N-Heterocyclic Carbene-Supported Aryl- and Alk- oxides of Beryllium and Magnesium

 , and
, and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
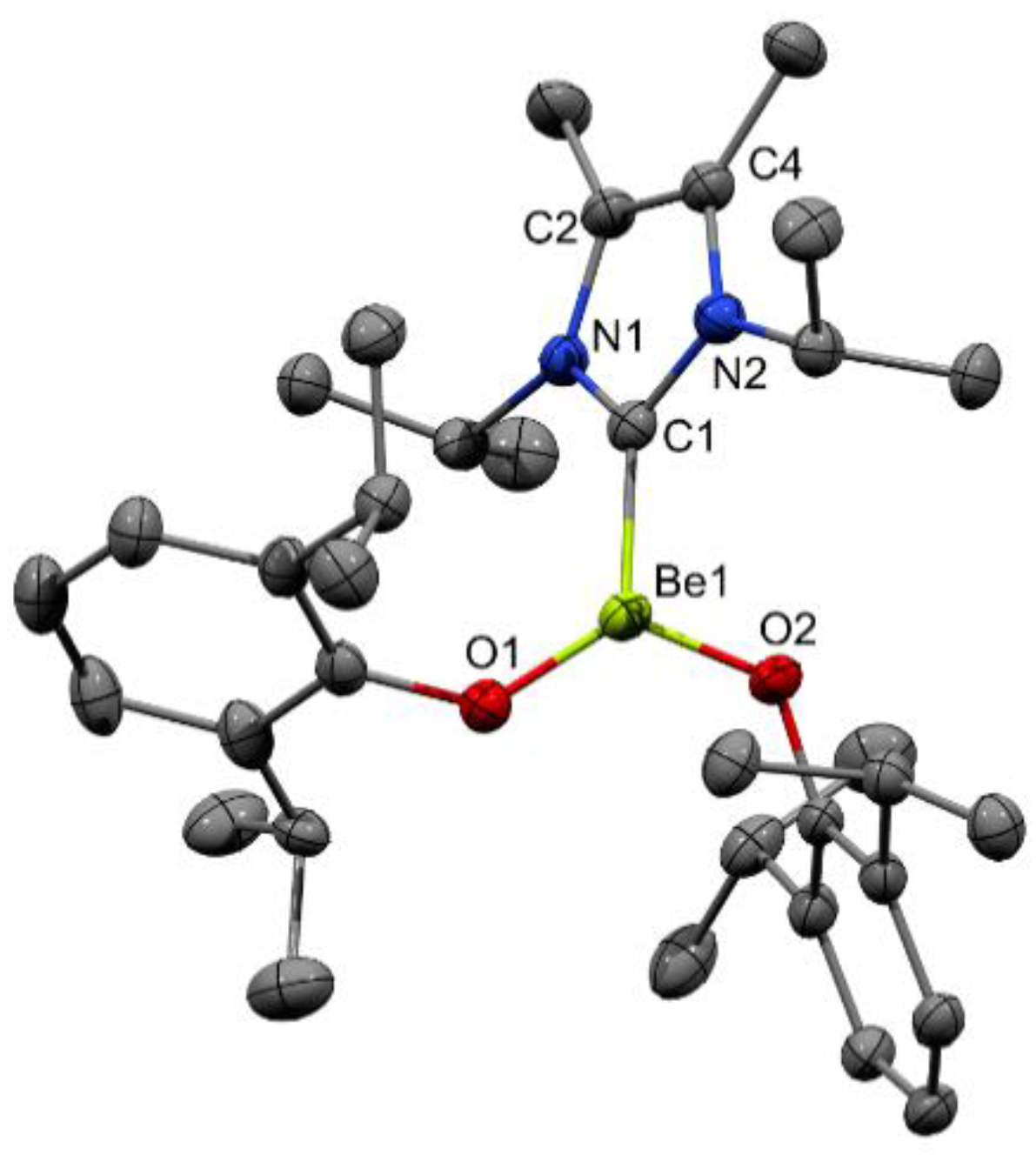
2. Results and Discussion
3. Materials and Methods
3.1. General Procedures
3.2. Synthesis of Lithium 2,6-Diisopropylphenolate
3.3. Synthesis of Compound 1
3.4. Synthesis of Compound 2
3.5. Synthesis of Compound 3
3.6. Synthesis of Compound 4
3.7. Synthesis of Compound 5
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mokkelbost, T.; Kaus, I.; Haugsrud, R.; Norby, T.; Grande, T.; Einarsrud, M.-A. High-Temperature Proton-Conducting Lanthanum Ortho-Niobate-Based Materials. Part II: Sintering Properties and Solubility of Alkaline Earth Oxides. J. Am. Chem. Soc. 2008, 91, 879–886. [Google Scholar] [CrossRef]
- Haynes, W.M. CRC Handbook of Chemistry and Physics: A Ready-Reference Book of Chemical and Physical Data: 2011–2012, 92nd ed.; CRC: Boca Raton, FL, USA, 2011. [Google Scholar]
- Diao, Y.; Walawender, W.P.; Sorensen, C.M.; Klabunde, K.J.; Ricker, T. Hydrolysis of Magnesium Methoxide. Effects of Toluene on Gel Structure and Gel Chemistry. Chem. Mater. 2002, 14, 362–368. [Google Scholar] [CrossRef]
- Perera, L.C.; Raymond, O.; Henderson, W.; Brothers, P.J.; Plieger, P.G. Advances in beryllium coordination chemistry. Coord. Chem. Rev. 2017, 352, 264–290. [Google Scholar] [CrossRef]
- Bell, N.A.; Shearer, H.M.M.; Twiss, J. Dimeric tert-butoxyberyllium bromide-diethyl ether adduct, [Be2Br2(C4H9O)2(C4H10O)2], and a comparison with the magnesium analogue. Acta Crystallogr. 1984, 40, 605–607. [Google Scholar] [CrossRef]
- Bell, N.A.; Coates, G.E.; Shearer, H.M.M.; Twiss, J. π-Bonding in three-co-ordinate beryllium compounds. Structure of tetra-µ-t-butoxydichlorotriberyllium. Chem. Commun. 1983, 15, 840–841. [Google Scholar] [CrossRef]
- Bell, N.A.; Coates, G.E.; Schneider, M.L.; Shearer, H.M.M. Terminal beryllium–hydrogen bonding. X-Ray crystal structure of the (2-dimethylamino-N-methylethylamido)hydridoberyllium dimer. Chem. Commun. 1983, 15, 828–829. [Google Scholar] [CrossRef]
- Naglav, D.; Tobey, B.; Wölper, C.; Bläser, D.; Jansen, G.; Schulz, S. On the Stability of Trimeric Beryllium Hydroxide Scorpionate Complexes. Eur. J. Inorg. Chem. 2016, 2016, 2424–2431. [Google Scholar] [CrossRef]
- Pietrzak, T.; Kubisiak, M.; Justyniak, I.; Zelga, K.; Bojarski, E.; Tratkiewicz, E.; Ochal, Z.; Lewiński, J. Oxygenation Chemistry of Magnesium Alkyls Incorporating β-Diketiminate Ligands Revisited. Chem. Eur. J. 2016, 22, 17776–17783. [Google Scholar] [CrossRef]
- Ruhlandt-Senge, K.; Bartlett, R.A.; Olmstead, M.M.; Power, P.P. Synthesis and structural characterization of the beryllium compounds [Be(2,4,6-Me3C6H2)2(OEt2)], [Be{O(2,4,6-tert-Bu3C6H2)}2(OEt2)], and [Be{S(2,4,6-tert-Bu3C6H2)}2(THF)].cntdot.PhMe and determination of the structure of [BeCl2(OEt2)2]. Inorg. Chem. 1993, 32, 1724–1728. [Google Scholar] [CrossRef]
- Arrowsmith, M.; Hill, M.S.; Kociok-Köhn, G.; MacDougall, D.J.; Mahon, M.F.; Mallov, I. Three-Coordinate Beryllium β-Diketiminates: Synthesis and Reduction Chemistry. Inorg. Chem. 2012, 51, 13408–13418. [Google Scholar] [CrossRef]
- Arrowsmith, M.; Crimmin, M.R.; Hill, M.S.; Kociok-Köhn, G. Beryllium derivatives of a phenyl-substituted β-diketiminate: A well-defined ring opening reaction of tetrahydrofuran. Dalton Trans. 2013, 42, 9720–9726. [Google Scholar] [CrossRef] [PubMed]
- Moseley, P.T.; Shearer, H.M.M. The crystal structure of the addition product of a Grignard reagent with a ketone. Chem. Commun. 1968, 5, 279–280. [Google Scholar] [CrossRef]
- Chisholm, M.H.; Huffman, J.C.; Phomphrai, K. Monomeric metal alkoxides and trialkyl siloxides: (BDI)Mg(OtBu)(THF) and (BDI)Zn(OSiPh3)(THF). Comments on single site catalysts for ring-opening polymerization of lactides. Dalton Trans. 2001, 3, 222–224. [Google Scholar] [CrossRef]
- Zechmann, C.A.; Boyle, T.J.; Rodriguez, M.A.; Kemp, R.A. Synthesis, characterization, and structural study of sterically hindered magnesium alkoxide and siloxide compounds. Inorg. Chim. Acta 2001, 319, 137–146. [Google Scholar] [CrossRef]
- Fedushkin, I.L.; Morozov, A.G.; Hummert, M.; Schumann, H. Alkylmagnesium Complexes with the Rigid dpp-bian Ligand {dpp-bian = 1,2-Bis[(2,6-diisopropylphenyl)imino]acenaphthene}. Eur. J. Inorg. Chem. 2008, 2008, 1584–1588. [Google Scholar] [CrossRef]
- Boutland, A.J.; Lamsfus, C.A.; Maitland, B.; Maron, L.; Stasch, A.; Jones, C. Accessing Stable Magnesium Acyl Compounds: Reductive Cleavage of Esters by Magnesium(I) Dimers. Chem. Eur. J. 2017, 23, 14049–14055. [Google Scholar] [CrossRef]
- Wilson, A.S.S.; Hill, M.S.; Mahon, M.F.; Dinoi, C.; Maron, L. Organocalcium-mediated nucleophilic alkylation of benzene. Science 2017, 358, 1168–1171. [Google Scholar] [CrossRef]
- Wilson, A.S.S.; Dinoi, C.; Hill, M.S.; Mahon, M.F.; Maron, L. Heterolysis of Dihydrogen by Nucleophilic Calcium Alkyls. Angew. Chem. Int. Ed. 2018, 57, 15500–15504. [Google Scholar] [CrossRef]
- Green, S.P.; Jones, C.; Stasch, A. Stable Magnesium(I) Compounds with Mg-Mg Bonds. Science 2007, 318, 1754–1757. [Google Scholar] [CrossRef]
- Jones, C. Dimeric magnesium(I) β-diketiminates: A new class of quasi-universal reducing agent. Nat. Chem. Rev. 2017, 1, 0059. [Google Scholar] [CrossRef]
- Bonyhady, S.J.; Collis, D.; Frenking, G.; Holzmann, N.; Jones, C.; Stasch, A. Synthesis of a stable adduct of dialane(4) (Al2H4) via hydrogenation of a magnesium(I) dimer. Nat. Chem. 2010, 2, 865. [Google Scholar] [CrossRef] [PubMed]
- Sidiropoulos, A.; Jones, C.; Stasch, A.; Klein, S.; Frenking, G. N-Heterocyclic Carbene Stabilized Digermanium(0). Angew. Chem. Int. Ed. 2009, 48, 9701–9704. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.; Sidiropoulos, A.; Holzmann, N.; Frenking, G.; Stasch, A. An N-heterocyclic carbene adduct of diatomic tin, Sn [double bond, length as m-dash] Sn. Chem. Commun. 2012, 48, 9855–9857. [Google Scholar] [CrossRef] [PubMed]
- Rit, A.; Campos, J.; Niu, H.; Aldridge, S. A stable heavier group 14 analogue of vinylidene. Nat. Chem. 2016, 8, 1022. [Google Scholar] [CrossRef]
- Arras, J.; Kruczyński, T.; Bresien, J.; Schulz, A.; Schnöckel, H. Magnesium(I) Halide versus Magnesium Metal: Differences in Reaction Energy and Reactivity Monitored in Reduction Processes of P−Cl Bonds. Angew. Chem. Int. Ed. 2019, 58, 716–721. [Google Scholar] [CrossRef] [PubMed]
- Bakewell, C.; White, A.J.P.; Crimmin, M.R. Addition of Carbon–Fluorine Bonds to a Mg(I)–Mg(I) Bond: An Equivalent of Grignard Formation in Solution. J. Am. Chem. Soc. 2016, 138, 12763–12766. [Google Scholar] [CrossRef] [PubMed]
- Stasch, A.; Jones, C. Stable dimeric magnesium(i) compounds: From chemical landmarks to versatile reagents. Dalton Trans. 2011, 40, 5659–5672. [Google Scholar] [CrossRef]
- Hill, M.S.; Liptrot, D.J.; Weetman, C. Alkaline earths as main group reagents in molecular catalysis. Chem. Soc. Rev. 2016, 45, 972–988. [Google Scholar] [CrossRef]
- Rosch, B.; Gentner, T.X.; Elsen, H.; Fischer, C.A.; Langer, J.; Wiesinger, M.; Harder, S. Nucleophilic Aromatic Substitution at Benzene with Powerful Strontium Hydride and Alkyl Complexes. Angew. Chem. Int. Ed. 2019, 58, 5396–5401. [Google Scholar] [CrossRef]
- de Bruin-Dickason, C.N.; Sutcliffe, T.; Alvarez Lamsfus, C.; Deacon, G.B.; Maron, L.; Jones, C. Kinetic stabilisation of a molecular strontium hydride complex using an extremely bulky amidinate ligand. Chem. Commun. 2018, 54, 786–789. [Google Scholar] [CrossRef]
- Causero, A.; Ballmann, G.; Pahl, J.; Zijlstra, H.; Färber, C.; Harder, S. Stabilization of Calcium Hydride Complexes by Fine Tuning of Amidinate Ligands. Organometallics 2016, 35, 3350–3360. [Google Scholar] [CrossRef]
- Pahl, J.; Brand, S.; Elsen, H.; Harder, S. Highly Lewis acidic cationic alkaline earth metal complexes. Chem. Commun. 2018, 54, 8685–8688. [Google Scholar] [CrossRef] [PubMed]
- Pahl, J.; Friedrich, A.; Elsen, H.; Harder, S. Cationic Magnesium π–Arene Complexes. Organometallics 2018, 37, 2901–2909. [Google Scholar] [CrossRef]
- Bayram, M.; Naglav, D.; Wölper, C.; Schulz, S. Syntheses and Structures of Homo- and Heteroleptic Beryllium Complexes Containing N,N′-Chelating Ligands. Organometallics 2017, 36, 467–473. [Google Scholar] [CrossRef]
- Nesterov, V.; Reiter, D.; Bag, P.; Frisch, P.; Holzner, R.; Porzelt, A.; Inoue, S. NHCs in Main Group Chemistry. Chem. Rev. 2018, 118, 9678–9842. [Google Scholar] [CrossRef] [PubMed]
- Bellemin-Laponnaz, S.; Dagorne, S. Group 1 and 2 and Early Transition Metal Complexes Bearing N-Heterocyclic Carbene Ligands: Coordination Chemistry, Reactivity, and Applications. Chem. Rev. 2014, 114, 8747–8774. [Google Scholar] [CrossRef]
- Arnold, P.L.; Casely, I.J.; Turner, Z.R.; Bellabarba, R.; Tooze, R.B. Magnesium and zinc complexes of functionalised, saturated N-heterocyclic carbene ligands: Carbene lability and functionalisation, and lactide polymerisation catalysis. Dalton Trans. 2009, 35, 7236–7247. [Google Scholar] [CrossRef]
- Zhang, D.; Kawaguchi, H. Deprotonation Attempts on Imidazolium Salt Tethered by Substituted Phenol and Construction of Its Magnesium Complex by Transmetalation. Organometallics 2006, 25, 5506–5509. [Google Scholar] [CrossRef]
- Drouin, F.; Whitehorne, T.J.J.; Schaper, F. Nacnac BnMgOtBu: A diketiminate-based catalyst for the polymerisation of rac-lactide with slight isotactic preference. Dalton Trans. 2011, 40, 1396–1400. [Google Scholar] [CrossRef]
- Chamberlain, B.M.; Cheng, M.; Moore, D.R.; Ovitt, T.M.; Lobkovsky, E.B.; Coates, G.W. Polymerization of Lactide with Zinc and Magnesium β-Diiminate Complexes: Stereocontrol and Mechanism. J. Am. Chem. Soc. 2001, 123, 3229–3238. [Google Scholar] [CrossRef]
- Dove, A.P.; Gibson, V.C.; Marshall, E.L.; White, A.J.P.; Williams, D.J. Magnesium and zinc complexes of a potentially tridentate β-diketiminate ligand. Dalton Trans. 2004, 4, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-Y.; Huang, B.-H.; Lin, C.-C. A Highly Efficient Initiator for the Ring-Opening Polymerization of Lactides and ε-Caprolactone: A Kinetic Study. Macromolecules 2005, 38, 5400–5405. [Google Scholar] [CrossRef]
- Chisholm, M.H.; Gallucci, J.C.; Phomphrai, K. Comparative Study of the Coordination Chemistry and Lactide Polymerization of Alkoxide and Amide Complexes of Zinc and Magnesium with a β-Diiminato Ligand Bearing Ether Substituents. Inorg. Chem. 2005, 44, 8004–8010. [Google Scholar] [CrossRef] [PubMed]
- Raghavendra, B.; Shashank, P.V.S.; Pandey, M.K.; Reddy, N.D. CO2/Epoxide Coupling and the ROP of ε-Caprolactone: Mg and Al Complexes of γ-Phosphino-ketiminates as Dual-Purpose Catalysts. Organometallics 2018, 37, 1656–1664. [Google Scholar] [CrossRef]
- Arrowsmith, M.; Hadlington, T.J.; Hill, M.S.; Kociok-Köhn, G. Magnesium-catalysed hydroboration of aldehydes and ketones. Chem. Commun. 2012, 48, 4567–4569. [Google Scholar] [CrossRef]
- Paparo, A.; Jones, C. Beryllium Halide Complexes Incorporating Neutral or Anionic Ligands: Potential Precursors for Beryllium Chemistry. Chem. Asian J. 2019, 14, 486–490. [Google Scholar] [CrossRef]
- Freeman, L.A.; Walley, J.E.; Obi, A.D.; Wang, G.; Dickie, D.A.; Molino, A.; Wilson, D.J.D.; Gilliard, R.J., Jr. Stepwise Reduction at Magnesium and Beryllium: Cooperative Effects of Carbenes with Redox Non-Innocent alpha-Diimines. Inorg. Chem. 2019, 58, 10554–10568. [Google Scholar] [CrossRef]
- Walley, J.E.; Obi, A.D.; Breiner, G.; Wang, G.; Dickie, D.A.; Molino, A.; Dutton, J.L.; Wilson, D.J.D.; Gilliard, R.J., Jr. Cyclic(alkyl)(amino) Carbene-Promoted Ring Expansion of a Carbodicarbene Beryllacycle. Inorg. Chem. 2019, 58, 11118–11126. [Google Scholar] [CrossRef]
- Schuster, J.K.; Roy, D.K.; Lenczyk, C.; Mies, J.; Braunschweig, H. New Outcomes of Beryllium Chemistry: Lewis Base Adducts for Salt Elimination Reactions. Inorg. Chem. 2019, 58, 2652–2658. [Google Scholar] [CrossRef]
- Arrowsmith, M.; Braunschweig, H.; Celik, M.A.; Dellermann, T.; Dewhurst, R.D.; Ewing, W.C.; Hammond, K.; Kramer, T.; Krummenacher, I.; Mies, J.; et al. Neutral zero-valent s-block complexes with strong multiple bonding. Nat. Chem. 2016, 8, 890. [Google Scholar] [CrossRef]
- Wang, G.; Freeman, L.A.; Dickie, D.A.; Mokrai, R.; Benkő, Z.; Gilliard, R.J., Jr. Isolation of Cyclic(Alkyl)(Amino) Carbene-Bismuthinidene Mediated by a Beryllium(0) Complex. Chem. Eur. J. 2019, 25, 4335–4339. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, W.A.; Runte, O.; Artus, G. Synthesis and structure of an ionic beryllium-“carbene” complex. J. Organomet. Chem. 1995, 501, C1–C4. [Google Scholar] [CrossRef]
- Gilliard, R.J.; Abraham, M.Y.; Wang, Y.; Wei, P.; Xie, Y.; Quillian, B.; Schaefer, H.F.; Schleyer, P.v.R.; Robinson, G.H. Carbene-Stabilized Beryllium Borohydride. J. Am. Chem. Soc. 2012, 134, 9953–9955. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, M.; Hill, M.S.; Kociok-Kohn, G.; MacDougall, D.J.; Mahon, M.F. Beryllium-induced C-N bond activation and ring opening of an N-heterocyclic carbene. Angew. Chem. Int. Ed. 2012, 51, 2098–2100. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, M.; Hill, M.S.; Kociok-Köhn, G. Activation of N-Heterocyclic Carbenes by {BeH2} and {Be(H)(Me)} Fragments. Organometallics 2015, 34, 653–662. [Google Scholar] [CrossRef]
- Walley, J.E.; Breiner, G.; Wang, G.; Dickie, D.A.; Molino, A.; Dutton, J.L.; Wilson, D.J.D.; Gilliard, R.J., Jr. s-Block carbodicarbene chemistry: C(sp(3))-H activation and cyclization mediated by a beryllium center. Chem. Commun. 2019, 55, 1967–1970. [Google Scholar] [CrossRef] [PubMed]
- Petz, W.; Dehnicke, K.; Holzmann, N.; Frenking, G.; Neumüller, B. The Reaction of BeCl2 with Carbodiphosphorane C(PPh3)2; Experimental and Theoretical Studies. Z. Anorg. Allg. Chem. 2011, 637, 1702–1710. [Google Scholar] [CrossRef]
- Buchner, M.R.; Spang, N.; Müller, M.; Rudel, S.S. Formation and Properties of the Trichloroberyllate Ion. Inorg. Chem. 2018, 57, 11314–11317. [Google Scholar] [CrossRef]
- Hopkinson, M.N.; Richter, C.; Schedler, M.; Glorius, F. An overview of N-heterocyclic carbenes. Nature 2014, 510, 485. [Google Scholar] [CrossRef]
- Arduengo, A.J.; Harlow, R.L.; Kline, M. A stable crystalline carbene. J. Am. Chem. Soc. 1991, 113, 361–363. [Google Scholar] [CrossRef]
- Wong, Y.O.; Freeman, L.A.; Agakidou, A.D.; Dickie, D.A.; Webster, C.E.; Gilliard, R.J. Two Carbenes versus One in Magnesium Chemistry: Synthesis of Terminal Dihalide, Dialkyl, and Grignard Reagents. Organometallics 2019, 38, 688–696. [Google Scholar] [CrossRef]
- Langer, J.; Krieck, S.; Fischer, R.; Görls, H.; Walther, D.; Westerhausen, M. 1,4-Dioxane Adducts of Grignard Reagents: Synthesis, Ether Fragmentation Reactions, and Structural Diversity of Grignard Reagent/1,4-Dioxane Complexes. Organometallics 2009, 28, 5814–5820. [Google Scholar] [CrossRef]
- Nöth, H.; Schlosser, D. The Aminolysis of Beryllium Dichloride with Diisopropylamine and Reactions of Some Aminoberyllium Chlorides. Eur. J. Inorg. Chem. 2003, 12, 2245–2254. [Google Scholar] [CrossRef]
- Hanna, T.A.; Ghosh, A.K.; Ibarra, C.; Zakharov, L.N.; Rheingold, A.L.; Watson, W.H. Facile Formation of Molybdenum(VI) Monooxo Aryloxides MoO(OAr)4-nCln from Molybdenum Dioxo Dichloride. Inorg. Chem. 2004, 43, 7567–7569. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walley, J.E.; Wong, Y.-O.; Freeman, L.A.; Dickie, D.A.; Gilliard, R.J., Jr. N-Heterocyclic Carbene-Supported Aryl- and Alk- oxides of Beryllium and Magnesium. Catalysts 2019, 9, 934. https://doi.org/10.3390/catal9110934
Walley JE, Wong Y-O, Freeman LA, Dickie DA, Gilliard RJ Jr. N-Heterocyclic Carbene-Supported Aryl- and Alk- oxides of Beryllium and Magnesium. Catalysts. 2019; 9(11):934. https://doi.org/10.3390/catal9110934
Chicago/Turabian StyleWalley, Jacob E., Yuen-Onn Wong, Lucas A. Freeman, Diane A. Dickie, and Robert J. Gilliard, Jr. 2019. "N-Heterocyclic Carbene-Supported Aryl- and Alk- oxides of Beryllium and Magnesium" Catalysts 9, no. 11: 934. https://doi.org/10.3390/catal9110934
APA StyleWalley, J. E., Wong, Y.-O., Freeman, L. A., Dickie, D. A., & Gilliard, R. J., Jr. (2019). N-Heterocyclic Carbene-Supported Aryl- and Alk- oxides of Beryllium and Magnesium. Catalysts, 9(11), 934. https://doi.org/10.3390/catal9110934