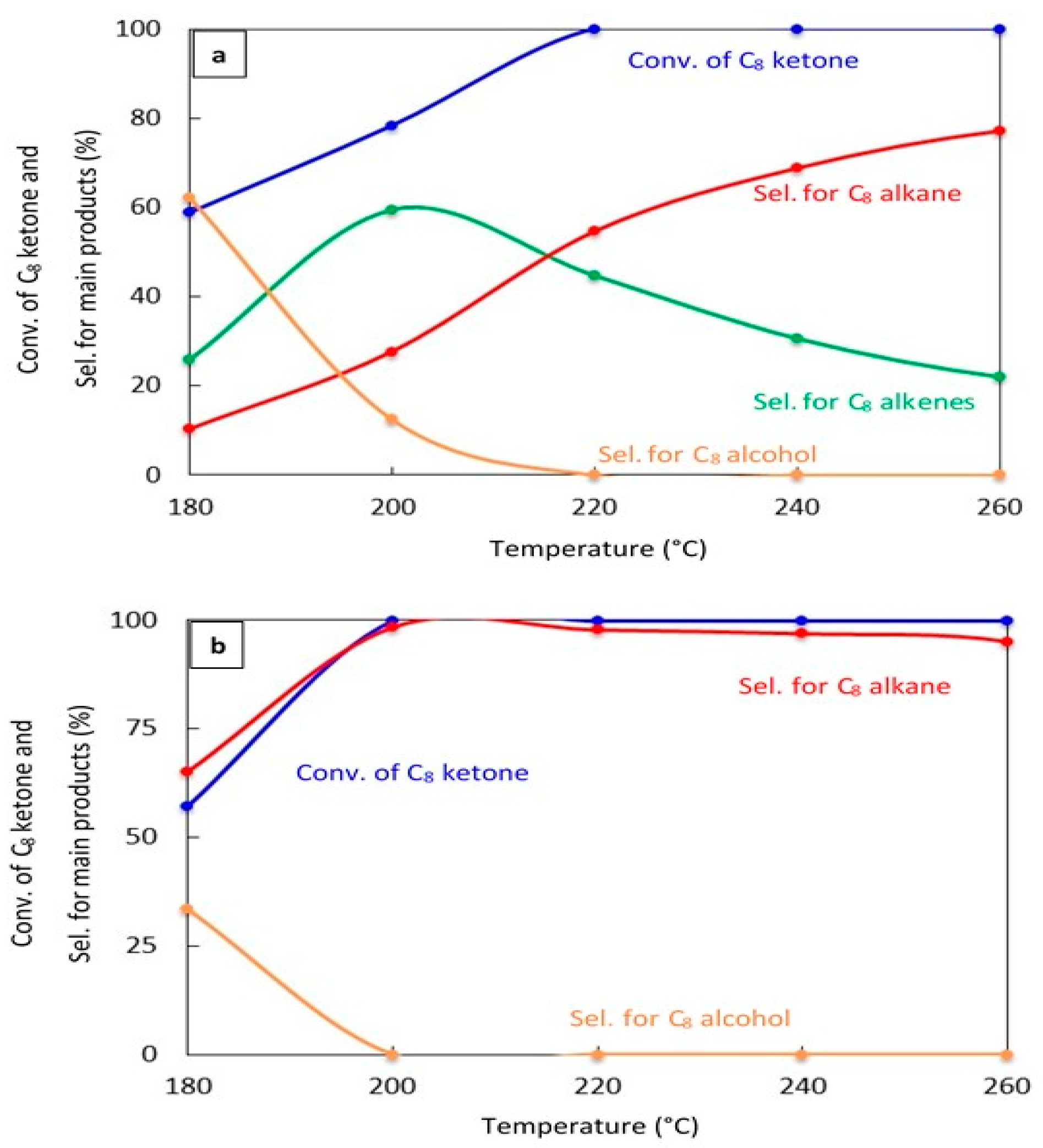
The effect of changing the reaction temperature on the conversion of the C8 ketone and the distribution of major products over 20% Cu–Al2O3 and 1% Pt–Al2O3. Figure 4a,b show the catalytic results after 1 h on stream for both catalysts, respectively. Over 20% Cu–Al2O3, the main products were a mixture of C8 alkenes and the C8 alkane, and the selectivity for the C8 alkane increased with increased temperatures resulting from further hydrogenation of C8 alkenes. Over 1% Pt–Al2O3, however, the main product was C8 alkane at all temperatures, although its selectivity decreased at low temperatures in favor of the C8 alcohol.
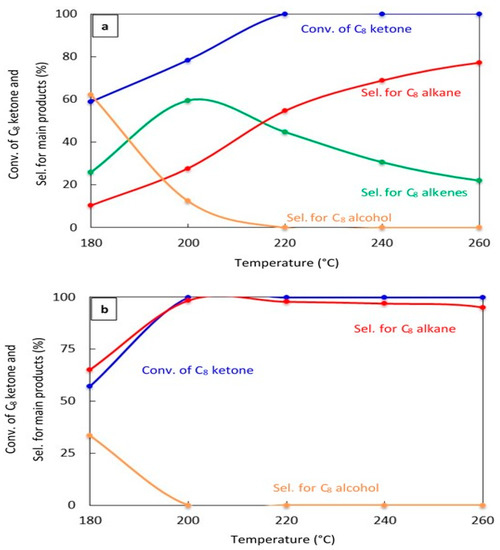
Figure 4.
Catalytic results for the conversion of the C8 ketone to several products with different temperatures using 0.5 g of catalyst: (a) (20% Cu-Al2O3), (b) (1% Pt-Al2O3). Reaction conditions: reaction temperatures ranged from 180 °C to 260 °C; the total flow rate of H2 and N2, 85 mL/min; the flow rate of H2 and N2, 68.5 and 16.5 mL/min, respectively; the flow rate of C8 ketone, 1 mL/h; and the molar ratio of H2/C8 ketone, 25. All catalysts were calcined for 4 h.
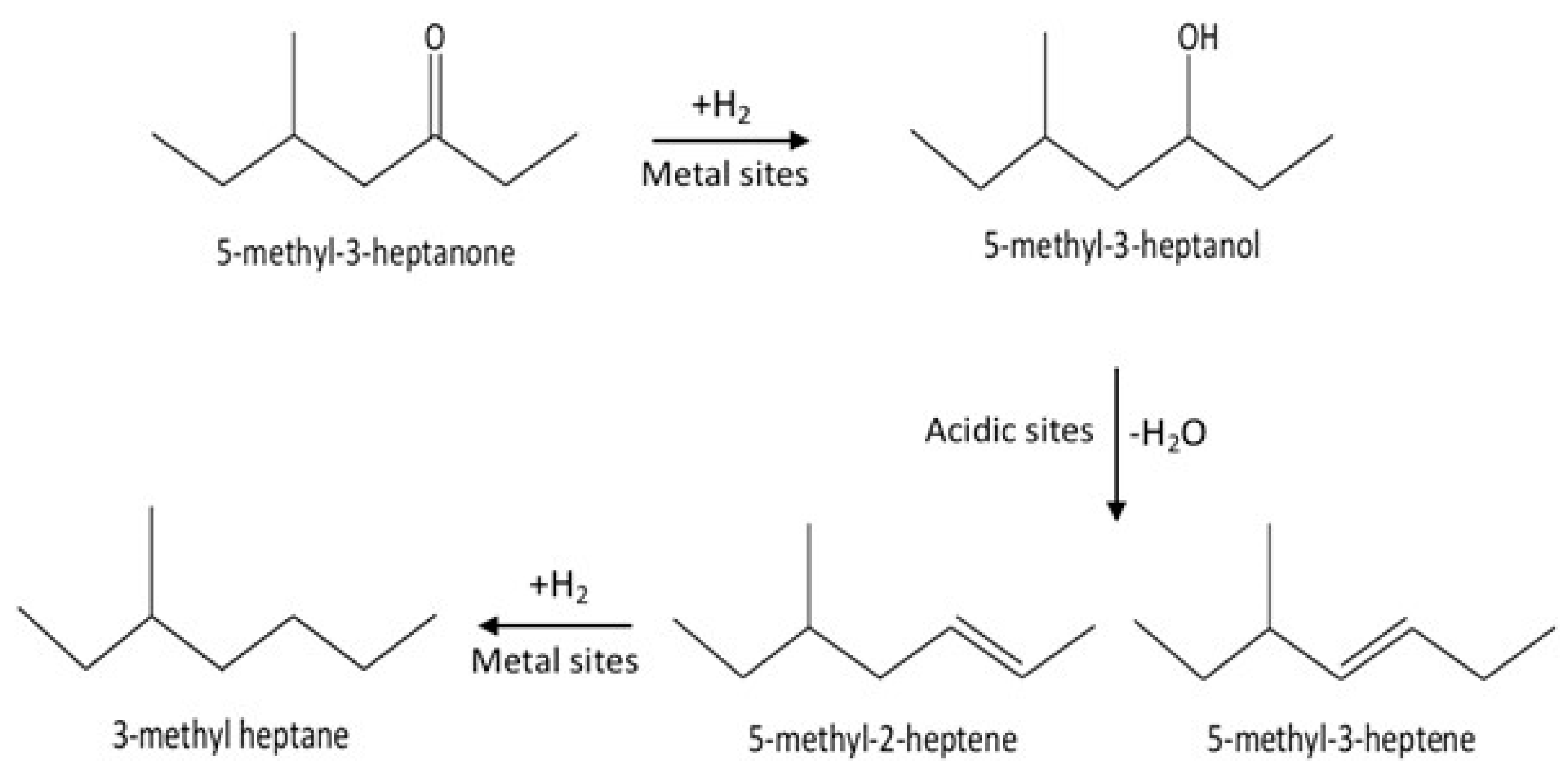
These results indicate that the conversion of C8 ketones to C8 alkane occurred by a series reaction, as summarized in Scheme 1. The first step in the reaction mechanism is the hydrogenation of the C8 ketone to the C8 alcohol over metal sites. The second step includes dehydration of the alcohol to C8 alkenes on acid sites (support). The alkenes can further be hydrogenated to the C8 alkane over metal sites [18,35,44]. Temperature controls the rate of all reaction pathways. At low temperatures, for both Cu–Al2O3 and Pt–Al2O3, significant amounts of C8 alcohols were formed because subsequent reactions did not proceed at a fast enough rate. It is interesting to note that even at the lowest temperature, Pt–Al2O3 produced no C8 alkenes, instead producing C8 alkane exclusively. This suggests that Pt–Al2O3 was extremely active for the hydrogenation of C8 alkenes to C8 alkanes. Higher temperatures allowed further reactions to occur, pushing the product selectivities toward C8 alkenes and C8 alkane.
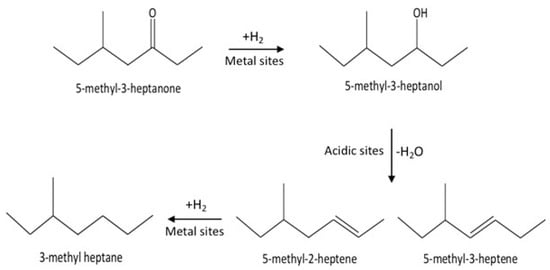
Scheme 1.
The mechanism of conversion for the C8 ketone to C8 alkenes and C8 alkane.
2.2.3. Effect of H2/C8 Ketone Molar Ratio
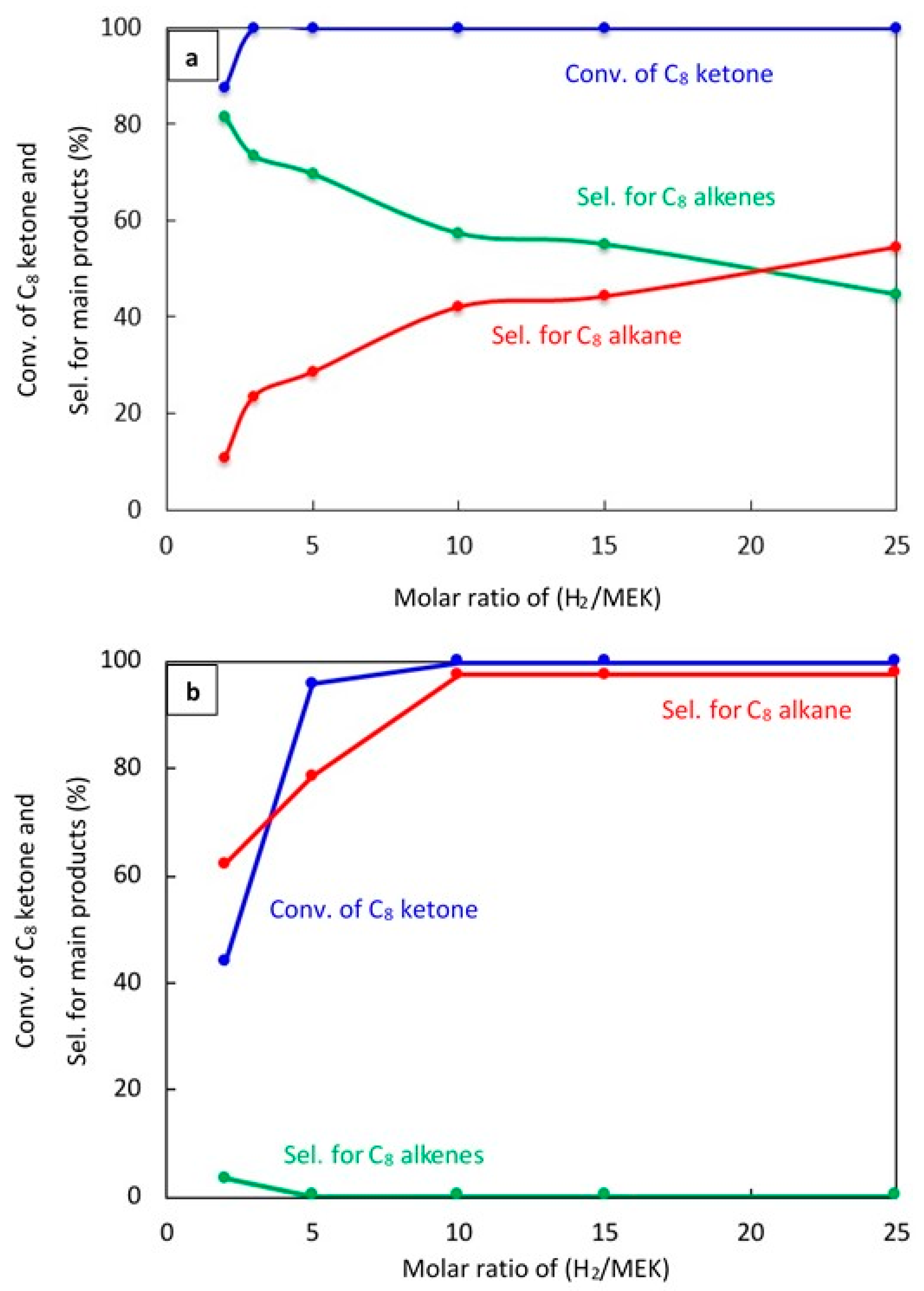
Experiments were performed to study how changing the molar ratio of H2 to C8 ketone affects the conversion of C8 ketone and the distribution of products over both 20% Cu–Al2O3 and 1% Pt–Al2O3 as shown in Figure 5a,b, respectively, after 1 h on stream.
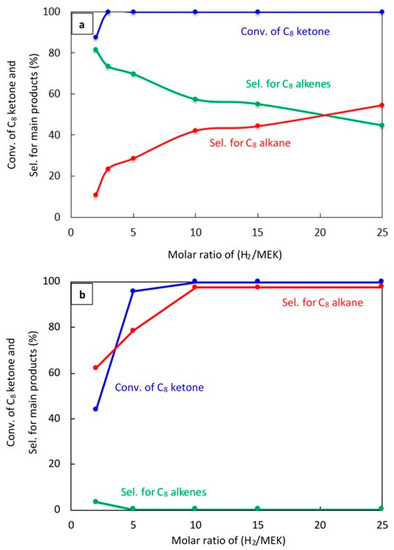
Figure 5.
Catalytic results for the conversion of C8 ketone to several products using different H2/C8 ketone molar ratios using 0.5 g of the catalysts: (a) (20% Cu–Al2O3), (b) (1% Pt–Al2O3). Reaction conditions: reaction temperature, 220 °C; the total flow rate of H2 and N2, 85 mL/min; the flow rate of H2 and N2, 68.5 and 16.5 mL/min, respectively; the flow rate of C8 ketone, 1 mL/h; the molar ratio of H2/C8 ketone was varied from 2 to 25.
Figure 5a shows that the H2/C8 ketone ratio directly impacted on both the C8 ketone conversion and product selectivity. A higher H2/C8 ketone ratio increased the C8 ketone conversion: the conversion was 100% at all ratios above two. In addition, as seen in this figure, the H2/C8 ketone molar ratio affected the selectivity for products. With an increase in ratio from two to 25, the selectivity for C8 alkenes decreased from 82% to 45%. This decrease was due to more hydrogenation of the C8 alkenes to C8 alkane. The highest selectivity for C8 alkenes was found at a H2/C8 ketone ratio of two with a selectivity of 82%.
Over 1% Pt–Al2O3, as shown in Figure 5b, the conversion of the C8 ketone increased from (44%) to (99.9%) when the H2/C8 ketone ratio increased from two to 25. The main product selectivity was still the C8 alkane, although the selectivity for this product decreased dramatically when the ratio was five or below. For those ratios, the selectivity for other products like the C8 unsaturated ketone, butane, xylene, aromatics, and condensation products became more important.
2.2.4. Effect of Space Time
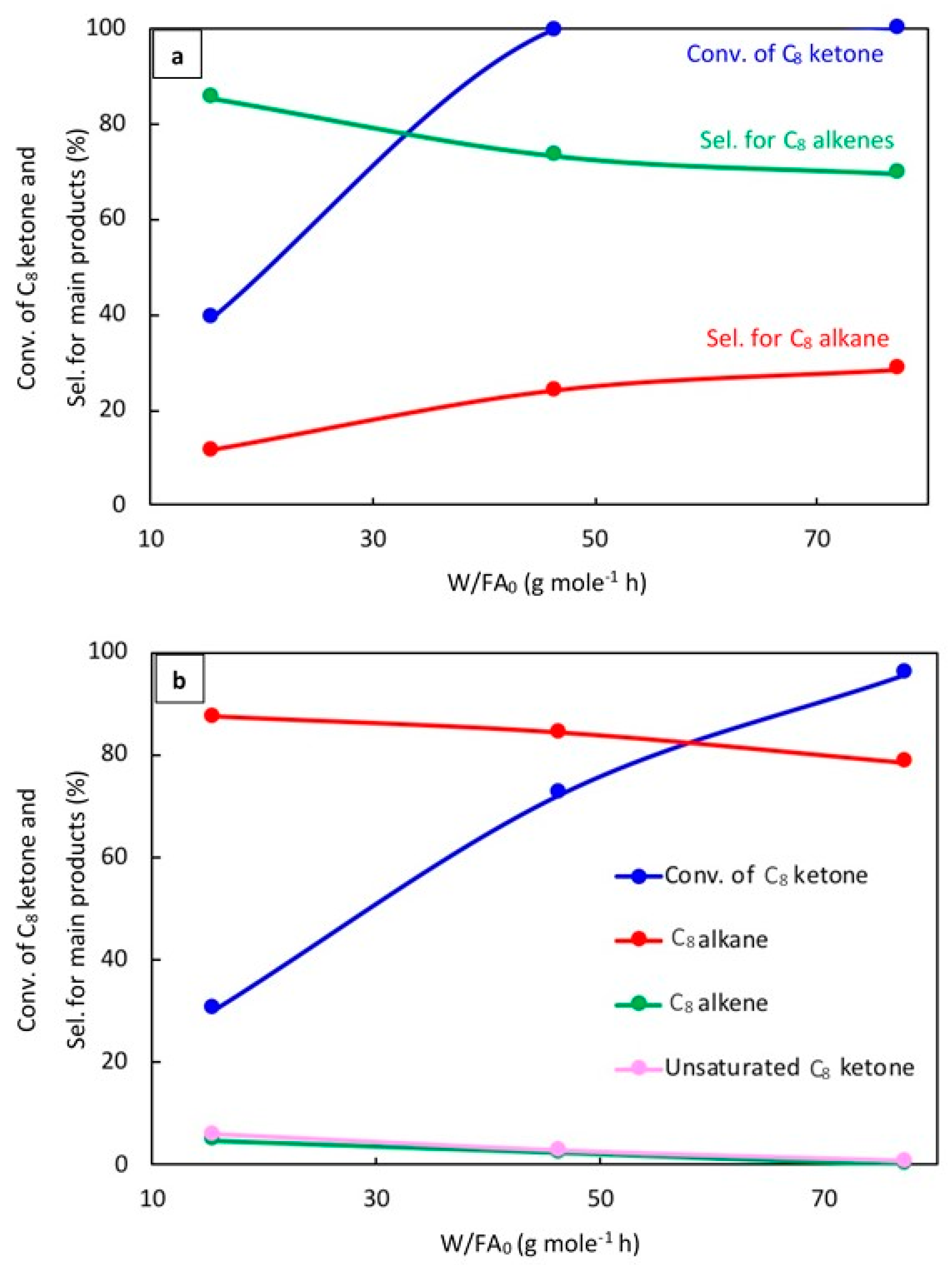
The effect of space time, W/FA0, where W is the weight of catalyst (g) and FA0 is the molar flow rate of C8 ketone (mol h−1), was evaluated to better understand the reaction mechanism. Results for 20% Cu–Al2O3 and 1% Pt–Al2O3 at 220 °C are as shown in Figure 6a,b, respectively, after 1 h of the reaction.
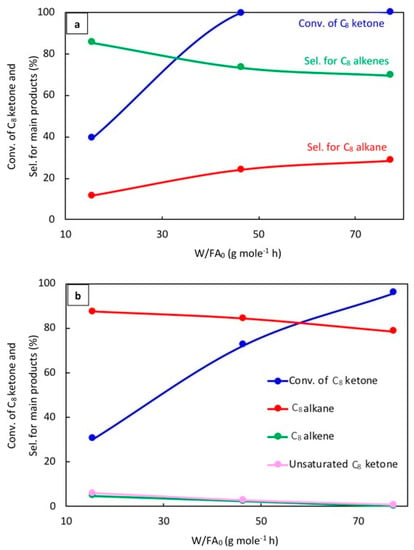
Figure 6.
Catalytic results for the conversion of the C8 ketone to several products with different space times after 1 h of the reaction over (a) (20% Cu-Al2O3), and (b) (1% Pt-Al2O3). Reaction conditions: reaction temperature of 220 °C; total flow rate of H2 and N2 of 85 mL/min; flow rate of C8 ketone of 1 mL/h; molar ratio of H2/C8 ketone of 5; and a calcination time of 4 h.
As can be seen in Figure 6a, the conversion of the C8 ketone increased from 39% to 100% when the space time increased from 15 to 77 g·mol−1·h. The selectivity for C8 alkenes decreased from 86% to 70%, while the selectivity for the C8 alkane increased from 12% to 29%. In addition, a small amount of C8 alcohol was detected at low space times.
The effect of space time on the conversion of the C8 ketone and the distribution of products over 1% Pt–Al2O3 is displayed in Figure 6b. As can be seen, the conversion of the C8 ketone increased from 31% to 96% when the space time increased from 15 to 77 g·mol−1·h, and the selectivity for the C8 alkane decreased from 87% to 79%. The drop-in alkane selectivity was associated with the formation of byproducts like aromatics and condensation products. C8 alkenes and unsaturated C8 ketones were detected at low space times and decreased with increased space times. Unsaturated C8 ketones were produced by dehydrogenation reactions on Pt.
The results for different W/FA0 for Cu and Pt showed a few important trends. First, it can be seen that 1% Pt–Al2O3 primarily produced alkanes even at the smallest space times. This indicates that the hydrogenation of alkenes to alkane (C=C bond) was quite fast. On the other hand, this reaction was much slower on 20% Cu–Al2O3. The conversion of C8 ketone over 20% Cu–Al2O3 (39%) was higher than over 1% Pt–Al2O3 (31%) at the same low space time, suggesting that hydrogenation of this ketone (C=O bond) to alcohol was faster on 20% Cu–Al2O3 than on 1% Pt–Al2O3. The results also showed that 1% Pt–Al2O3 could catalyze dehydrogenation reactions under some conditions, yielding unsaturated ketones. Again, this was not noted for 20% Cu–Al2O3 due to its lower dehydrogenation activity.
2.2.5. Catalyst Stability
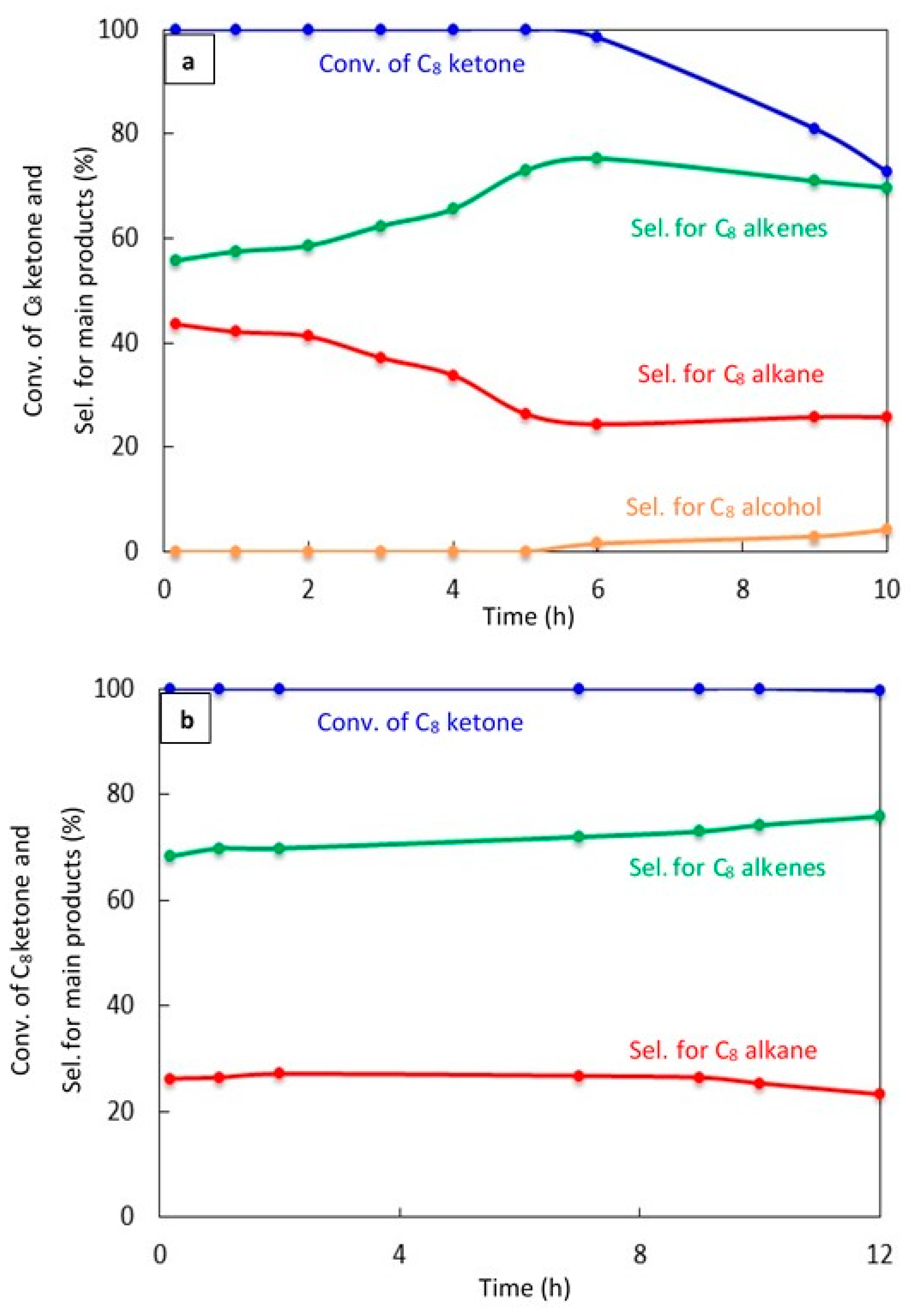
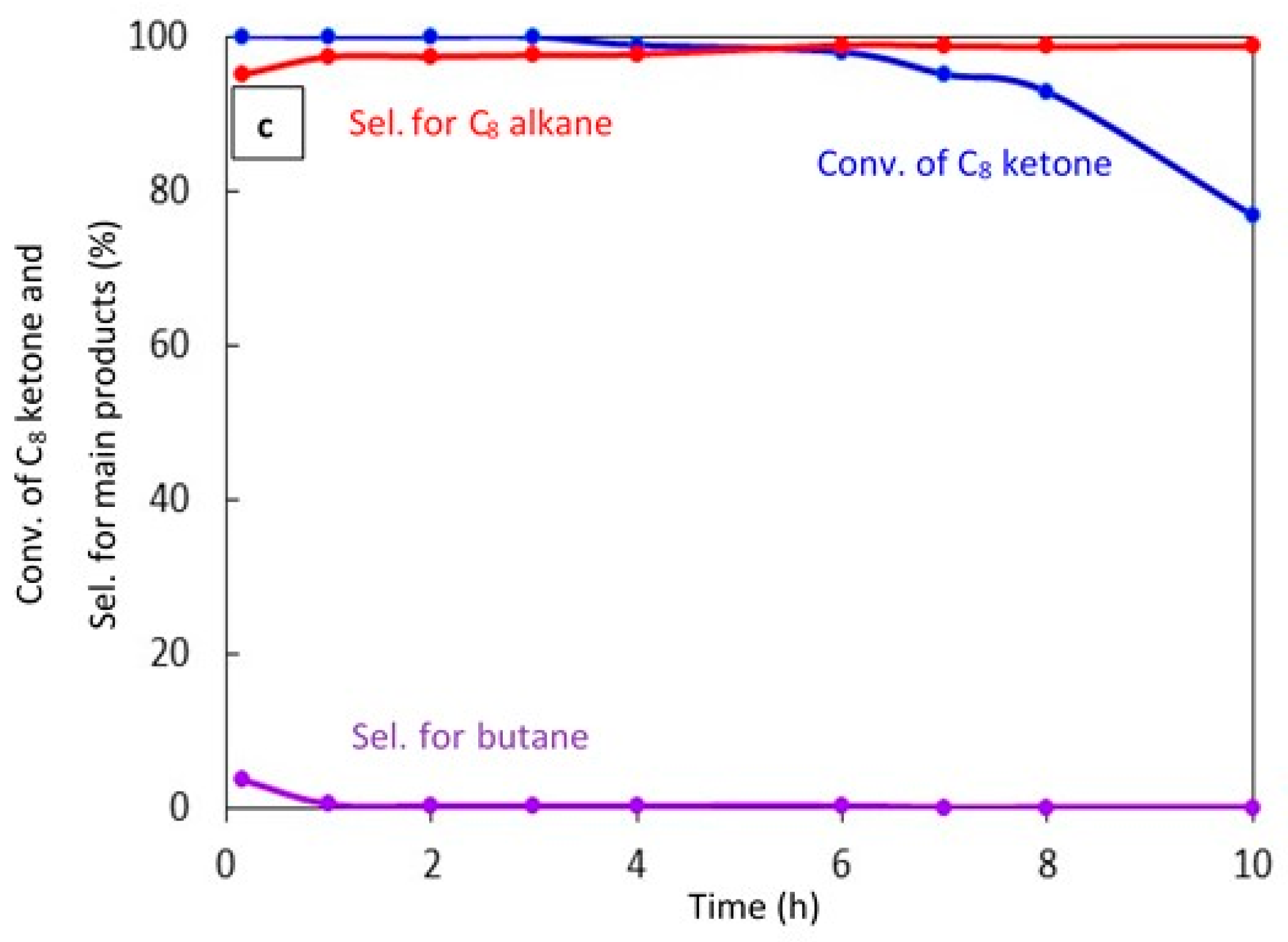
The catalyst stability was studied for 20% Cu–Al2O3 with different calcination times (4 h and 8 h) and for 1% Pt–Al2O3 calcined for 4 h, as shown in Figure 7a–c, respectively.
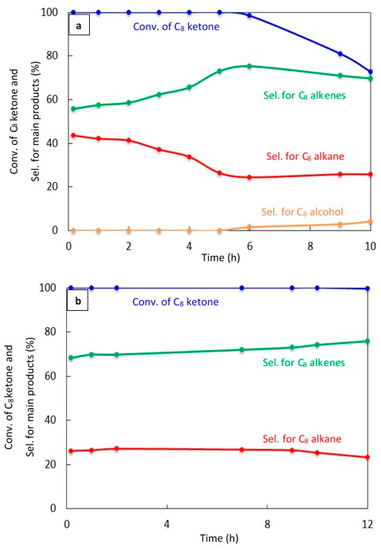
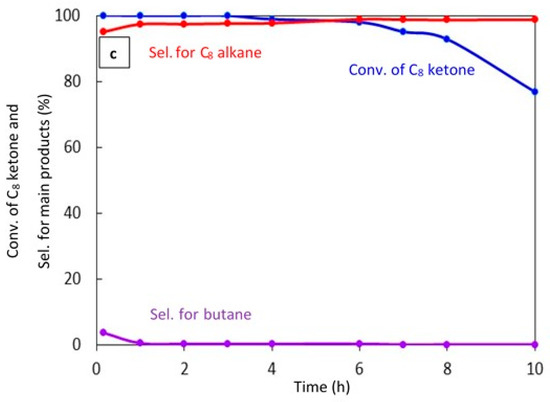
Figure 7.
Catalytic results for the conversion of the C8 ketone to several products with time using 0.5 g of catalysts: (a) (20% Cu–Al2O3), calcination time (4 h); (b) (20% Cu–Al2O3), calcination time (8 h); (c) (1% Pt–Al2O3), calcination time (4 h). Reaction conditions: reaction temperature, 220 °C; the total flow rate of H2 and N2, 85 mL/min; the flow rate of C8 ketone, 1 mL/h; the molar ratio of H2/C8 ketone, 10.
Figure 7a shows a significant deactivation for 20% Cu–Al2O3 calcined for 4 h. The conversion of C8 ketone was 100% during the first 5 h, but decreased to 73% after 10 h. C8 alkene selectivity increased with time to reach above 75% at 6 h, then decreased slightly with time to 70% after 10 h, which was associated with an increase in the production of C8 alcohol, an intermediate product. In addition, the selectivity for the C8 alkane decreased with time.
Figure 7b displays the conversion of C8 ketone and the distribution of products during 12 h of reaction over 20% Cu–Al2O3 calcined for 8 h. As can be seen, the conversion was maintained at near 100% for nearly the entire time studied, with a slight decrease to 99.5% at 12 h. The selectivity for C8 alkenes increased with time from 68% at the start of the reaction to 76% after 12 h, while the selectivity for the C8 alkane decreased from 26% to 23% with time. From the results in Figure 7a,b, it appears that longer calcination times led to more stable catalysts. This may be because the concentration of acidic sites was lower on 20% Cu–Al2O3 with longer calcination times than with short calcination times (see Table 1, NH3 uptake results). A larger number of acidic sites would likely increase the rate of deactivation [45].
Figure 7c shows the conversion of the C8 ketone and the distribution of major products during 10 h of reaction over 1% Pt–Al2O3. It is clear that the conversion of the C8 ketone was 99.8% during the first 3 h, then decreased at 4 h from 98.8% to 77% at 10 h. However, the selectivity for C8 alkane as the main product was approximately constant at over 97%, and small amounts of butane were detected during the first 10 min of the reaction. In addition, an experiment was conducted with a high H2/C8 ketone ratio (25) (data not shown). The results showed that both the conversion of the C8 ketone and the selectivity for the C8 alkane were approximately constant at above 99% and 97%, respectively, over 24 h. This suggests that hydrogen helps to maintain the activity of the catalyst, most likely by preventing coke formation.
3. Materials and Methods
3.1. Materials
5-methyl-3-heptanone was purchased from Tokyo Chemical Industry (TCI). The copper precursor (copper(II) nitrate tri-hydrate [Cu(NO3)2·3H2O (99%)]) was obtained from Fisher Scientific, while the Pt precursor (tetraammineplatinum(II) nitrate [Pt(NH3)4(NO3)2]) and aluminum oxide (catalyst support, high surface area, 1/8” pellet) were obtained from Alfa Aesar (Haverhill, MA, USA).
3.2. Preparation of Supported Catalysts
Both catalysts (Cu–Al2O3 and Pt–Al2O3) were synthesized using the incipient wetness impregnation method, and the metal precursors were Cu(NO3)2·3H2O (99%) and Pt(NH3)4(NO3)2, respectively. Prior to loading the metal precursor, the support (Al2O3) was crushed and sieved to obtain particles ≤ 0.15 mm in size (mesh 100). The precursor solution of a metal was prepared through dissolving the metal precursor in an amount of water just sufficient to fill the pores of the support by means of dropwise addition with manual mixing. Typically, 5 g of Al2O3 was impregnated in 5 mL of metal precursor solution containing the calculated amount of Cu(NO3)2·3H2O and Pt(NH3)4(NO3)2, respectively, to give approximately 1 wt% Cu–Al2O3, 20 wt% Cu–Al2O3, and 1 wt% Pt–Al2O3. The volume of precursor solution needed to fill these pores was determined by adding small quantities of the solvent slowly to the weighed amount of support with stirring until the mixture became slightly liquid. After that, this ratio of weight:volume was used to prepare a metal precursor solution with a suitable concentration to achieve the desired metal loading [46]. The catalyst was dried in an oven overnight at 100 °C, heated in a furnace at 110 °C for 2 h, and calcined at 550 °C for 4 h. A ramp rate of 2 °C/min was used for temperatures up to 110 °C and 1 °C/min up to 550 °C. The resulting catalyst was crushed and sieved to <0.15 mm for Pt–Al2O3 and to ≤0.18 mm for Cu–Al2O3.
3.3. Catalytic Reaction
Catalysts prepared as described above were used for the conversion of the C8 ketone to C8 alkenes and the C8 alkane. The catalytic conversion of the C8 ketone was performed in a fixed-bed reactor constructed of stainless steel (id = 0.85 cm) at atmospheric pressure. Prior to the reaction, 0.5 g of catalyst was reduced in the reactor at 300 °C for 1 h with flow rates of hydrogen and nitrogen equal to 68.5 and 16.5 mL/min, respectively. After this reduction process, the C8 ketone was mixed with nitrogen and hydrogen in a preheater at the desired reaction temperature before flowing into the reactor. The C8 ketone was fed at a feed rate of 1 (mL/h) via a micro pump (Eldex 1SMP) along with H2 and N2 with flow rates of 68.5 and 16.5 mL/min, respectively. The molar ratio of the H2/C8 ketone was maintained at around 25, and the temperature of the reaction was kept at 220 °C using heating tape as a heating source. The product compositions were analyzed using an on-line gas chromatograph (SRI 8610C) with an MXT-1 column (100% dimethyl polysiloxane (nonpolar phase), 60 m, ID 0.53 mm) equipped with Flame Ionization Detector (FID) and Thermal conductivity detector (TCD). The temperature of the effluents from the reactor was maintained above 230 °C to avoid condensation of the liquid products. The gas chromatograph oven was maintained at 40 °C for 5 min, and then increased to 120 °C at a ramp rate of 40 °C/min, before being finally raised to 250 °C at a rate of 20 °C/min and kept at this temperature for 10 min.
An Agilent 7890A GC-MS equipped with an Agilent 5975C MS detector was utilized to identify the products detected by the Gas Chromatography (GC). The conversion of MEK and the selectivity for the products were determined using Equations (2) and (3):
Repeat trials showed that the conversions and selectivities were repeatable within 5%.
Two different C8 alkenes were detected (5-methyl-3-heptene and 5-methyl-2-heptene), while a single C8 alkane (3-methyl heptane) was detected in the product. For brevity, these will be referred to as C8 alkenes and C8 alkane in the rest of the paper.
3.4. Catalyst Characterization
3.4.1. X-Ray Diffraction (XRD)
X-ray diffraction data were obtained with a Rigaku Miniflex II desktop x-ray diffractometer (Tokyo, Japan) with Cu Kα radiation (λ = 0.15406 nm) operated at 30 kV and 15 mA. A range of two theta angles from 6° to 70° were used for all catalysts with a step size of 0.02° and scan speed of 2°/min.
3.4.2. Brunauer–Emmett–Teller (BET)
Surface areas of all catalysts were found by the Brunauer–Emmett–Teller (BET) gas (nitrogen) adsorption method. N2 adsorption/desorption analyses were run on a Quantachrome Autosorb-1 instrument (Boynton Beach, FL, USA) at −196 °C. Data were analyzed with Autosorb-1 software. The samples were degassed at 350 °C for 4 h before beginning the adsorption analysis. The BET method was used to calculate the total surface area at relative pressures from 0.007 to 0.03.
3.4.3. Transmission Electron Microscopy (TEM)
Regarding transmission electron microscopy (TEM) imaging, a small amount of the supported catalysts was dispersed in ethanol with a 30 min sonication. A drop of the homogeneous suspension was deposited on a lacey carbon TEM grid, then tested by TEM using an FEI Talos TEM at 160 and 200 kV.
3.4.4. Temperature Programmed Desorption (NH3–TPD and CO2–TPD)
Temperature programmed desorption of ammonia (NH3–TPD) and carbon dioxide (CO2–TPD) were performed to investigate the surface acidity and basicity of the catalysts. An Altamira AMI-200 system was used for all TPD studies. Before adsorption, 0.1 g of supported metal catalyst was placed in a quartz U-tube reactor and pre-treated at 550 °C under helium for 1 h, followed by cooling to 100 °C. After that, the catalyst was reduced by passing H2/Ar (10 v/v %) with a flow of 40 mL/min and heating up to 300 °C at a ramp rate of 10 °C/min, held at 300 °C for 2 h, and finally cooled to 100 °C. Next, 1% NH3/He at a flow rate of 50 mL/min was introduced at 100 °C for 1 h. The sample was flushed with He at 100 °C for 2 h to remove physically adsorbed NH3 molecules. In the end, the temperature was raised to 600 °C at a ramp rate of 10 °C/min. For CO2–TPD experiments, 0.1 g of the sample was pretreated in the same as that for NH3–TPD except that the cooling temperature after reducing the catalyst was 50 °C. After reducing, the sample was treated with 10% CO2 in helium at a flow of 50 mL/min at 50 °C. The sample was flushed with He at 50 °C for 2 h to remove physisorbed CO2 molecules. Finally, the temperature was raised at a ramp rate of 10 °C/min to 600 °C. Any NH3 and CO2 desorbed from the samples were detected by a thermal conductivity detector (TCD).
4. Conclusions
Hydrodeoxygenation of the C8 ketone (5-methyl-3-heptanone) to C8 alkenes (5-methyl-3-heptene and 5-methyl-2-heptene) and C8 alkane (3-methyl heptane) was investigated using bifunctional catalysts composed of one of the transition metals (Cu or Pt) loaded on Al2O3 to provide both hydrogenation and dehydration sites. Over 20% Cu–Al2O3, a mixture of C8 alkenes and a C8 alkane was produced, while 1% Pt–Al2O3 produced nearly all of the C8 alkane because Pt was a more active hydrogenation catalyst for the C8 alkene. The ratio of the H2/C8 ketone had a significant impact on the catalytic chemistry. Higher ratios led to a higher production of the C8 alkane and higher C8 ketone conversions. Higher reaction temperatures also enhanced C8 ketone conversion and selectivity for the C8 alkane. These results are consistent with a pathway consisting of a series of reactions where the C8 ketone is hydrogenated to a C8 alcohol on metal sites, dehydrated to C8 alkenes on acidic sites, and further hydrogenated to a C8 alkane. Catalyst deactivation over 10 h was noted for the Cu catalyst calcined for a relatively short time and the Pt catalyst, but little deactivation was noted for the Cu catalyst calcined for a longer period of time.
Author Contributions
Conceptualization, Z.A.-A.; Formal analysis, Z.A.-A., H.A.-A., X.L., P.T. and K.H.; Investigation, H.A.-A., X.L. and P.T.; Supervision, K.H.; Writing—original draft, Z.A.-A.; Writing—review and editing, K.H.
Funding
This research was funded by [the Higher Committee for Education Development in Iraq (HCED)], and [the Navy SBIR Phase II] [Contr #NG8335], in collaboration with [TekHolding Inc.].
Conflicts of Interest
The authors declare no conflict of interest.
References
- Yu, E.K.; Saddler, J.N. Fed-batch approach to production of 2,3-butanediol by Klebsiella pneumoniae grown on high substrate concentrations. Appl. Environ. Microbiol. 1983, 46, 630–635. [Google Scholar] [PubMed]
- Zeng, A.P.; Biebl, H.; Deckwer, W.D. Effect of pH and acetic acid on growth and 2,3-butanediol production of Enterobacter aerogenes in continuous culture. Appl. Microbiol. Biotechnol. 1990, 33, 485–489. [Google Scholar] [CrossRef]
- De Mas, C.; Jansen, N.B.; Tsao, G.T. Production of optically active 2,3-butanediol by Bacillus polymyxa. Biotechnol. Bioeng. 1988, 31, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Ledingham, G.A.; Neish, A.C. Fermentative Production of 2,3-Butanediol. In Industrial Fermentations; Underkofler, L.A., Hickey, R.J., Eds.; Chemical Publishing Co: New York, NY, USA, 1954; Volume 2, pp. 27–93. [Google Scholar]
- Jansen, N.B.; Flickinger, M.C.; Tsao, G.T. Production of 2,3-butanediol from D-xylose by Klebsiella oxytoca ATCC 8724. Biotechnol. Bioeng. 1984, 26, 362–369. [Google Scholar] [CrossRef]
- Qureshi, N.; Cheryan, M. Effects of aeration on 2,3-butanediol production from glucose by Klebsiella oxytoca. J. Ferment. Bioeng. 1989, 67, 415–418. [Google Scholar] [CrossRef]
- Ji, X.J.; Huang, H.; Du, J.; Zhu, J.G.; Ren, L.J.; Hu, N.; Li, S. Enhanced 2,3-butanediol production by Klebsiella oxytoca using a two-stage agitation speed control strategy. Bioresour. Technol. 2009, 100, 3410–3414. [Google Scholar] [CrossRef]
- Saha, B.C.; Bothast, R.J. Production of 2,3-butanediol by newly isolated Enterobacter cloacae. Appl. Microbiol. Biotechnol. 1999, 52, 321–326. [Google Scholar] [CrossRef]
- Molnár, Á.; Bucsi, I.; Bartók, M. Pinacol Rearrangement on Zeolites. Stud. Surf. Sci. Catal. 1988, 41, 203–210. [Google Scholar]
- Multer, A.; McGraw, N.; Hohn, K.; Vadlani, P. Production of methyl ethyl ketone from biomass using a hybrid biochemical/catalytic approach. Ind. Eng. Chem. Res. 2012, 52, 56–60. [Google Scholar] [CrossRef]
- Emerson, R.R.; Flickinger, M.C.; Tsao, G.T. Kinetics of dehydration of aqueous 2,3-butanediol to methyl ethyl ketone. Ind. Eng. Chem. Prod. Res. Dev. 1982, 21, 473–477. [Google Scholar] [CrossRef]
- Zhang, W.; Yu, D.; Ji, X.; Huang, H. Efficient dehydration of bio-based 2,3-butanediol to butanone over boric acid modified HZSM-5 zeolites. Green Chem. 2012, 14, 3441–3450. [Google Scholar] [CrossRef]
- Bucsi, I.; Molnár, Á.; Bartók, M.; Olah, G.A. Transformation of 1,2-diols over perfluorinated resinsulfonic acids (Nafion-H). Tetrahedron 1994, 50, 8195–8202. [Google Scholar] [CrossRef]
- Török, B.; Bucsi, I.; Beregszászi, T.; Kapocsi, I.; Molnár, Á. Transformation of diols in the presence of heteropoly acids under homogeneous and heterogeneous conditions. J. Mol. Catal. A Chem. 1996, 107, 305–311. [Google Scholar] [CrossRef]
- Nikitina, M.A.; Ivanova, I.I. Conversion of 2,3-Butanediol over Phosphate Catalysts. ChemCatChem 2016, 8, 1346–1353. [Google Scholar] [CrossRef]
- Lee, J.; Grutzner, J.B.; Walters, W.E.; Delgass, W.N. The conversion of 2,3-butanediol to methyl ethyl ketone over zeolites. Stud. Surf. Sci. Catal. 2000, 130, 2603–2608. [Google Scholar]
- Kelly, G.J. Aldol Condensation Reaction and Catalyst Therefor. U.S. Patent 6,706,928, 16 March 2004. [Google Scholar]
- Zheng, Q.; Wales, M.D.; Heidlage, M.G.; Rezac, M.; Wang, H.; Bossmann, S.H.; Hohn, K.L. Conversion of 2,3-butanediol to butenes over bifunctional catalysts in a single reactor. J. Catal. 2015, 330, 222–237. [Google Scholar] [CrossRef]
- Zheng, Q.; Grossardt, J.; Almkhelfe, H.; Xu, J.; Grady, B.P.; Douglas, J.T.; Amama, P.B.; Hohn, K.L. Study of mesoporous catalysts for conversion of 2,3-butanediol to butenes. J. Catal. 2017, 354, 182–196. [Google Scholar] [CrossRef]
- Al-Auda, Z.; Al-Atabi, H.; Li, X.; Zheng, Q.; Hohn, K.L. Conversion of methyl ethyl ketone to butenes over bifunctional catalysts. Appl. Catal. A Gen. 2019, 570, 173–182. [Google Scholar] [CrossRef]
- Al-Auda, Z.; Al-Atabi, H.; Hohn, K. Metals on ZrO2: Catalysts for the Aldol Condensation of Methyl Ethyl Ketone (MEK) to C8 Ketones. Catalysts 2018, 8, 622. [Google Scholar] [CrossRef]
- Brands, D.S.; Poels, E.K.; Bliek, A. Ester hydrogenolysis over promoted Cu/SiO2 catalysts. Appl. Catal. A Gen. 1999, 184, 279–289. [Google Scholar] [CrossRef]
- Fan, X.; Liu, S.; Yan, X.; Du, X.; Chen, L. A continuous process for the production of 2,2,6,6-tetramethylpiperidin-4-ol catalyzed by Cu–Cr/γ-Al2O3. Catal. Commun. 2010, 11, 960–963. [Google Scholar] [CrossRef]
- Saadi, A.; Rassoul, Z.; Bettahar, M.M. Gas phase hydrogenation of benzaldehyde over supported copper catalysts. J. Mol. Catal. A Chem. 2000, 164, 205–216. [Google Scholar] [CrossRef]
- Nagaraja, B.M.; Kumar, V.S.; Shasikala, V.; Padmasri, A.H.; Sreedhar, B.; Raju, B.D.; Rao, K.S.R. A highly efficient Cu/MgO catalyst for vapour phase hydrogenation of furfural to furfuryl alcohol. Catal. Commun. 2003, 4, 287–293. [Google Scholar] [CrossRef]
- Chambers, A.; Jackson, S.D.; Stirling, D.; Webb, G. Selective hydrogenation of cinnamaldehyde over supported copper catalysts. J. Catal. 1997, 168, 301–314. [Google Scholar] [CrossRef]
- Poondi, D.; Vannice, M.A. The influence of MSI (metal-support interactions) on phenylacetaldehyde hydrogenation over Pt catalysts. J. Mol. Catal. A Chem. 1997, 124, 79–89. [Google Scholar] [CrossRef]
- Vannice, M.A.; Sen, B. Metal-support effects on the intramolecular selectivity of crotonaldehyde hydrogenation over platinum. J. Catal. 1989, 115, 65–78. [Google Scholar] [CrossRef]
- Vannice, M.A.; Poondi, D. The effect of metal-support interactions on the hydrogenation of benzaldehyde and benzyl alcohol. J. Catal. 1997, 169, 166–175. [Google Scholar] [CrossRef]
- Kijeński, J.; Winiarek, P.; Paryjczak, T.; Lewicki, A.; Mikołajska, A. Platinum deposited on monolayer supports in selective hydrogenation of furfural to furfuryl alcohol. Appl. Catal. A Gen. 2002, 233, 171–182. [Google Scholar] [CrossRef]
- Shi, B.C.; Davis, B.H. Alcohol dehydration: Mechanism of ether formation using an alumina catalyst. J. Catal. 1995, 157, 359–367. [Google Scholar] [CrossRef]
- Kostestkyy, P.; Yu, J.; Gorte, R.J.; Mpourmpakis, G. Structure–activity relationships on metal-oxides: Alcohol dehydration. Catal. Sci. Technol. 2014, 4, 3861–3869. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Liu, S.; Tamura, M.; Tomishige, K. Catalytic total hydrodeoxygenation of biomass-derived polyfunctionalized substrates to alkanes. ChemSusChem 2015, 8, 1114–1132. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Liu, S.; Kong, X.; Fan, X.; Yan, X.; Chen, L. A continuous process for the reductive deoxygenation of aromatic ketones over Cu30Cr10/γ-Al2O3. Res. Chem. Intermed. 2012, 38, 1341–1349. [Google Scholar] [CrossRef]
- Alotaibi, M.A.; Kozhevnikova, E.F.; Kozhevnikov, I.V. Efficient hydrodeoxygenation of biomass-derived ketones over bifunctional Pt-polyoxometalate catalyst. Chem. Commun. 2012, 48, 7194–7196. [Google Scholar] [CrossRef] [PubMed]
- Alharbi, K.; Kozhevnikova, E.F.; Kozhevnikov, I.V. Hydrogenation of ketones over bifunctional Pt-heteropoly acid catalyst in the gas phase. Appl. Catal. A Gen. 2015, 504, 457–462. [Google Scholar] [CrossRef]
- Yang, J.; Li, N.; Li, G.; Wang, W.; Wang, A.; Wang, X.; Cong, Y.; Zhang, T. Synthesis of renewable high-density fuels using cyclopentanone derived from lignocellulose. Chem. Commun. 2014, 50, 2572–2574. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Zhu, H.; Niu, L.; Xiao, G.; Xiao, R. Catalytic hydroprocessing of furfural to cyclopentanol over Ni/CNTs catalysts: Model reaction for upgrading of bio-oil. Catal. Lett. 2014, 144, 235–241. [Google Scholar] [CrossRef]
- Li, X.L.; Deng, J.; Shi, J.; Pan, T.; Yu, C.G.; Xu, H.J.; Fu, Y. Selective conversion of furfural to cyclopentanone or cyclopentanol using different preparation methods of Cu–Co catalysts. Green Chem. 2015, 17, 1038–1046. [Google Scholar] [CrossRef]
- Guo, J.; Xu, G.; Han, Z.; Zhang, Y.; Fu, Y.; Guo, Q. Selective conversion of furfural to cyclopentanone with CuZnAl catalysts. ACS Sustain. Chem. Eng. 2014, 2, 2259–2266. [Google Scholar] [CrossRef]
- Chary, K.V.R.; Sagar, G.V.; Srikanth, C.S.; Rao, V.V. Characterization and catalytic functionalities of copper oxide catalysts supported on zirconia. J. Phys. Chem. B. 2007, 111, 543–550. [Google Scholar] [CrossRef]
- Sagar, G.V.; Rao, P.V.R.; Srikanth, C.S.; Chary, K.V.R. Dispersion and Reactivity of Copper Catalysts Supported on Al2O3−ZrO2. J. Phys. Chem. B 2006, 110, 13881–13888. [Google Scholar] [CrossRef]
- Marella, R.K.; Koppadi, K.S.; Jyothi, Y.; Rao, K.S.R.; Burri, D.R. Selective gas-phase hydrogenation of benzonitrile into benzylamine over Cu–MgO catalysts without using any additives. New J. Chem. 2013, 37, 3229–3235. [Google Scholar] [CrossRef]
- Zheng, Q.; Xu, J.; Liu, B.; Hohn, K.L. Mechanistic study of the catalytic conversion of 2,3-butanediol to butenes. J. Catal. 2018, 360, 221–239. [Google Scholar] [CrossRef]
- Bartholomew, C.H. Mechanisms of catalyst deactivation. Appl. Catal. A Gen. 2001, 212, 17–60. [Google Scholar] [CrossRef]
- Augustine, R.L. Heterogeneous Catalysis for the Synthetic Chemist; CRC Press: Boca Raton, FL, USA, 1995. [Google Scholar]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).