Role of the Outer Metal of Double Metal Cyanides on the Catalytic Efficiency in Styrene Oxidation

Abstract
:
1. Introduction
2. Results and Discussion
2.1. Catalyst Characterization
2.1.1. Infrared (IR) Spectroscopy
2.1.2. Energy-Dispersed X-ray Spectrometry (EDS) and Hydration Degree
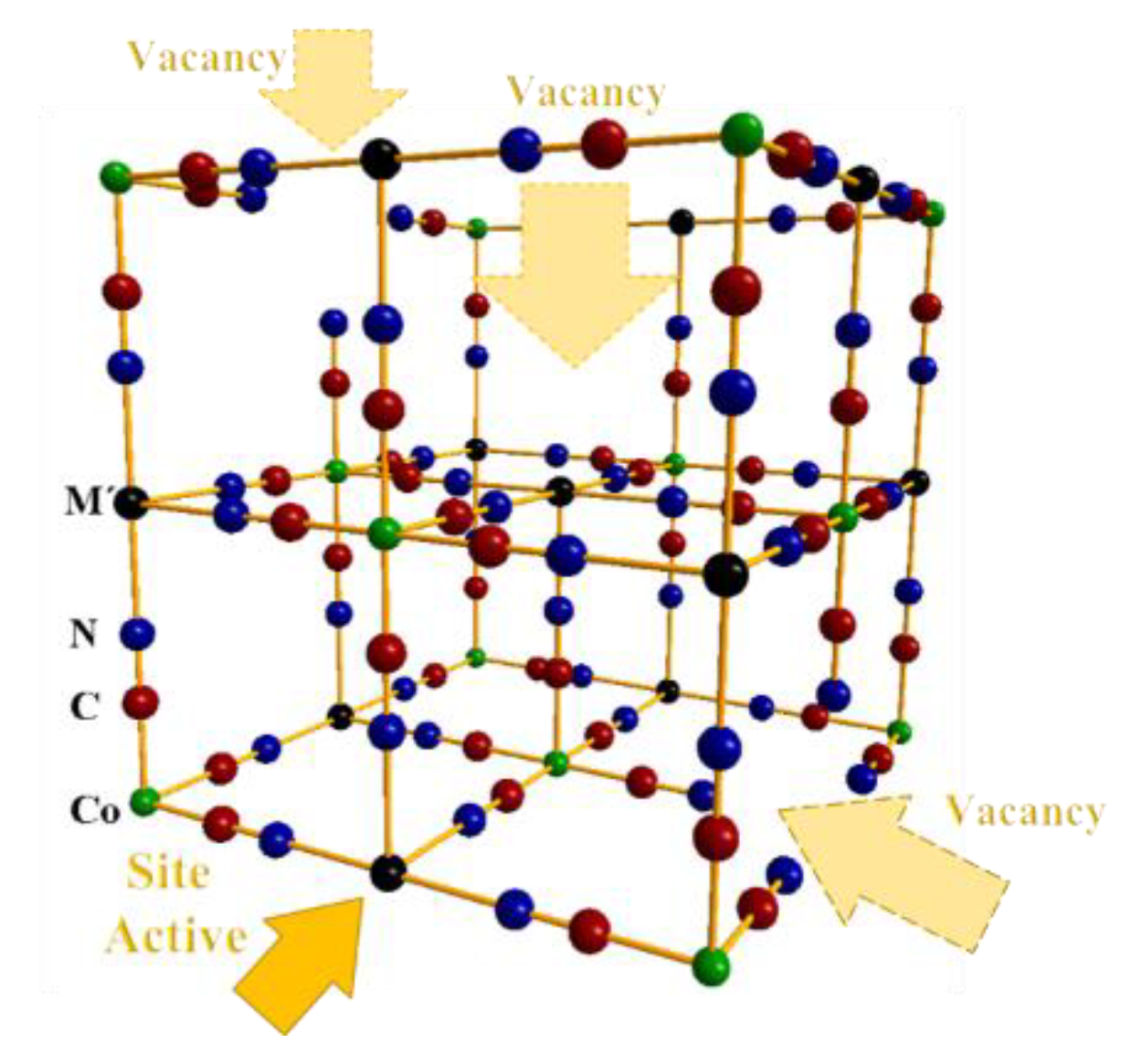
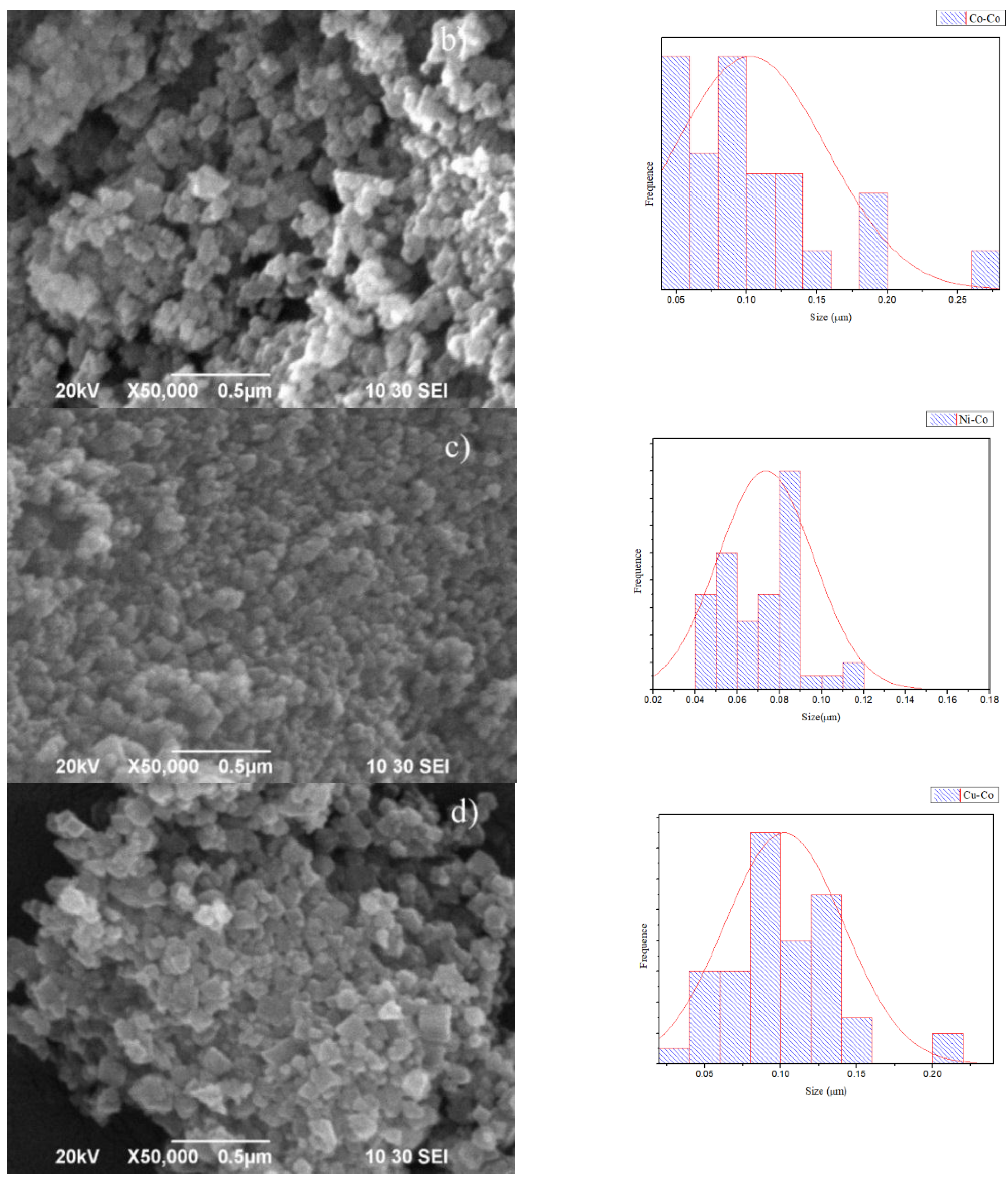
2.1.3. X-ray Diffraction (XRD) and Scanning Electron Microscopy (SEM) Images
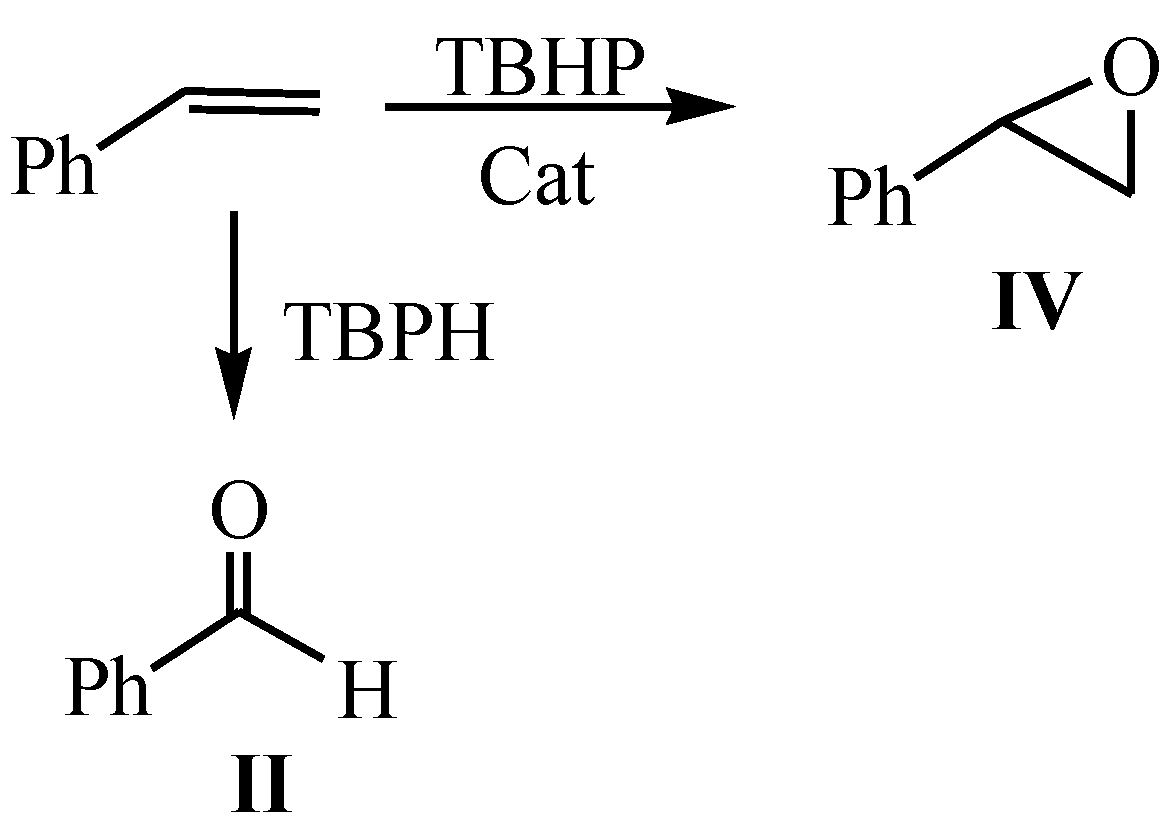
2.2. Catalytic Performance of DMC in the Styrene Oxidation and Proposed Mechanism
2.3. Impact of Outer Cation in DMC in Presence of TBHP
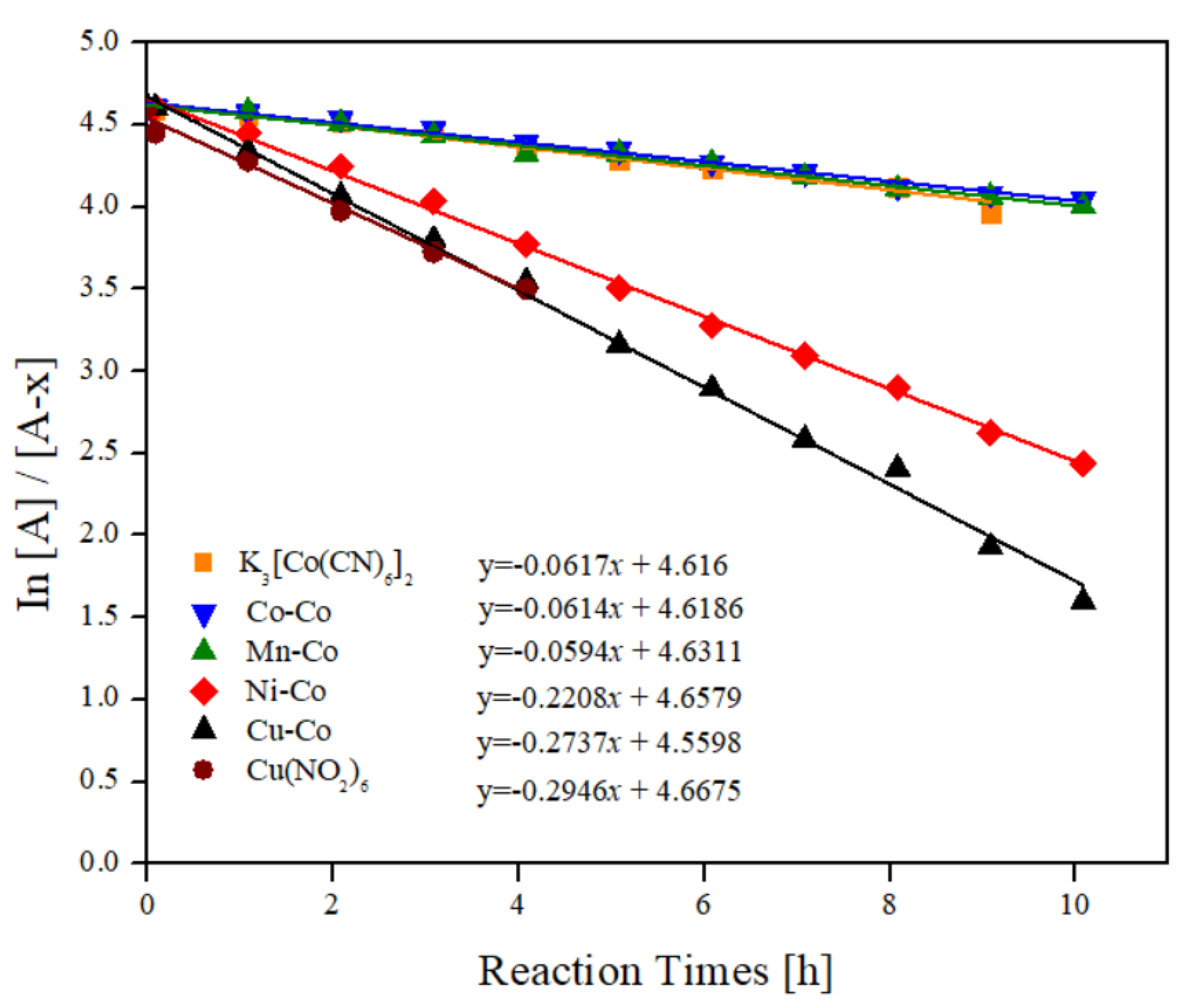
2.4. Kinetic Analysis and Effect of the Outer Cation in Catalysis
3. Experimental
3.1. Materials
3.2. Catalyst Preparation
3.3. Characterization Apparatus
3.4. Catalytic Reactions
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sato, O.; Iyoda, T.; Fujishima, A.; Hashimoto, K. Photoinduced Magnetization of a Cobalt-Iron Cyanide. Science 1996, 272, 704–705. [Google Scholar] [CrossRef] [PubMed]
- Shimamoto, N.; Ohkoshi, S.I.; Sato, O.; Hashimoto, K. Control of Charge-Transfer-Induced Spin Transition Temperature on Cobalt−Iron Prussian Blue Analogues. Inorg. Chem. 2002, 41, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Ohkoshi, S.-I.; Abe, Y.; Fujishima, A.; Hashimoto, K. Design and Preparation of a Novel Magnet Exhibiting Two Compensation Temperatures Based on Molecular Field Theory. Phys. Rev. Lett. 1999, 82, 1285–1288. [Google Scholar] [CrossRef]
- Yusuf, S.M.; Kumar, A.; Yakhmi, J.V. Temperature- and magnetic-field-controlled magnetic pole reversal in a molecular magnetic compound Temperature- and magnetic-field-controlled magnetic pole reversal. Appl. Phys. Lett. 2013, 182506, 11–14. [Google Scholar]
- Reguera, L.; Balmaseda, J.; Del Castillo, L.F.; Reguera, E. Hydrogen Storage in Porous Cyanometalates: Role of the Exchangeable Alkali Metal. J. Phys. Chem. C 2008, 112, 5589–5597. [Google Scholar] [CrossRef]
- Xie, X.; Ye, M.; Liu, C.; Hsu, P.-C.; Criddle, C.S.; Cui, Y. Use of low cost and easily regenerated Prussian Blue cathodes for efficient electrical energy recovery in a microbial battery. Energy Environ. Sci. 2015, 8, 546–551. [Google Scholar] [CrossRef]
- Chen, Y.-Z.; Zhang, R.; Jiao, L.; Jiang, H.-L. Metal–organic framework-derived porous materials for catalysis. Coord. Chem. Rev. 2018, 362, 1–23. [Google Scholar] [CrossRef]
- Zhang, A.; Li, L.; Li, J.; Zhang, Y.; Gao, S. Epoxidation of olefins with O2 and isobutyraldehyde catalyzed by cobalt (II)-containing zeolitic imidazolate framework material. Catal. Commun. 2011, 12, 1183–1187. [Google Scholar] [CrossRef]
- Saikia, L.; Satyarthi, J.K.; Gonnade, R.; Srinivas, D.; Ratnasamy, P. Double Metal Cyanides as Efficient Solid Acid Catalysts for Synthesis of β-Amino Alcohols Under Solvent-Free Conditions. Catal. Lett. 2008, 123, 24–31. [Google Scholar] [CrossRef]
- Sebastian, J.; Darbha, S. Structure-induced catalytic activity of Co–Zn double-metal cyanide complexes for terpolymerization of propylene oxide, cyclohexene oxide and CO2. RSC Adv. 2015, 5, 18196–18203. [Google Scholar] [CrossRef]
- Balmaseda, J.; Reguera, E.; Gomez, A.; Roque, J.; Vazquez, C.; Autie, M. On the Microporous Nature of Transition Metal Nitroprussides. J. Phys. Chem. B 2003, 107, 11360–11369. [Google Scholar] [CrossRef]
- Paolella, A.; Faure, C.; Timoshevskii, V.; Marras, S.; Bertoni, G.; Guerfi, A.; Vijh, A.; Armand, M.; Zaghib, K. A review on hexacyanoferrate-based materials for energy storage and smart windows: challenges and perspectives. J. Mater. Chem. A 2017, 5, 18919–18932. [Google Scholar] [CrossRef]
- Ali, S.; Bansal, V.; Khan, A.; Jain, S.; Ansari, M. Growth of zinc hexacyanoferrate nanocubes and their potential as heterogeneous catalyst for solvent-free oxidation of benzyl alcohol. J. Mol. Catal. A: Chem. 2009, 303, 60–64. [Google Scholar] [CrossRef]
- Parrott, L.K.; Erasmus, E. Metal Hexacyanometallate Nanoparticles: Spectroscopic Investigation on the Influence of Oxidation State of Metals on Catalytic Activity. Catal. Lett. 2018, 148, 2008–2018. [Google Scholar] [CrossRef]
- Valvekens, P.; De Vos, D. Catalysts for Organic Reactions; Elsevier B.V.: Amsterdam, The Netherlands, 2016; Volume 2, no. Ii. [Google Scholar]
- Zhang, K.; Kang, S.; Yao, Z.-S.; Nakamura, K.; Yamamoto, T.; Einaga, Y.; Azuma, N.; Miyazaki, Y.; Nakano, M.; Kanegawa, S.; et al. Charge-Transfer Phase Transition of a Cyanide-Bridged FeII /FeIII Coordination Polymer. Angew. Chem. Int. Ed. 2016, 55, 6047–6050. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Seong, S.; Kim, H.W.; Kim, D.H.; Thakur, N.; Yusuf, S.M.; Kim, B.; Min, B.I.; Kim, Y.; Kim, J.-Y.; et al. Soft X-ray absorption spectroscopy study of the electronic structures of the MnFe Prussian blue analogs (Rbx Bay) Mn[3-(x+2y)]/2 [Fe(CN)6] H2O. Phys. Rev. B 2017, 96, 1–8. [Google Scholar] [CrossRef]
- Shirakawa, T.; Higashi, M.; Tomita, O.; Abe, R. Surface-modified metal sulfides as stable H2-evolving photocatalysts in Z-scheme water splitting with a [Fe(CN)6]3-/4-redox mediator under visible-light irradiation. Sustain. Energy Fuels 2017, 1, 1065–1073. [Google Scholar] [CrossRef]
- Liang, Y.; Yi, C.; Tricard, S.; Fang, J.; Zhao, J.; Shen, W. Prussian blue analogues as heterogeneous catalysts for epoxidation of styrene. RSC Adv. 2015, 5, 17993–17999. [Google Scholar] [CrossRef]
- Nakamoto, K.; Brisdon, A. Infrared and Raman Spectra of Inorganic and Coordination Compounds Part B, 6th ed.; Wiely: Hoboken, NJ, USA, 2009; pp. 110–122. [Google Scholar]
- Basolo, F.; Busch, R.H.; Johnson, R. Química de los compuestos de coordinación; Reverté: Barcelona, Spain, 1964. [Google Scholar]
- Irving, H.; Williams, R.J.P. The Stability of Transition-metal Complexes. J. Chem. Soc. 1949, 637, 3192. [Google Scholar] [CrossRef]
- Reguera, E.; Hernández, J.R.; Champi, A.; Duque, J.G.; Granado, E.; Rettori, C. Unique Coordination of Copper in Hexacyanometallates. Zeitschrift für Physikalische Chemie 2006, 220, 1609–1619. [Google Scholar] [CrossRef]
- Roque, J.; Reguera, E.; Balmaseda, J.; Rodríguez-Hernández, J.; Reguera, L.; Del Castillo, L.F. Porous hexacyanocobaltates(III): Role of the metal on the framework properties. Microporous Mesoporous Mater. 2007, 103, 57–71. [Google Scholar] [CrossRef]
- Krap, C.P.; Balmaseda, J.; Del Castillo, L.F.; Zamora, B.; Reguera, E. Hydrogen Storage in Prussian Blue Analogues: H2Interaction with the Metal Found at the Cavity Surface. Energy Fuels 2010, 24, 581–589. [Google Scholar] [CrossRef]
- Tong, J.; Li, W.; Bo, L.; Wang, H.; Hu, Y.; Zhang, Z.; Mahboob, A. Selective oxidation of styrene catalyzed by cerium-doped cobalt ferrite nanocrystals with greatly enhanced catalytic performance. J. Catal. 2016, 344, 474–481. [Google Scholar] [CrossRef]
- Yin, Z.; Sun, P. Palladium-Catalyzed Direct ortho-Acylation through an Oxidative Coupling of Acetanilides with Toluene Derivatives. J. Org. Chem. 2012, 77, 11339–11344. [Google Scholar] [CrossRef] [PubMed]
- Fraile, J.M.; García, N.; Mayoral, J.A.; Santomauro, F.G.; Guidotti, M. Multifunctional catalysis promoted by solvent effects: Ti-mcm41 for a one-pot, four-step, epoxidation-rearrangement-oxidation-decarboxylation reaction sequence on stilbenes and styrenes. ACS Catal. 2015, 5, 3552–3561. [Google Scholar] [CrossRef]
- Tan, J.; Zheng, T.; Yu, Y.; Xu, K. TBHP-promoted direct oxidation reaction of benzylic Csp3–H bonds to ketones. RSC Adv. 2017, 7, 15176–15180. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst a | Identification | Vibrations | |||
|---|---|---|---|---|---|
| M–Co | δ(OH) c cm−1 | δ(Co–CN) cm–1 | |||
| K3[Co(CN)6]* b | K–Co | 2127 | - | 564 | 416 |
| Mn3[Co(CN)6]2·11H2O | Mn–Co | 2159 | 1611 | 443 | - |
| Co3[Co(CN)6]2·13H2O | Co–Co | 2171 | 1608 | 453 | - |
| Ni3[Co(CN)6]2·H2O | Ni–Co | 2184 | 1610 | 466 | - |
| Cu3[Co(CN)6]2·9H2O | Cu–Co | 2187 | 1604 | 465 | - |
| Catalyst | Scale | Crystallite Size Å a | Adsorbate | Vp [25] (cm3/g) |
|---|---|---|---|---|
| Mn–Co | Bulk | 1087 ± 0.50453 | N2 | 0.448 ± 0.012 |
| CO2 | 0.281 ± 0.012 | |||
| Co–Co | Nanometer | 414 ± 0.0542 | N2 | 0.381 ± 0.002 |
| CO2 | 0.224 ± 0.002 | |||
| Ni–Co | Nanometer | 632 ± 0.02268 | N2 | - |
| CO2 | 0.181 ± 0.003 | |||
| Cu–Co | Nanometer | 392 ± 0.03907 | N2 | 0.234 ± 0.003 |
| CO2 | 0.202 ± 0.001 |
| Entry | Catalyst | Reaction Time (h) | Conversion (%) | Selectivity | Yield of SO (%) | |||
|---|---|---|---|---|---|---|---|---|
| Styrene | TBHP | SO | Bzh | Acid c | ||||
| 1 | Mn-Co a | 4 | 30.8 | 30.5 | 2.2 | 32.1 | 65.7 | 0.6 |
| 2 | Mn-Co a | 10 | 35.7 | 35.6 | 2.8 | 39.3 | 57.9 | 0.8 |
| 3 | Co-Co a | 10 | 35.6 | 35.3 | 2.5 | 33.9 | 63.6 | 0.8 |
| 4 | Ni-Co a | 10 | 42.3 | 51.2 | 10.2 | 49.9 | 39.9 | 4.2 |
| 5 | Cu-Co a | 10 | 43 | 71 | 36.3 | 47.9 | 15.8 | 20.5 |
| 6 | Mn-Co b | 10 | 45.3 | 30.5 | 26.6 | 73.4 | - | 12 |
| 7 | Co-Co b | 10 | 43 | 28.9 | 24.9 | 75.1 | - | 10.7 |
| 8 | Ni-Co b | 10 | 88.6 | 61 | 52.4 | 47.6 | - | 46.4 |
| 9 | Cu-Co b | 10 | 95.1 | 89.6 | 67.4 | 32.6 | - | 64 |
| 10 | K3[Co(CN)6] b | 10 | 44.1 | 32 | 21.1 | 78.8 | >1 | 10 |
| 11 | Cu(NO2)6· 6H2O b | 4 | 68.3 | 100 | 30.3 | 58.7 | 11 | 21.5 |
| 12 | Blank | 8 | 34 | 23.5 | - | 77.1 | 22.9 | 0.0 |
| Entry | Catalyst | Slope (k) | Intercept | R-Square |
|---|---|---|---|---|
| 1 | Mn–Co | 0.06 | 4.63 | 0.99 |
| 2 | Co–Co | 0.06 | 4.62 | 0.99 |
| 3 | Ni–Co | 0.22 | 4.66 | 0.99 |
| 4 | Cu–Co | 0.27 | 4.67 | 0.99 |
| 5 | K3[Co(CN)6] | 0.06 | 4.62 | 0.99 |
| 6 | Cu(NO2)6·6H2O | 0.29 | 4.56 | 0.99 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molina-Maldonado, P.; Ruíz-Guerrero, R.; Hernández-Fuentes, C. Role of the Outer Metal of Double Metal Cyanides on the Catalytic Efficiency in Styrene Oxidation. Catalysts 2019, 9, 905. https://doi.org/10.3390/catal9110905
Molina-Maldonado P, Ruíz-Guerrero R, Hernández-Fuentes C. Role of the Outer Metal of Double Metal Cyanides on the Catalytic Efficiency in Styrene Oxidation. Catalysts. 2019; 9(11):905. https://doi.org/10.3390/catal9110905
Chicago/Turabian StyleMolina-Maldonado, Paulina, Rosario Ruíz-Guerrero, and Carlos Hernández-Fuentes. 2019. "Role of the Outer Metal of Double Metal Cyanides on the Catalytic Efficiency in Styrene Oxidation" Catalysts 9, no. 11: 905. https://doi.org/10.3390/catal9110905
APA StyleMolina-Maldonado, P., Ruíz-Guerrero, R., & Hernández-Fuentes, C. (2019). Role of the Outer Metal of Double Metal Cyanides on the Catalytic Efficiency in Styrene Oxidation. Catalysts, 9(11), 905. https://doi.org/10.3390/catal9110905