1. Introduction
Global energy consumption is rising rapidly due to continuous economic and population growth. The vast majority of energy required is produced from nonrenewable fossil fuels and resources, which causes considerable emissions of greenhouse gases such as carbon dioxide (CO
2). Chemical conversion strategies, such as the hydrogenation of CO
2 to methanol (CH
3OH) via heterogeneous catalysis, can play an important role in reducing CO
2 emissions [
1,
2,
3,
4,
5]. For this purpose, the development of highly selective, active, and stable catalysts is essential.
CuO/ZnO/Al
2O
3 is a conventional composite catalyst which is widely used for methanol synthesis from syngas. Copper species are considered to be the most important active component for the hydrogenation of CO
2 to CH
3OH. It is generally proposed that the coordination, chemisorption, and activation of carbon dioxide takes place at Cu
0 or Cu
+ sites. The H
δ+ and H
δ-ions required for the catalytic process are provided by the homogeneous splitting of hydrogen at the promoter, usually zinc oxide (ZnO) [
6]. Some research groups claim that metallic Cu atoms act as active sites in methanol synthesis [
7,
8,
9]. Pan et al. [
10] and Rasmussen et al. [
11,
12] were able to demonstrate a direct proportional relationship between the activity of the catalyst and the surface of metallic copper. In contrast, Jansen et al. [
13] and Chinchen et al. [
14] identified Cu
+ as the active site for methanol synthesis by investigations on Cu/ZnO/SiO
2 and Cu/ZnO/Al
2O
3 materials. Other research groups were able to show that both metallic copper and copper with a low valence (Cu
δ+ and Cu
+) can influence the catalytic activity of Cu-based oxide catalysts [
15,
16,
17,
18]. Chen et al. [
19] suggested that the activity of copper-based catalysts for methanol synthesis is directly proportional to the total exposed copper (CuO and Cu
+) and thus ultimately independent from the specific Cu sites.
In general, the resolution of the electronic and geometrical structures of the active sites is an important step towards the rational and effective catalyst design. Since methanol synthesis on a Cu-based catalyst is a structurally sensitive reaction, it is suggested to modify its performance with various carrier components. In addition, a suitable carrier component is capable for tuning or influencing the interactions between the catalyst active component and the promoter.
Many research groups discuss ZrO
2 as an additional component for methanol synthesis [
20,
21,
22] as it has a high stability under reducing or oxidizing conditions. One reason for the increased activity compared to Al
2O
3 used in conventional catalyst systems is the stronger affinity of ZrO
2 to water, which can act as poison on active metal sites [
23,
24]. Several research groups observed that the catalytic activity and selectivity with respect to methanol can be increased due to an improved copper dispersion in the presence of ZrO
2 [
25,
26,
27,
28,
29,
30,
31]. This resulted in a specific interface which favors methanol synthesis. Liu et al. [
16] found out that the binding energy of Cu 2p
3/2 is higher than that of pure CuO in CuO/ZrO
2 catalysts. Furthermore, the authors could show that CuO and ZrO
2 are not only physically mixed, but also interact with each other. XRD measurements have demonstrated the dominance of the monoclinic crystal phase for unloaded ZrO
2, whereas copper-loaded ZrO
2 additionally consist of a tetragonal phase. The proportion of tetragonal crystal phase increases at higher Cu loadings [
20]. The monoclinic phase even disappears completely if the Cu load exceeds a value of 10 wt.%. Moreover, the proven interactions between CuO and ZrO
2 inhibit the phase transformation of ZrO
2. Oxygen vacancies in ZrO
2 can be the reason for this metal–carrier interaction, leading to geometric effects which influence or change the dispersion and morphology of the introduced metal. Koeppel and Baiker [
32] also observed morphological changes in the investigated catalysts. They concluded that efficient methanol synthesis catalysts should consist of microcrystalline Cu particles which are stabilized by interaction with an amorphous ZrO
2 matrix, resulting in a large specific interface. Suh et al. [
33] supposed that the addition of ZrO
2 favors the formation of Cu
+ ions and thus changes the Cu
+/Cu
0 ratio at the catalyst surface, which is indicated by an increased activity of the catalyst at low temperatures and pressures.
In addition to the individual components of the catalyst system itself, the preparation method can also influence the structural, textural, solid state surface, and catalytic properties of the proposed materials. Precipitation reactions are widely used in synthesis of copper-based catalysts. Precipitants such as sodium carbonates and oxalates are added to a solution of the desired salt concentration to form catalyst precursors. Frei et al. investigated influence of the precipitation and ageing temperature on Cu/ZnO/ZrO
2 catalyst samples [
34]. Chen et al. prepared CuO/ZnO/ZrO
2 catalyst by the polymeric precursor method [
35].
Impregnation technology can also be used for preparation of methanol synthesis catalysts. The performance of the materials synthesized by this method is less competitive than the catalyst synthesized by the coprecipitation method [
36].
The focus of this work is the synthesis of a series of CuO/ZnO/ZrO2 catalyst samples prepared by coprecipitation, one-pot synthesis, and wet impregnation, as well as a comprehensive characterization of them in order to study synthesis-to-property relations. Cetyltrimethylammonium bromide (CTAB) was used in the surfactant-supported coprecipitation to improve the structural or textural characteristics of the catalytic samples. Therefore, the main objectives and expected results of this work are the preparation and characterization of doubly promoted nanocomposite catalysts based on ZrO2, which are suitable to catalyze the hydrogenation of carbon dioxide. For this reason, a series of ZrO2-based supported catalysts were synthesized leading to the general formulation of M-P/ZrO2 (M and P denotes the active metal, the surface, or structural modifiers). In addition to the preparation and comprehensive characterization of potential catalyst systems in different compositions, the aim of this work is to realize their targeted modification with regard to their properties, e.g., the specific surface area or surface acidity. In addition to structural investigations, spectroscopic in situ measurements were carried out on a selected catalyst system to observe the interaction of CO2 or CO2/H2 mixtures with the catalyst surface during the reaction.
2. Results and Discussion
2.1. Powder X-ray Diffraction (XRD)
The catalyst systems synthesized according to the description in the experimental section were characterized by XRD with respect to their composition. The diffraction patterns of the individual samples are shown in
Figure 1.
Very sharp, clear reflections of the synthesized samples indicate a crystalline structure of all catalyst systems. The reflexes that occur at 2θ = 32.5°, 35.5°, 38.6°, 48.8°, 53.7°, 58.16°, 61.6°, and 66.29° in all X-ray diffraction patterns are characteristic for CuO (JCPDS card No. 48-1548) [
1,
27,
36,
37,
38,
39,
40,
41,
42]. In some samples (F-CZZ-631-1, F-CZZ-631-2, and F-CZZ-631-3), the reflex occurring at 2θ = 32.5° is partially superimposed by the 2θ = 31.8° reflex characteristic for ZnO. Further reflexes could be detected in the diffracted image patterns of the samples N-CZZ-631, E-CZZ-631, F-CZZ-631-1, F-CZZ-361-2, and F-CZZ-631-3 at 2θ = 34.3°, 36.2°, 47.54°, 56.6°, 62.8°, and 67.9°, which can be clearly assigned to ZnO (JCPDS card No. 43-0002) [
27,
36,
38,
39,
40,
41,
42,
43]. All samples showed reflexes at 2θ = 30.3° and 50.7°. These could be clearly assigned to the tetragonal phase of ZrO
2 (JCPDS card No. 17-923) [
37,
38,
44]. The weak reflex occurring at 2θ = 50.7°, which is assigned to the t-ZrO
2 phase, is overlaid by a reflex at 2θ = 50.5° in the samples synthesized by means of coprecipitation. This is generally assigned to metallic copper [
1,
38,
45].
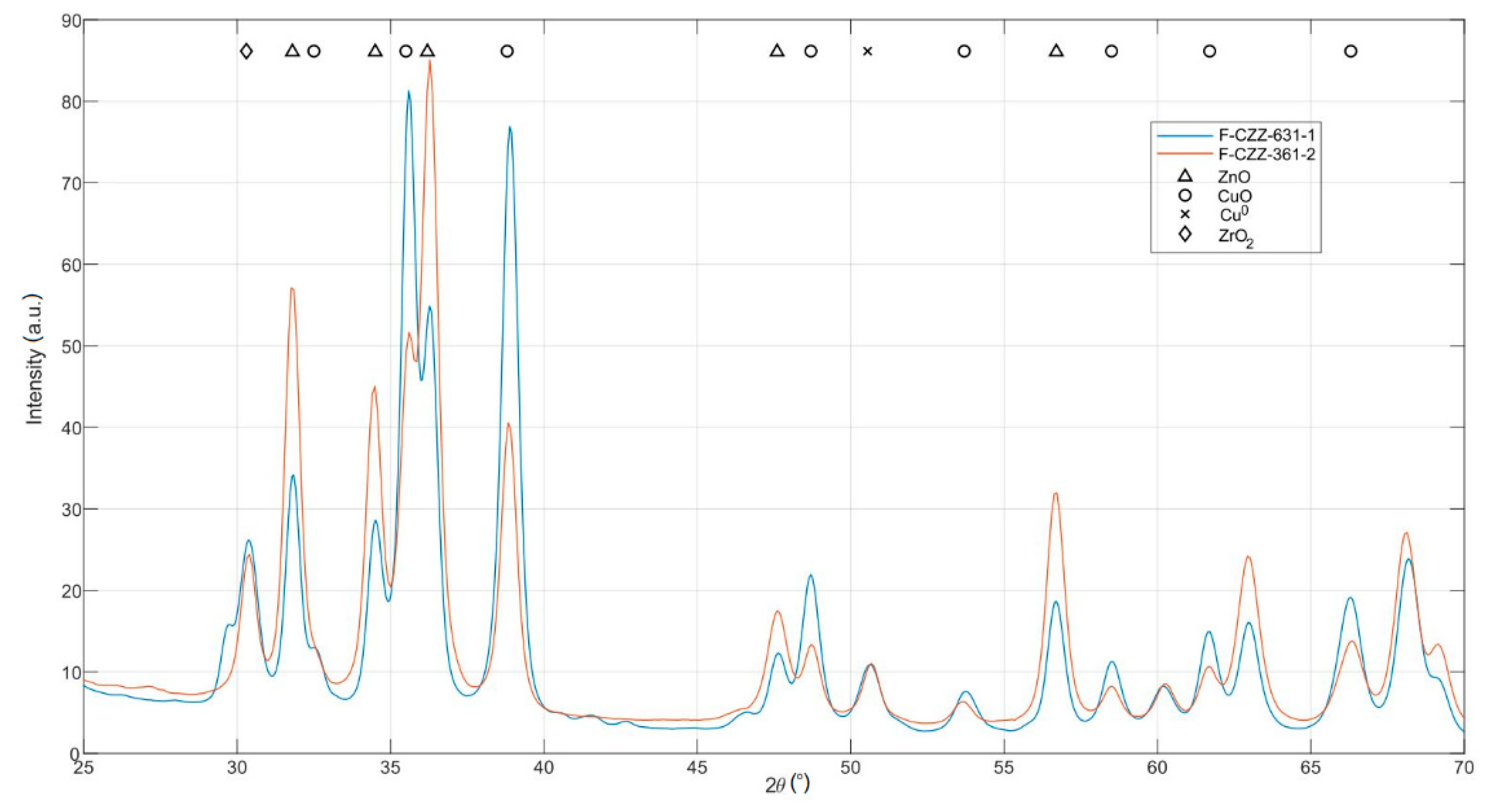
The comparison of X-ray diffraction patterns of the samples F-CZZ-631-1 and F-CZZ-361-2 is of particular interest. According to experimental section, both catalyst systems should have a different composition of the components CuO/ZnO/ZrO
2 (60/30/10 to 30/60/10 wt.%). The diffraction patterns of these samples (
Figure 2) show characteristic reflex positions of CuO, ZnO, and ZrO
2. Reflexes of CuO and ZnO show strongly different intensities, while those of ZrO
2 are almost identical. However, only limited quantitative information can be obtained from the XRD. Nevertheless, a general trend can be observed from the peak heights. The sample F-CZZ-361-2 has a smaller CuO amount, a higher ZnO amount compared to the sample F-CZZ-631-1, or at least a varying dispersity of both components.
To get deeper insights, the crystal size of the CuO-components was determined based on the Debye-Scherrer Equation (1) [
46,
47].
Di—the mean crystal size of the ordered domains
K—the Scherrer form factor
λ—the wavelength of X-rays
β—the full width at half maximum
θ0—the Bragg angle
The Scherrer form factor is set to K = 0.94, according to literature [
48]. The wavelength of the X-ray radiation is 0.1541 nm. The full half-width was determined using analysis software. In order to obtain more accurate results, reflections between 2θ = 20° and 50° should be applied [
48]. Therefore, the CuO reflex was used at 2θ = 38.6°, since it is strongly pronounced in all X-ray diffraction patterns and can be evaluated. The values determined in this way are summarized in
Table 1.
The crystal sizes of the copper oxide in the samples synthesized by wet impregnation and one-pot synthesis are in the range of 8 to 10 nm. The crystal sizes of CuO in the samples synthesized by means of coprecipitation are about 10–15 nm. They are significantly larger than those in the remaining samples and allow the assumption that the synthesis method had a significant influence on the CuO crystal size. In addition, the respective ZnO content also influences the CuO crystal sizes. For example, the ZnO content was increased and led to smaller CuO crystals with a size of 10 nm for the same synthesis procedure. Surfactant addition leads to a decrease in the CuO crystallite size, which is apparently related to the structural or textural characteristics of the resulting sample.
2.2. Temperature-Programmed Reduction (TPR)
TPR enables observation of different species on the catalyst surface and the determination of reducibility of catalyst components. The detected TPR profiles of the catalyst systems obtained by wet impregnation, one-pot synthesis, and coprecipitation are shown in
Figure 3.
Each of the TPR profiles of the catalyst systems investigated shows characteristic peaks in the temperature range 150–330 °C, which can be assigned to the reduction of CuO to Cu
0 [
49].
It is noticeable that the TPR profiles of the samples synthesized by wet impregnation N-CZZ-631 have only one defined reduction peak. Therefore, only one CuO-type of reducible species was formed in this sample. The reduction peaks of the samples E-CZZ-631 and F-CZZ-631-1 show clear shoulders which indicate the existence of different CuO species. In the course of a more detailed analysis, these can be referred to as a low-temperature peak and a high-temperature peak or as α- and β-peaks [
27,
40,
50]. The low temperature peak α could be assigned to the reduction of highly dispersed CuO species and the high temperature peak β to the reduction of so-called bulk CuO species [
27,
40,
49].
The TPR of the sample N-CZZ-631-2 also shows a clear reduction peak at a relative high temperature (T = 278 °C), which can be assigned to the presence of bulk CuO phase. The TPR profile of the sample E-CZZ-631-3 indicates a peak in the range of lower temperatures (T = 255 °C) with one shoulder at higher temperature (T = 275 °C). For precise analysis, the reduction peaks obtained were divided into two peaks by means of a Gaussian adjustment (
Figure 4).
In this sample, a highly dispersed CuO phase (α-peak) and a bulk-CuO (β-peak) can be observed. The β-peak lies in an almost identical temperature range as the reduction peak in the TPR profile of the sample N-CZZ-631-2. Therefore, it can be assumed that in both cases the β-peak belongs to the bulk CuO phase. From the respective peak heights or their share in the total reduction peak, the proportion of the highly dispersed CuO phase is increased in the sample E-CZZ-631-3 leading to a decreased amount of bulk CuO.
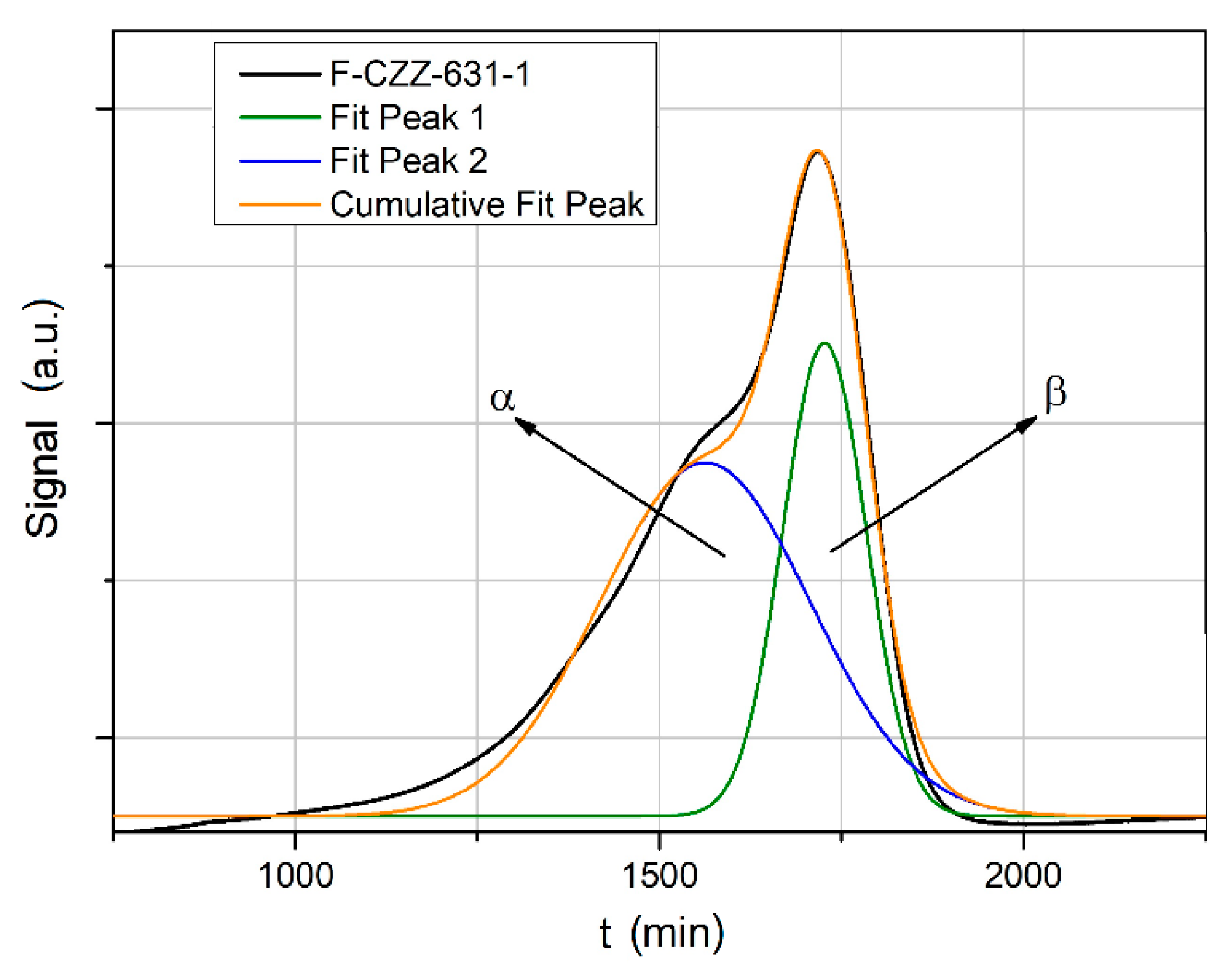
The deconvolution of the TPR profiles was also performed for the sample F-CZZ-631-1 (
Figure 5).
The proportions of the highly dispersed and bulk phases of the CuO, in contrast to the sample obtained by one-pot synthesis are almost identical. Both peaks occur at significantly higher temperatures (α-peak at T = 290 °C or β-peak at T = 325 °C). Since ZrO
2 was used as a carrier component in all synthesized samples, differences in the modification of ZrO
2 could explain the different reducibility. Although the dispersion of CuO on t-ZrO
2 is higher than that on m-ZrO
2, CuO can be reduced at significantly higher temperatures [
51]. However, since there were no reflex layers in the X-ray diffraction patterns indicating an m-ZrO
2 phase, the different crystal size of the CuO on the catalyst surface can cause the reducibility shift. It is generally assumed that the smaller the CuO particles on the catalyst surface, the lower the reduction temperature [
52,
53,
54].
The CuO crystal sizes summarized in
Table 1 are much higher in the samples obtained by coprecipitation. This supports the assumption that the higher CuO crystal sizes of the catalyst system F-CZZ-631-1 synthesized by coprecipitation lead to an increase of the reduction temperature. The deconvolution of the TPR profiles also shows that the proportion of highly dispersed CuO species in the catalyst system prepared by one-pot synthesis is much higher compared to the system synthesized by coprecipitation.
The consumption of H2 during the TPR analysis of the sample F-CZZ-631-1 was much lower than that of all other catalyst systems. This confirms the results obtained by the XRD that a Cu0 phase was formed in the coprecipitated catalyst systems in addition to the formation of the CuO phase, in contrast to all other samples. Assuming that all samples formed approximately the same number of Cu species during the synthesis, the reduced consumption of H2 can be explained by the reduced number of CuO sites which can be reduced to Cu0.
The TPR profiles of the samples F-CZZ-631-5 and F-CZZ-361-6 with a CuO/ZnO/ZrO
2 content of 60/30/10 and 30/60/10 wt.%, respectively, are shown in
Figure 6. The influence of the composition of these catalyst systems is to be examined more closely, since both samples were synthesized by means of coprecipitation and differ only in composition.
The form of the TPR profile is only slightly influenced by the altered composition of the catalyst system. However, the catalyst system F-CZZ-361-2 has a reduction peak at slightly lower temperatures in the range of T = 290 °C. As discussed before, this can be assigned to the α-peak of the highly dispersed CuO phase. The temperature region in which a reduction occurred (T = 290–305 °C) is very small. In the area of the β-peak, no reduction peak could be detected. Thus, the phase composition of the CuO catalyst system could be influenced in such a way that an increase in the ZnO content or a decrease in the CuO content almost exclusively provides the formation of a highly dispersed CuO phase. This confirms the observations of other research groups, that adjacent ZnO can improve the CuO dispersity by weakening the tendency of the CuO particles to form agglomerates [
55,
56,
57,
58]. Against this background, the relationship between copper reducibility and the portion of the highly dispersed or bulk CuO phase could be observed. As a consequence, the catalyst system F-CZZ-631-1 showed an increased CuO crystal size compared to the catalyst system F-CZZ-361-2 and, hence, is more difficult to reduce.
2.3. Nitrogen Physisorption
In addition to phase analysis using powder XRD and TPR, nitrogen physisorption and the BET method were used to characterize the catalyst samples. The specific surface areas determined in this way are summarized in
Table 2.
The specific surface areas of the investigated catalyst systems with identical composition (N-CZZ-631, E-CZZ-631, and F-CZZ-631-1) do not differ significantly for varying synthesis methods. The sample F-CZZ-361-2 (SBET = 33 m2/g) has an increased specific surface area compared to the sample F-CZZ-631-1 (SBET = 22 m2/g). As already discussed, a higher CuO content in the catalyst system leads to formation of agglomerated CuO phase and, therefore, to a smaller specific surface area.
Surprisingly, a very small specific surface area of S
BET = 10 m
2/g was obtained for the sample F-CZZ-631-3 produced by the surfactant-supported coprecipitation. Since the use of surfactants in synthesis did not increase the specific surface area, further samples were synthesized by surfactant-supported coprecipitation using an adapted hydrothermal treatment. These materials were identical in their CuO/ZnO/ZrO
2 composition as well as in the amount of CTAB used. However, the duration of the hydrothermal treatment and the subsequent calcination was varied. An overview of the parameters of the synthesis and pretreatment as well as the resulting specific surface areas are shown in
Table 3.
Since a high temperature is required to remove the surfactants and to form a mesoporous structure, the duration of the calcination was shortened in order to reduce the thermal load of the sample. In addition, an extended hydrothermal treatment was performed during synthesis to increase the specific surface area. The shortened calcination duration and the extended hydrothermal treatment showed a positive effect. The specific surface area of the catalyst system synthesized by surfactant-supported coprecipitation could be approximately doubled by shortening the calcination (SBET, F-CZZ-631-3 = 10 m2/g to SBET, F-CZZ-631-4 = 22 m2/g). It can be concluded that due to the high thermal load, the porous structures of the sample F-CZZ-631-1 collapsed, resulting in a low specific surface area. By doubling the hydrothermal pretreatment time, the specific surface could also be significantly increased (SBET, F-CZZ-631-5 = 31 m2/g).
The sorption isotherms of the investigated catalyst systems were analyzed in order to gain further insights into the textural properties.
Figure 7 shows the isotherms of the samples N-CZZ-631 and E-CZZ-631.
The catalyst systems show adsorption isotherms of type IV (according to IUPAC). The isotherm curves indicate weakly porous substances with microporous/mesoporous pore structure. A type H3 hysteresis characteristic for layered solids with narrow pore networks not completely filled with condensate is observed. There is a strong similarity of the catalyst samples regarding porous properties, independent of the synthetic routes for CuO/ZnO/ZrO2 (60/30/10 wt.%) composition.
In
Figure 8, the sorption isotherms of the catalyst samples synthesized by coprecipitation (with and without surfactant addition) and of the wet-impregnated samples are compared.
The sorption isotherms of the sample F-CZZ-631-1 (green) synthesized by coprecipitation (without surfactant) are at a similar level to the sorption isotherms of the sample N-CZZ-631 (red). The sample F-CZZ-631-1 shows a larger slope at low relative pressures, which results from higher amount of micropores. The sorption isotherms of the catalyst sample F-CZZ-631-9 (blue) synthesized by surfactant-supported coprecipitation are far below those of other systems. Much lower specific surface area of this sample provides less adsorptive capacity, which indicate mesoporous systems resulting from particle aggregates. In addition, to an adsorption isotherm of type IV, all samples show hysteresis loops of type H3 which indicate mesoporous systems resulting from particle aggregates.
Sorption isotherms of the catalyst samples F-CZZ-631-3, F-CZZ-631-4, and F-CZZ-631-5 are compared in
Figure 9. The already discussed adsorption isotherm for the sample F-CZZ-631-9 serves as a comparison regarding the influence of the changed pretreatment and its effects on the pore structure.
The sorption isotherm of the sample F-CZZ-631-10 (calcination time of 5 h) is higher than that of the sample F-CZZ-631-9 (calcination time of 12 h). This coincides with the results of the specific surface areas. The sample F-CZZ-631-11 showed a further increased specific surface area (SBET,F-CZZ-631-11 = 31 m²/g), leading to higher sorption isotherms. The adsorption isotherms of the samples F-CZZ-631-10 and F-CZZ-631-11 also show a significant increase in the middle range of the relative pressure (0.4 ≤ p/p0 ≤ 0.8), which indicates an increased proportion of mesopores. The fact that this increase starts slightly earlier with the F-CZZ-631-11 catalyst system than with the F-CZZ-631-10 catalyst system reveals that the mesopores formed have smaller pore radii.
Both catalyst samples (F-CZZ-631-10 and F-CZZ-631-11) show type IV adsorption isotherms. These are characteristic for mesoporous solids. The sample F-CZZ-631-10 forms a hysteresis of type H2b, which indicates a rather wide pore radius distribution. In contrast, the catalyst system F-CZZ-631-11 produces a hysteresis loop of type H2a, which is characteristic for a rather narrow pore radius distribution. Consequently, the size of the pore radii can be influenced by the duration of the hydrothermal pretreatment.
2.4. Temperature-Programmed Ammonia Desorption (TPAD)
Besides the specific surface area, the acidity of catalytic materials plays an important role in the investigated hydrogenation reaction. By means of TPAD the surface acidity of a solid sample can be examined. The position of desorption maxima in the TPAD profile provides information about the strength of the acid sites.
The TPAD profiles of the selected samples N-CZZ-631, E-CZZ-631, F-CZZ-631-1, F-CZZ-631-5 are presented in
Figure 10. The investigated samples possess a desorption maximum at low temperatures of about 200–250 °C but of different intensity. The different width of the signals of these samples can be attributed to acid sites of various strengths. The TPAD profile of the sample F-CZZ-631-5 displays two desorption peaks. The high desorption peak at low temperature indicates presence of weak acid sites. The samples N-CZZ-631 and E-CZZ-631 show a very weak desorption profile of the same form. Nevertheless, two weak desorption peaks can be detected. The sample F-CZZ-631-1 takes an intermediate position among the investigated samples. It shows weak acidic properties. The intensity of desorption peak at higher temperature is much higher which indicates the presence of a number of strong acid sites.
Given the fact that the sample F-CZZ-631-1 has a significantly higher amount of strong acidic sites, it can be assumed that the observed formation of larger CuO particles after the thermal treatment as well as the more difficult reducibility of this sample compared to other synthesized samples depends mainly on the nucleophilicity / electrophilicity of the support surface. Therefore, it is of particular interest to study the nature of the interaction of the CO2/H2 mixture for methanol synthesis with the support surface in order to make statements about possible catalytically active sites and the formation of intermediates. For this purpose, in situ diffuse reflection Fourier transform infrared spectroscopy (DRIFTS) investigations were performed on this selected sample.
2.5. In Situ Diffuse Reflection Fourier Transform Infrared Spectroscopy (DRIFTS)
DRIFTS was used to gain precise insights into the reaction mechanism and the interactions of CO2 and CO2/H2 with the catalyst surface. Before loading the sample with carbon dioxide or CO2/H2, a thermal and reductive treatment was carried out to remove water and other surface-bond species.
Figure 11 shows the DRIFT spectra after adsorption of pure CO
2 on F-CZZ-631-1 at various temperatures.
ZrO
2 has oxygen vacancies which can be further increased by doping with metal oxides. This is reflected in the IR spectra of adsorbed CO
2 (
Figure 11). In contrast to typical Al
2O
3-based methanol synthesis catalysts, where CO
2 is first bound to the solid surface via a carboxylate structure [
59], sample F-CCZ-631-1 does not form this surface structure. It forms another structure, which overlaps and has more intense bond strength. The form of CO
2 bonding changes with increasing temperature. After CO
2 adsorption at room temperature, monodentate carbonate species in the form of double bands in the range of 1420–1540 cm
−1, 1330–1390 cm
−1, and 980–1050 cm
−1 can be observed [
59,
60,
61,
62,
63]. Furthermore, clear bands in the range of 1600–1670 cm
−1 and 1280–1310 cm
−1 were detected, which are characteristic for the bidentate carbonate species [
59,
60,
61,
62,
63]. These are thermally unstable compared to the monodentate carbonate species and are completely degraded at 250 °C. The band around 1250 cm
−1 can, therefore, be assigned to carbonate species adsorbed on ZrO
2 [
59].
Due to the kind of ZrO
2 preparation (precipitation and calcination of ZrO(OH)
2), formate structures can be formed in the presence of CO
2, which is indicated by the superimposed bands in the range 1580–1620 cm
−1 and 1340–1390 cm
−1, as well as by the typical bands about 2500 cm
−1 [
64,
65,
66]. In addition, hydrocarbonate species (CO
3H
−) may also be present (bands in the range of 1225, 1400–1500, 1615–1630, and 3600 cm
−1), which are formed from CO
2 with the participation of hydroxyl groups and surface oxygen atoms [
59]. However, they are thermally unstable and decompose at lower temperatures.
It can be concluded from the DRIFT spectra of the sample F-CCZ-631-1 that in the temperature range interesting for catalysis (T = 200–250 °C), the interaction of CO2 with the solid surface occurs predominantly in form of monodentate carbonate species at lower temperatures and formate species at higher temperatures.
The DRIFT spectra of the adsorption of the CO
2/H
2 gas mixture on the catalyst sample F-CZZ-631-1 at various temperatures show some differences compared to the results with pure CO
2 (
Figure 12).
In presence of H
2, the formation of formate species was identified, as well as the transformation of monodentate into bidentate carbonate species (significant increase of the intensity of the absorption band at 1620 cm
−1 accompanied by strong decrease of the band intensity at 1480 cm
−1) from the beginning [
59,
60,
61,
62]. The very weak absorption bands around 2800 cm
−1 indicate the presence of C-H stretching vibrations or hydrocarbonlike fragments at the solid surface. At the same time, more intensive absorption bands in the range 3500–3700 cm
−1 can be observed, which could be caused by both the corresponding OH valence vibrations or the overtone vibrations of the CO-bonds [
67]. Based on known vibrational frequencies for different molecular components, a possible methanol formation was observed [
68].
To support this assumption, difference spectra for the adsorption of the CO
2/H
2 gas mixture at different temperatures were determined (
Figure 13). Regarding the methanol formation, investigations of the sample F-CCZ-631-1 with the CO
2/H
2 gas mixture at temperature T = 200–250 °C confirm the assumption from the results of the static system during the adsorption of pure CO
2 and the CO
2/H
2 mixture. The negative absorption bands in the range 1300–1600 cm
−1 where characteristic bands for the formate species can be observed indicate that these species are continuously consumed as a result of the CO
2 hydrogenation to methanol, which enters the gas phase [
64,
65,
66]. The water formed is identifiable by the OH valence vibrations in the range of about 3500 cm
−1 [
67]. The higher the reaction temperature, the more clearly this effect is visible.
Based on the results obtained from the DRIFT spectra, it can be confirmed that the (monodentate and bidentate) carbonate species are mainly responsible for the CO
2 adsorption mechanism [
59,
62]. The assumed formate reaction pathway for the methanol formation can be reconstructed by the characteristic bands of formate intermediates as well [
69,
70]. Taking into account literature results and the findings from the structural and surface chemical studies in this work, it can be supposed that the CO
2/H
2 conversion to methanol presupposes the presence of catalytic active Cu/CuO pairs in the interfaces of CuO/ZnO/ZrO
2 mixed metal oxide catalysts. The formation of such pairs with Cu
0 or Cu
+ species is evidently promoted by ZnO (improving the reducibility of CuO phase to metallic copper) with the coparticipation of the acid support ZrO
2 (generation of the electrophilic metal particles).
3. Materials and Methods
3.1. Catalysts Preparation
The synthesis of the catalyst systems investigated in this work was carried out by (surfactant-supported) coprecipitation, one-pot synthesis, and wet impregnation.
In conventional coprecipitation, an aqueous solution (at a total cation concentration of 1 mol/L) of Cu(NO3)2 xH2O (Puratronic®, 99.999%, metals basis, Alfa Aesar, Kandel, Germany), Zn(NO3)2 6H2O (Puratronic®, 99.998%, metals basis, Alfa Aesar, Kandel, Germany), and ZrO2(NO3)2 xH2O (Puratronic®, 99.994%, metals basis, Alfa Aesar, Kandel, Germany) was prepared. A 1 M Na2CO3 solution was then added dropwise at a temperature of 65 °C up to a pH value of 7.
In order to increase the specific surface area of the catalyst systems, surfactant-supported coprecipitation was also used. Cetyltrimethylammonium bromide (CTAB, Sigma-Aldrich, Taufkirchen, Germany) was dissolved in 300 mL distilled water and stirred for 30 min. The different metal nitrates were added. The amount of CTAB was determined by the ratio 6:10 (mol CTAB: mol metal cations). A 0.5 M NaOH solution was then added dropwise to the prepared solution, which consists of metal nitrates and CTAB, up to a pH of 10 while stirring continuously, and then stirred at 90 °C for 12 or 48 h. The resulting precipitates were then filtered, washed, and dried for 24 h.
It should be mentioned that for the starting chemicals whose water content is not explicitly specified by the providers (Cu(NO3)2 xH2O and ZrO2(NO3)2 xH2O), a thermogravimetric analysis (TGA) was carried out to determine the exact water content.
The nitrates Cu(NO3)2 xH2O (Puratronic®, 99.999%, metals basis), Zn(NO3)2 6H2O (Puratronic®, 99.998%, metals basis), and ZrO2(NO3)2 xH2O (Puratronic®, 99.994%, metals basis) used in the one-pot synthesis were weighed and dissolved in water, analogous to the procedure described above. The resulting solution was then evaporated (100 °C for 5 h). The residue formed was dried in drying oven in air for 24 h.
In wet impregnation, the synthesis was analogous to the one-pot synthesis. However, in contrast to the other syntheses, the zirconium oxide was dissolved in advance and then added gradually in the form of ZrO(OH)2 to the nitrates Cu(NO3)2 xH2O (Puratronic®, 99.999%, metals basis) and Zn(NO3)2 6H2O (Puratronic®, 99.998%, metals basis).
The samples prepared in this way were named and numbered. The naming scheme consists of the synthesis method used (F—precipitation, N—wet impregnation, or E—one-pot synthesis), the components used (CuO, ZnO, ZrO
2), and their proportion (in % by weight). For example, the designation F-CZZ-631-5 means: the sample was synthesized by the precipitation (F) and contains 60 wt.% CuO (C), 30 wt.% ZnO (Z), and 10 wt.% ZrO
2 (Z) followed by the preparation number of sample. An overview of all samples is shown in
Table 4.
3.2. Catalysts Characterization
XRD patterns were obtained with a SuperNova crystal diffraction instrument using Cu Kα radiation. Nitrogen adsorption–desorption isotherms were measured at 77 K on a Sorptomatic 1990 (Carlo Erba Instruments, Egelsbach, Germany). Acidic properties were analyzed by temperature-programmed desorption of ammonia. After pretreatment—including calcination, ammonia adsorption, and removal of physisorbed ammonia—the desorption was analyzed by in situ IR-spectroscopy (Nicolet Impact 400, Germany) and mass spectrometry in the range of 373 to 823 K. The reducibility of metal oxides and the nature of the CuO phase was characterized by TPR (Thermo Scientific, Dreieich, Germany). DRIFTS (Bruker, Karlsruhe, Germany) measurements enable in situ investigations of heterogeneous catalyzed gas-phase reactions under realistic reaction conditions. For these measurements, both pure CO2 (0.04 L/min) and a H2/CO2 gas mixture (in a ratio of H2/CO2 = 3) were used in two separate measurements. In order to characterize adsorption/desorption processes, spectra were collected during the measurement. For this purpose, the measuring cell was heated to 300 °C at 5 K/min). In addition, investigations were carried out with the CO2/H2 gas mixture in a temperature range of T = 200–250 °C, which is typical in the case of low temperature synthesis of methanol.
4. Conclusions
The focus of this work was the preparation and modification of potential catalyst systems CuO/ZnO/ZrO2 in different ways (wet impregnation, one-pot synthesis, coprecipitation) and compositions to achieve a catalytically active CO2/H2 conversion to methanol. Therefore, individual catalyst systems were characterized with regard to their structural, textural, and surface-acid properties to identify the surface intermediates species which may possibly occur during the methanol synthesis.
By means of XRD, it could be shown that all catalyst systems contained the CuO, ZnO, and t-ZrO2 species crucial for the catalytic hydrogenation of CO2 to CH3OH. In addition, however, differences between the selected synthesis routes could also be observed. The powder X-ray diffraction pattern of the samples synthesized by coprecipitation showed additional reflex layers characteristic of metallic copper. In general, copper is considered the most important active component in the hydrogenation of CO2 to CH3OH. The role of different Cu species is being controversially discussed. Nevertheless, many research groups agree that metallic copper as an active site has a significant positive influence on the catalytic activity of methanol synthesis. In addition, by varying the CuO or ZnO content of individual catalyst systems, the role of ZnO as H2 reservoir could be confirmed based on investigations in a reducing atmosphere. Thus, the catalyst system with a proportion of 60 wt.% ZnO exhibited an increased reducibility compared to the catalyst system with a proportion of 30 wt.% ZnO. This could be shown by the increased Cu-reflex layers in the X-ray diffraction pattern of the sample with the higher ZnO content after 2 h exposure in a reducing atmosphere.
Subsequent TPR studies illustrated that different CuO phases on the individual catalyst surfaces were formed. It could be revealed that the sample produced by wet impregnation mainly forms the so-called bulk CuO phase. In addition to the bulk CuO, highly dispersed CuO was identified for the samples obtained by the one-pot synthesis and coprecipitation. However, these differed in their respective proportion of bulk or highly dispersed CuO. Moreover, it was possible to establish a relationship between the crystal sizes of the CuO determined by the Debye-Scherrer equation and the TPR profiles. Thus, it is generally concluded that the smaller the CuO particles on the catalyst surface, the lower the reduction temperature, resulting in a shift of the reduction peak to lower temperatures. This also explains the increased temperatures of the reduction peaks of the sample obtained by coprecipitation and surfactant-supported coprecipitation, since the crystal size of the sample was much higher than that of the other systems.
Here, synergistic effects could also be observed when comparing the catalyst systems with different ZnO contents. As a consequence, the sample with a proportion of 60 wt.% produced only highly dispersed CuO, whereas the sample with a ZnO proportion of 30 wt.% produced both highly dispersed and bulk CuO on the catalyst surface. This supports the observation of other research groups that the ZnO promoter improves CuO dispersity, and thus the reducibility of CuO phase to metallic cooper, by attenuating the agglomeration of Cu particles. On the other hand, the support ZrO2 also contributes to the stabilization of the CuO phase and thus decreases the metallic surface, but at the same time—due to the strong metal-carrier interaction—generates electron-deficient metal particles, which apparently provide high methanol selectivity.
The specific surface areas of the individual catalyst systems were also determined by means of nitrogen physisorption. The catalyst systems of identical composition showed similar specific surface areas (SBET = from 17 up to 24 m²/g), independent of the synthesis pathway. A surfactant-supported coprecipitation for the synthesis of individual catalyst systems was also used to increase the specific surface area. Initially, only catalyst systems with a small specific surface area could be synthesized (SBET = 10 m²/g). It was assumed that the surfactant structures collapsed due to the high thermal pretreatment during calcination, resulting in the conspicuously low specific surface area. Therefore, the thermal load was first reduced by shortening the calcination duration. As a result, the specific surface area of the catalyst system could be doubled (SBET = 22 m²/g). Due to the reduced thermal load, structured pore networks could be formed. In addition, the duration of hydrothermal pretreatment during synthesis was increased with the aim of improving micelle formation. In such a way, a further increase of the specific surface was achieved and the specific surface area could be tripled (SBET = 10 m²/g to SBET = 31 m²/g).
The investigations of the surface-acid properties by TPAD showed that the sample synthesized by surfactant-supported coprecipitation (at a calcination time of 5 h and 48 h hydrothermal pretreatment) showed the highest acidity of all catalyst systems synthesized in this study. It could be shown that this is almost exclusively based on rather weak acid sites. The more uniform distribution of acidic sites enables an ordered surface acidity, which can lead to an increased total acidity. In the remaining catalyst systems, however, the acidic sites of varying strength were formed.
In addition, in situ DRIFTS was applied to investigate the interaction of CO2 and CO2/H2 gas mixtures with the catalyst. The adsorption of CO2 takes place mainly via monodentate and bidentate carbonate species. Formate species, which are generally regarded as essential intermediates for the formation of CH3OH, could also be detected. Based on the results obtained, it could also be assumed that CH3OH was formed, wherein the Cu/CuO pairs in the interfaces of CuO/ZnO/ZrO2 mixed oxide catalysts are discussed as catalytic active sites.
In the future, the in situ spectroscopic results obtained within this work with regard to the reaction mechanism of methanol synthesis, which could not yet be conclusively confirmed, could be improved to the extent that they are carried out closer to the reaction parameters used in practice. A catalytic testing, which would serve to compare the catalytic activity of the investigated catalyst systems with that of systems commonly used in methanol synthesis or those of other research groups, is planned. In addition, yttrium doping of the support material ZrO2 to increase structural stability can also improve the catalytic performance, since ZrO2 is a promising catalyst support for methanol synthesis due to its stability under reducing and oxidizing atmospheres or its strong affinity for water (which acts as an inhibitor of the active metal sites).













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}