Recent Advances in C–H Bond Functionalization with Ruthenium-Based Catalysts

Bioorganic Chemistry Laboratory, CSIR-National Institute of Oceanography, Dona Paula-403004, Goa, India
Catalysts 2019, 9(2), 173; https://doi.org/10.3390/catal9020173
Submission received: 4 January 2019
/
Revised: 2 February 2019
/
Accepted: 5 February 2019
/
Published: 12 February 2019
(This article belongs to the Section Catalysis in Organic and Polymer Chemistry)
Abstract
:The past decades have witnessed rapid development in organic synthesis via catalysis, particularly the reactions through C–H bond functionalization. Transition metals such as Pd, Rh and Ru constitute a crucial catalyst in these C–H bond functionalization reactions. This process is highly attractive not only because it saves reaction time and reduces waste,but also, more importantly, it allows the reaction to be performed in a highly region specific manner. Indeed, several organic compounds could be readily accessed via C–H bond functionalization with transition metals. In the recent past, tremendous progress has been made on C–H bond functionalization via ruthenium catalysis, including less expensive but more stable ruthenium(II) catalysts. The ruthenium-catalysed C–H bond functionalization, viz. arylation, alkenylation, annulation, oxygenation, and halogenation involving C–C, C–O, C–N, and C–X bond forming reactions, has been described and presented in numerous reviews. This review discusses the recent development of C–H bond functionalization with various ruthenium-based catalysts. The first section of the review presents arylation reactions covering arylation directed by N–Heteroaryl groups, oxidative arylation, dehydrative arylation and arylation involving decarboxylative and sp3-C–H bond functionalization. Subsequently, the ruthenium-catalysed alkenylation, alkylation, allylation including oxidative alkenylation and meta-selective C–H bond alkylation has been presented. Finally, the oxidative annulation of various arenes with alkynes involving C–H/O–H or C–H/N–H bond cleavage reactions has been discussed.
1. Introduction
Synthesis of organic compounds with a heterocyclic ring and natural product scaffolds are extremely important as they form an indispensable structural unit of bioactive compounds and pharmacophores [1,2]. Tremendous effort has been made in the designing and development of organic synthesis that could provide ready access to these valuable organic compounds. The traditional organic synthesis often requires multistep reactions with the use of several hazardous reagents, and, thus, the process is neither environmentally friendly nor economically viable. To overcome these problems, an alternative synthetic method with better atom and step economical process is highly desirable. In the recent past, in order to overcome the above shortcomings, organic synthesis has steadily shifted towards a catalytic pathway. It is noteworthy that, in organic molecules, C–H bonds often outnumber the other type of bonds; therefore, a direct functionalization of C–H bonds could drastically shorten the synthetic route of any organic synthesis. Indeed, a direct C–H bond functionalization pathway is expected to provide a greener and more economical pathway for ready access to various functional organic compounds.
Classical C–C cross-coupling was first studied with Ni and Pd catalysts [3,4]. Since this classical cross-coupling requires pre-functionalized starting materials and produces a stoichiometric amount of metal salt as the site product, the process is not attractive (Scheme 1a). During the past few decades, organic synthesis through C–H bond functionalization has emerged as one of the most viable methods not only for selective organic synthesis [5] but also for ready access to avariety of functional molecules including pharmaceutical relevant compounds [6] and natural products [7,8]. The cross-coupling through direct C–H bond functionalization is highly attractive as the process not only provides a greener reaction protocol but also reduces waste. Generally, three types of direct C–Hfunctionalization strategies are known in the transition metal-catalysed C–H bond functionalization [9]. The direct C–H bond functionalization with aryl halides or alkyl halides usually requires an addition of a base to neutralize the generated acid, HX (Scheme 1b). On the other hand, the coupling of arene or alkene with an organometallic reagent requires an oxidant to generate the metal species (Scheme 1c).
In both the direct C–H functionalization methods, apre-functionalised starting material is required. On the contrary, the dehydrogenative method involving the coupling of two C–H bonds, first discovered by Moritani and Fujiwara with a Pd catalyst, is the most step-and atom economical process, as it does not require any pre-functionalized starting materials (Scheme 1d).
During the last decade, transition metal-catalysed C–H bond functionalization has rapidly developed and its utility in the synthesis of organic compounds is well documented [10]. Initially, transition metals such as Pd [11,12] and Rh [13,14] have contributed enormously in these transition metal-catalysed C–H bond functionalizations. In the recent past, the reactions have been explored with much cheaper ruthenium complexes, particularly with the more stable ruthenium(II) catalysts [15]. In general, the ruthenium-catalysed C–H bond functionalization has been done in two pathways, namely the oxidative addition pathway and the deprotonation pathway. C–H bond functionalization via the oxidative pathway is usually performed with low valent ruthenium complexes, such as [RuH2(CO)(PPh3)3] or [Ru3(CO)12], whereas ruthenium(II) complexes, viz. [RuCl2(PPh3)3] and [RuCl2(p-cymene)]2 in association with acetate or carbonate bases, are used for the C–H bond functionalization via the deprotonation pathway [16]. In the deprotonation pathways, transition metals with low valent oxidation states such as Pd(0), Rh(I) or Ru(0) are required to oxidize to the higher oxidation state, viz. Pd(II), Rh(III) and Ru(II), in the presence of organic or inorganic oxidants to regenerate the active species.
In recent years, the ruthenium-catalysed C–H bond functionalization has been explored with a varietyof N–Heteroaryl directing groups [15] as well as other directing groups such as ketones [17], esters [18], amides [19], carboxylic acids [20], etc. Generally, the cationic ruthenium species, [RuCl2(p-cymene)]2/AgSbF6 or [RuCl2(p-cymene)]2/KPF6 have been used for C–H bond functionalization, directed by a weakly coordinatedoxygen and π-bond donor group. The C–H bond functionalization of weakly coordinated directing groups has been extensively studied by the research groups of Jeganmohan [21,22], Ackermann [23] and others [24]. Recently, the benefits of the carboxylic acid ligand in C–H bond functionalization reaction have been explored. Indeed, the carboxylic acid-assisted reaction revolutionized the C–H bond functionalization by transition metals including ruthenium catalysts [25]. The carboxylate-assisted C–H bond functionalization with ruthenium(II)-catalysts has been studied extensively by Ackermann [26,27], Dixneuf [15,28] and later by others [29]. It is noteworthy that ruthenium(II) catalysts partner with carboxylate ligandsto offer an attractive pathway for the C–H bond functionalization, allowing the reaction to operate under mild conditions and even in water [30,31].
Recently, ruthenium-catalysed C–H bond functionalization has been studied in various reactions, namely arylation [15], alkenylation [16], and annulation[26]. Subsequently, several new reactions have been discovered, such as direct C–H bond oxygenation [32], amidation [33], amination [34], cyanation [35], halogenations [36] and selenylation [37]. These reactions on ruthenium-catalysed C–H bond functionalization have been presented in numerous research papers and review articles by several groups [15,16,25,26,38,39,40]. Furthermore, the mechanistic study of ruthenium-catalysed C–H bond functionalization was recently described by the research group of Lan [41] in the context of arylation, alkylation and alkenylation,and by Nelson and co-workers on selective ortho-arylation directed by Lewis basic groups [42]. However, new reactions and methodologies ofC–H bond functionalization with ruthenium-based catalysts are being discovered. Keeping this in mind, this review describes the recent development on the C–H bond functionalization reactions covering arylation, alkenylation, alkylation and annulation reactions with ruthenium-based catalysts. The review focusses on the developments in this field over the past eight years and includes reports that have appeared up until 2018.
2. Ruthenium-Catalysed C–H Bond Arylation
2.1. C–H Bond Arylation Directed by N–Heteroarenes
Ruthenium-catalysed C–H bond arylation has been studied with various substrates, in particular with substrates with an N–Heteroaryl ring as the directing group such as 2-phenylpyridine, 1-phenylpyrazole, quinoline, tetrazole, imidazole, pyrimedyl, etc. The catalytic C–H bond arylation has been progressing steadily and covers several other substrates apart from the nitrogen heteroaryl directing groups and also reactions that have employed a variety of ruthenium catalysts. In 2001, Oi et al. described the arylation of 2-phenylpyridine with aryl halide using [RuCl2(η6-C6H6)]2 in the presence of 4 equiv. of PPh3 and a base (Scheme 2a) [43]. In subsequent years, they reported the arylation of aromatic 2-alkenylpyridines and aryl bromides with [RuCl2(η6-C6H6)]2 in the presence of a base (Scheme 2b) [44].
In 2008, Ackerman’s research group studied for the first time the beneficial effects of carboxylic acid in the ruthenium-catalysed direct arylation of arene in toluene, a less coordinating and apolar solvent [45]. They performed the arylation of several arenes with nitrogen-containing directing groups including 2-pyridyl, oxazolin-2-yl, N-pyrazolyl and N-triazolyl with aryl bromide using [RuCl2(p-cymene)]2 (2.5 mol%) in the presence of mesitylic acid (30 mol%) to give arylated products (Scheme 3). They have shown that the reaction could be performed using cheaper but less reactive aryl chlorides. The reaction scope has extended to 1,4-disubstituted 1,2,3-triazoles leading to ortho-arylated products. However, under this reaction condition, direct arylation did not occur with ortho-substituted aryl chloride; instead, the reaction led to a homo-coupling product.
Christakakou et al. reported the arylation of 2-phenylpyridine using aryl bromide in the presence of [RuCl2(p-cymene)]2, DBU and PPh3 (Scheme 4) [46]. The arylated product formed depending on the nature of the aryl bromide employed. Thus, employing bromobenzene as an arylating agent gave 26% and 71% yield of mono- and diarylated products, whereas bromo-anisaldehyde gave only the monoarylated product. However, aryl bromides such as p-bromotoulune and p-bromotriflurotolune gave a mixture of mono- and diarylated products; in contrast, no arylated product was observed for p-nitrobromobenzene.
Pozgan and co-workers demonstrated a ruthenium-catalysed multiple C–H bonds arylation of 2-phenylpyridine with 2-bromopyridine promoted by microwave radiation (Scheme 5) [47].
The reaction proceeded using [RuCl2(p-cymene)]2 in the presence of an additive heated at 200 °C under microwave radiation. The reaction performed in water without additive resulted in a mixture of multiarylated products comprising mono-, di-, tri- tetra- and pentarylated products with the predominant formation of hexa(2-pyridyl)benzene. The reaction was best performed employing [RuCl2(p-cymene)]2 (10 mol %), KOPiv (40 mol%) and PPh3(20 mol%) giving hexa(2-pyridyl)ben- zene in quantitative yields (Scheme 5).
The arylation of heteroarene containing an oxazoline ring as the directing group was reported by the research group of Dixneuf using [RuCl2(p-cymene)]2 in association with PPh3 in the presence of cheaply available potassium acetate as an additive [48]. This catalytic system was successfully used for the direct arylation of aldimines or ketimines as well. Furthermore, it was shown that the diarylated aldimines could be readily hydrolyzed under acidic conditions to give the arylated benzaldehydes; thus, the process proved to be an efficient method forthe synthesis of functionalized aldehydes (Scheme 6, [48]).
Initially, the ruthenium-catalysed arylation was performed mostly using [RuCl2(arene)]2 catalysts. Later, the reaction was explored with several other ruthenium catalysts. Ackermann’s research group reported an arylation of 2-phenylpyridine with aryl halides directly employing [Ru(MesCO2)2(p-cymene)] using the aprotic solvent toluene [49]. Subsequently, the arylation reaction of 2-phenylpyridine was performed by Dixneuf and co-workers using the ruthenium catalyst [Ru(OAc)2(p-cymene)] in NMP [50] followed by [Ru(tBuCO2)2(p-cymene)] in water [51]. They have also performed the direct diarylation of 2-phenylpyridine with aryl chloride including hetero arylchloride using [RuCl2(p-cymene)]2/KOPiv catalytic system in diethyl carbonate [52]. This catalytic system has produced a diarylated product tolerant to various functional groups such as cyanide and esters. The arylation reaction of 2-phenylpyridine has been studied with several other ruthenium(II) catalysts such as O^O and N^O chelate ruthenium(II) complexes as well. Singh and Dixneuf described the catalytic activity of water-soluble (O^O) and (N^O) chelate ruthenium(II) catalysts towards arylation of 2-phenylpyridine with aryl chlorides or aryl bromides (Scheme 7) [53]. These water-soluble ruthenium(II) complexes were efficient for the arylation of 2-phenylpyridine affording predominantly diarylated products in the presence of a catalytic amount of KOPiv or KOAc in water.
Recently, Binnani et al. employed ruthenium(II) complexes with N^O donor pyridine ligands for the arylation of 2-phenylpyridine with aryl halides (Scheme 8) [54]. The reaction gave both diarylated and monoarylated products, with diarylated as the major products. They have even tested cationic ruthenium(II) complexes with a N^N donor pyridine ligand as a catalyst in the arylation of 2-phenylpyridine, but it was shown to be less effective as compared to the neutral ruthenium(II) complexes with N^O donor pyridine ligands.
2.2. Ruthenium-Catalysed Selective Monoarylation
Doherty et al. achieved the selective monoarylation of 2-phenylpyridine and phenylpyrazoles using ruthenium(II) catalysts, [RuCl2(p-cymene)(PPh2L)], which gave predominantly monoarylated products with a minor or insignificant amount of diarylated products (Scheme 9) [55]. The reaction gave 76% of monoarylation and 9% diarylation with chlorobenzene as the arylating agent. Other arylating agents such as ethyl-4-chlorobenzene also gave good selectivity, affording 64% monoarylation and only 3% diarylation product.
In 2013, Dixneuf and co-workers reported a highly attractive selective monoarylation of 2-phenylpyridine and phenylpyrazoles using aryl chlorides and heteroaryl halides in water [56] (Scheme 10). They screened the monoarylation using various ruthenium(II) catalysts and found that a ruthenium catalyst with the association of bulky phosphine ligands or phosphine containing ruthenium compound [RuCl2(PPh3)(p-cymene)] preferentially formed a monoarylated product providing mono/diarylated product ratios up to 96/4.
In 2014, Luise et al. reported a chelated-assisted ruthenium(II)-catalysed arylation of 2-phenylpyridine with Ph2IOTf as arylating agent thatgave a high yield ofthe monoarylated product ofup to 95% (Scheme 11) [57].
They proposed that the arylation reaction proceeded by a mechanism similar to the Pd-catalysed C–H arylation reaction (Scheme 12). The reaction proceeded efficiently with [Ru(OAc)2(p-cymene)] in the presence of K2CO3 in toluene at 80 °C.
Following this, Reddy et al. described a versatile monoarylation of 2-phenylpyridine and phenylpyrazole using aryl boronic acids as arylating agent with [RuCl2(p-cymene)]2 in the presence of PhI(OCOCF3)2 (Scheme 13) [58].
The selective monoarylation of 2-phenylpyridine has been studied with various ruthenium catalysts by several research groups. Recently, Binnani et al. demonstrated a selective monoarylation of 2-phenylpyridine with aryl halide using the water-soluble ruthenium(II) aniline complex, [RuCl2(NH2-Ar)(p-cymene)] (Scheme 14) [59]. This C–H bond ortho-arylation reaction gave predominantly monoarylated products showing that the electronic and steric properties of the coordinated aniline ligands have a significant effect on the catalytic activity.
In 2011, a highly efficient catalytic system for the arylation of phenyl tetrazole with aryl bromide was reported by Seki using the ruthenium(III) catalyst [RuCl3·xH2O] [60]. This catalytic process provided a greener and more sustainable approach to the access of Angiotensin II Receptor Blockers (ARBs), which are considered among the most efficient antihypertensives [61]. Recently, Ackermann performed this arylation using the less expensive aryl chloride and stable ruthenium(II) catalyst [RuCl2(p-cymene)]2 in association with amino acid ligands in the presence of potassium carbonate in 1,4-dioxane (Scheme 15) [62].
Ackermann and co-workers described the arylation of 2-phenoxypyridine with aryl halides providing ready access to arylated phenols [63]. The reaction was performed with [RuCl2(p-cymene)]2 in the presence of an additive and a base. The presence of a base is essential for this reaction as no reaction occurred when performed without a base. They observed that the reaction conducted in N-Methyl-2-Pyrrolidone (NMP), a solvent usually used in ruthenium-catalysed direct arylation of arenes, gave a predominantly diarylated product. However, when the reaction was performed in toluene, improvement in the reaction efficacy and mono selectivity was observed. Thus, the best condition was obtained when the reaction was performed in toluene at 120 °C using mesitylic acid and K2CO3. They proposed that the reaction proceeds through a six-member ruthenacycle cyclometallated species. Furthermore, the arylated 2-phenoxypyridine gave a monoarylated pyridine on treatment of the arylated product of 2-phenoxypyridine with MeOTf followed by sodium in methanol (Scheme 16).
In 2014, a selective monoarylation of 2-pyridylketones with aryl bromide via six-membered ruthenium cyclometalated species was described by Dixneuf and co-workers, employing [RuCl2(p-cymene)]2 in the presence of a base and additive (Scheme 17) [64]. The reaction was performed in NMP, employing K2CO3 as a base and KOAC as an additive, leading to 82:18 mono/diarylated products. The selectivity was further enhanced by employing Na2CO3 as the base in the presence of 30 mol% of p-CF3C6H4CO2H, affording 90:10 mono/diarylated products.
The direct C–H arylation of 2-phenylpyridine with aryl chlorides in water has been reported by Kaloğlu et al. employing a ruthenium‒NHC complex as catalyst (Scheme 18) [65].
The reaction was performed in the presence of KOAc and a base, producing only themonoarylated product. The selective monoarylation of 2-phenylpyridine was also performed with [RuCl3·xH2O] using mesitylic acid as the additive in nontoxic PEG-2000 by Ackermann [9,66] (Scheme 19), followed by Luo and Yu [67], then by the research group of Gimeno in water [68].
2.3. Arylation of Arenes with Imine and Diazine as Directing Groups
The arylation of the alkenylic C–H bond containing diazine as a directing group has been reported by Dori et al. using aryl halide as an arylating agent with[RuCl2(p-cymene)]2 in the presence of a base (Scheme 20) [69]. The reaction gave isomeric products in some of the cases, which were further hydrogenated using the ruthenium catalyst that remained from the reaction of the first step, resulting in sp3C–H bond functionalization product. Thus, this catalytic process provided an alternative approach to the more difficult sp3C-H bond functionalization.
Recently, Poli and co-workers demonstrated a C-3 arylation of furfural imines with [Ru3(CO)12] in the presence of benzylidene acetone as a sacrificial hydride acceptor[70]. This arylation was performed using aryl boronates as an arylating agent in 1,4-dioxane, leading to C–H arylated furfural imines, which on subsequent acid hydrolysis gave C-3 arylated furfural (Scheme 21).
2.4. Arylation Involving Dehydrative, Oxidative and Weakly Coordinated Directing Groups
In 2008, Ackermann’s research group demonstrated a highly attractive ruthenium-catalysed dehydrative arylation of phenylpyrazole with phenol using [RuCl2(p-cymene)]2 first in DMA [71], subsequently in water employing [Ru(MesCO2)2(p-cymene)]2 as the catalyst [72]. This reaction protocol gave a direct C–H/O–H arylation with phenol in the presence of TsCl and a base (Scheme 22).
Recently, Roger et al. described the arylation of phenylpyrazole using a phenol derivative of aryl triflate as the arylating agent with [RuCl2(p-cymene)]2 in the presence of a base [73]. The reaction produced a mixture of monoarylated and diarylated products. However, when the reaction was conducted in the presence of additive, predominantly diarylated product was formed. The reaction was best performed with PivOH as an additive in trifluorotoluene, affording only diarylated azole in 99% (Scheme 23).
In 2011, Ackermann’s research group reported the arylation of indole and pyrrole containing a pyrimidyl group with aryl bromide, employing [RuCl2(p-cymene)]2 in the presence of (1-Ad)CO2H and a base [74]. In later years, Sollertet al. performed this arylation using the same catalyst but in the presence of an oxidant [75]. This oxidative arylation method gave slightly higher yields and a shorter reaction time (Scheme 24).
The ruthenium-catalysed ortho-arylation of N-alkyl benzamide with phenylboronic acid was described by Jagenmohan’s group using [RuCl2(p-cymene)]2 in the presence of AgSbF6 and the oxidant, Ag2O (Scheme 25) [76]. They found that the presence of Ag2O is essential for the arylation to occur as the absence of it resulted in no coupling reaction, even when using a stoichiometric amount of AgSbF6. Furthermore, they have shown that the ortho-arylated N-alkyl benzamide on treatment with trifluoroacetic acid and HCl could lead to fluorenones.
Recently, Szostak and co-workers described the first ruthenium-catalysed arylation of heteroarene containing a pyridyl ring as the directing group with an organosilane as the arylating agent using [RuCl2(p-cymene)]2 in the presence of AgSbF6 and an oxidant, CuF2 [77]. The oxidant CuF2 served a dual role-in both the arylsilane activation and oxidisation of Ru(0) to an active Ru(II) species. The reaction produced exclusively diarylated product (94%) as compared to monoarylated product (6%) when employing 2-phenylpyridine in DCE, but a modest yield of mono-selectivity was observed with other solvents such as toluene, THF, iPrOH, and DMF. Surprisingly, when the reaction was performed using Ag2O as an oxidant, it resulted in high monoselectivity with 95:5 monoarylated/diarylated products, but the overall yield was reduced to 40% (Scheme 26, Table 1).
In 2016, the research group of Szostak demonstrated the ortho-selective direct arylation ofcyclic and N,N-dialkylbenzamides containing a weakly coordinated group, using aryl boronic acid as arylating agent [78]. They performed the reaction with [RuCl2(p-cymene)]2 in the presence of AgSbF6 and Ag2O to give ortho-arylated products. The reaction has a wide substrate scope with a high selectivity of monoarylation, over 95%, thus providing the valuable tertiary amide biaryl compounds (Scheme 27).
Subsequently, they have conducted this direct arylation reaction employing arylsilanes as the arylating agent instead of aryl boronic acid [79]. This reaction showed a high chemo-selectivity and effective when [RuCl2(p-cymene)]2 was employed in association with AgSbF6 in the presence of CuF2 (Scheme 28). The reaction was best performed in THF or DCE affording only the monoarylated products.
Furthermore, in a continuation of the oxidative arylation using aryl silanes as the arylating agent, Szostak and co-workers have reported the ruthenium-catalysed arylation of pyrimidyl indole in water [80]. The reaction was performed with a [RuCl2(p-cymene)]2/AgSbF6 catalytic system in the presence of an oxidant, leading to selective arylation at the C-2 position (Scheme 29).
The direct ortho-C–H arylation of arenes bearing a pyrimidine ring as the directing group has been reported by Nakamura [81], Gu [82] and Cheng [83], and later by Pozgan [84]. Recently, Pozgan and co-workers demonstrated theruthenium-catalysed direct arylation of arenes with aryl bromides directed by quinozolene [85]. The arylation proceeded with [RuCl2(p-cymene)]2 in the presence of a base and an additive, affording ortho-diarylated arene as the major product along with minor monoarylated arene. The reaction was most effectively performed when 1-phenylcyclopentane-1-carboxylic acid (PCCA) was used as an additive in 1, 4-dioxane, affording only diarylated product in 98% yield (Scheme 30).
In 2014, Zhao et al. demonstrated the arylation of tertiary amide, viz.furan-3-carboxamide with [Ru(H2)(CO)(PPh3)3], to give 2-aryl-3-furanamides [86]. This direct arylation of carboxamide mostly employed directing groups containing hetero atoms such as pyridine, pyrazole and oxazoline. In the same year, Ackermann and co-workers demonstrated a triazole-assisted C–H bond arylation of an amide with aryl bromide using [RuCl2(PPh3)3] in the presence of Na2CO3 (Scheme 31) [87]. The reaction was favourably proceeded with secondary amide having an acidic –NH free group, while the tertiary amide with no free –NH group failed to give the desired product. They found that other amide derivatives such as pyridyl substituted amide or the substrate derived from 8-aminoquinoline gave inferior results, which indicated a superior directing-group power of the traiazole auxiliary for this reaction. The screening of the reaction showed that an electron donating substituent on the 1, 2, 3-triazole was most suitable for this amide C–H arylation.
The ruthenium-catalysed sp2-C–H bond arylation of arenes directed by ketones has been studied by several research groups. In 2015, Yamamoto and Yamakawa described the arylation of acylarene with halogenated aryl boronic acids using various ruthenium(II) catalysts, viz., [RuH2(CO)(PPh3)3], [RuHCl(CO)(PPh3)3], [RuCl2(PPh3)3] and [RuH2(PPh3)4], in the presence of Cs2CO3 [88]. They found that the ruthenium complexes [RuH2(CO)(PPh3)3] and [RuHCl(CO)(PPh3)3] were effective for the arylation in the presence of a base (10 mol%). When the reaction was performed in the absence of a base it resulted in extremely low yields. Moreover, the reaction was best performed when using a halogenated aryl boronic acid (Scheme 32).
Recently, Kakiuchi’s research group successfully utilized a cyano (–CN) group as the directing group in the ruthenium-catalysed C–H bond arylation reaction [89]. They performed the arylation of aromatic nitrile with aryl boronic esters using [RuH2(CO)(PAr3)3] in the presence of KHCO3, resulting in aortho-arylated product along with a minor para-diarylated product (Scheme 33).
In 2016, Huang et al. demonstrated the arylation of a carboxylic acid with aryl and heteroaryl halides leading to ortho-arylated products [90]. The reaction was conducted employing [RuCl2(p-cymene)]2 in the presence of K2CO3 and PCy3 ligand in NMP at 80 °C. This protocol was successfully used for the ortho-arylation of aromatic or heteroaromatic carboxylic acids with diverse aryl and heteroaryl halides. Interestingly, the reaction gave an annulation product when iodophenol was used in the reaction instead of aryl halide (Scheme 34).
2.5. Arylation of C–H Bond without a Directing Group
In 2016, Larrosa and co-workers described the C–H bond arylation of fluoroarenes with aryl halides using various ruthenium(II) complexes as the catalyst in the presence of a base [91]. They first performed the reaction with [Ru(OPiv)2(p-cymene)] in the presence of a base, but no desired product was formed. They observed a quantitative dissociation of p-cymene ligand at the end of the reaction. Therefore, in order to prevent the dissociation of the arene ligand, they performed the reaction withan analogous ruthenium complex [Ru(OPiv)2(C6Me6)] containing a less labile arene ligand, C6Me6. However, in this case only 32% of arylated product was observed despite only 14% of C6Me6 ligand being found to be dissociated during the reaction. From this finding, they hypothesized that, under the reaction conditions, the dissociation of arene ligand might be necessary for the reaction to occur. Furthermore, they proposed that a ruthenium complex without arene ligand could also promote a cross-coupling reaction. Previously, it was reported that the presence of p-cymene ligand was necessary for the cross-coupling of cycloruthenated phenylpyridine complexes [92]. Interestingly, they found a significant improvement in the reaction when an arene-free cationic ruthenium complex, [Ru(tBuCN)6](BF4)2, was employed, obtaining a good yield of the desired arylated product (Scheme 35).
Subsequently, they demonstrated ruthenium-catalysed arylation of aromatic acids with aryl iodides using the same cationic ruthenium catalyst, [Ru(tBuCN)6](BF4)2 [93]. The reaction was performed in the presence of K2CO3 and an additive such as KOPiv or KOC(CF3)3 in 1,4-dioxane, affording ortho-arylation of carboxylic acids without decarboxylation (Scheme 36).
2.6. Arylation Involving Decarboxylative and sp3 C–H Bond Functionalization
In 2006, Sames’s research group demonstrated the direct arylation of cyclic amines, especially pyrrolidines involving an sp3C–H bond functionalization, using ruthenium(0) catalyst, [Ru3(CO)12] and aryl boronic acid as arylating reagents [94]. In subsequent years, Peschiulli et al. performed a similararylation of a saturated cyclic amine with aryl boronic esters in the presence of [Ru3(CO)12] [95]. Following this early discovery, Mihovilovic and co-workers have shown that a ruthenium(II) catalyst could efficiently activate the sp3C–H bond of benzylic amine [96]. They performed sp3C–H bond arylation of benzylic amine using an aryl halide with [RuCl2(p-cymene)]2 in the presence of a catalytic amount of KOPiv and a base. In later year, this arylation reaction was performed by Ackermann and co-workers, employing [Ru(MesCO2)2(p-cymene)] without an additional carboxylic acid but in the presence of a base (Scheme 37) [97].
Arylation of a carboxylic acid with transition metals such as Pd or Ru often involved decarboxylation. In 2007, a new ruthenium(0)-catalysed reaction involving a decarboxylative arylation of cyclic-2-aminoester was described by Sames and co-workers [98]. The arylation was performed using Ru3(CO)12 with boronates in xylene at 180 °C, resulting in decarboxylative arylation at the sp3 carbon centre. Recently, Moselage et al. reported a highly efficient ruthenium(II)-catalysed arylation involving decarboxylative C–C bond functionalization (Scheme 38) [99]. They performed the reaction in the presence of a carboxylic acid and a base. The presence of both the carboxylic acid and a base is essential as the absence of either of them resulted in extremely poor yield or no reaction.
This reaction proceeded effectively using [RuCl2(p-cymene)]2 in association with mesitylic acid or employing [Ru(MesCO2)2(p-cymene)] directly without the additional use of mesitylic acid with compatible results; this clearly suggests that the reaction proceeded through a carboxylate-assisted route (Scheme 39). Among the carboxylic acids tested, mesitylic acid gave the most beneficial effects. However, the reaction performed with RuCl3(H2O)3 or Ru3(CO)12 did not give satisfactory results even in the presence of mesitylic acid under similar reaction conditions, indicating that Ru(III) and Ru(0) species were not suitable for this arylation [99].
3. Ruthenium-Catalysed C–H Bond Alkenylation
The direct alkenylation of the sp2 or sp3C–H bond is a powerful method in C–C cross-coupling reactions. Many aromatic and heteroaromatic C–H bonds could be readily alkenylated through transition metal catalysts, leading to various alkenylated products. Heck coupling is the most effective method in the alkenylation process. However, it requires a pre-functionalization of starting materials, which limits the scope of the reaction. The oxidative alkenylation of the C–H bond is also known as the Fujiwara‒Moritani reaction or oxidative Heck reaction, and was reported as early as 1967, long before the Heck reaction. The reaction involved the coupling of arenes with an alkene in the presence of palladium catalyst and oxidant to give an alkenylated product. Initially, palladium and rhodium catalysts contributed enormously tothis oxidative alkenylation; however, with the pioneering work of Milstein et al. [100] in 2001, followed by Yi and Lee [101], the ruthenium-catalysed oxidative alkenylation reaction has progressed rapidly [15]. The oxidative alkenylation of the C–H bond has been performed successfully using stable ruthenium(II)-catalysts from 2011 onwards by the groups of Satoh and Miura [102], Ackermann [103], Bruneau and Dixneuf [104], Jeganmohan [32] and several others [105,106].
3.1. Alkenylation Involving sp2C–H Bond Directed by Nitrogen-Containing Hetroaryl Ring
The ruthenium(II)-catalysed alkenylation of sp2C–H bond directed by the nitrogen N–Heteroaryl group has been studied by several groups from 2011 onwards. Dixneuf and co-workers described the alkenylation of 1-phenyl-1-pyrazole with acrylates using [Ru(OAc)2(p-cymene)] in the presence of an oxidant in acetic acid at 100 °C [104]. The reaction gave predominantly monoalkenylated product along with minor homocoupling product with alkyl acrylates, but a mixture of monoalkenylated and homocoupling product wasobserved in 56:44 ratios with styrene (Scheme 40). However, homocoupling product was formed exclusively when aless reactive alkene, such as N,N-dimethyl acrylamide, was employed [104].
Later, Dixneuf’s research group described the alkenylation of 2-phenyloxazoline with an alkene to give ortho-monoalkenylated and dialkenylated products [107]. The reaction was performed with [RuCl2(p-cymene)]2 in the presence of an additive and an oxidant in ethanol at 80 °C. They screened several additives, of which binaphthyl phosphoric acid (BNPAH) was found to be the most effective, leading to 85/14 mono/dialkenylated products with methyl acrylate, whereas mono-alkenylated product formed exclusively with styrene (Scheme 41).
Alkenyl esters and ethers were also successfully used in the alkenylation of 2-phenylpyridine derivative in the presence of ruthenium(0) catalyst [Ru(COD)(COT)] in xylene at 150 °C, affording exclusively monoalkenylated product [108]. In 2013, Ackermann and Ma demonstrated the ruthenium-catalysed oxidative C–H bond alkenylation of 2-phenoxypyridine possessing a removable directing group [109]. They performed the reaction using [RuCl2(p-cymene)]2 in association with AgSbF6 in the presence of Cu(OAc)2·H2O at 120 °C for 16h. The reaction involved C–H bond functionalization via the formation of a six-membered ruthenacycle as the key intermediate, affording a monoalkenylated product (Scheme 42).
Ruthenium-catalysed alkenylation has been studied with several other nitrogen heteroaryl directing groups. For instance, alkenylation of 2-phenylimidazo[1,2,a]pyridine leading to a monoalkenylated product was described by Sawant et al. employing [RuCl2(p-cymene)]2 in association with AgSbF6 in the presence of Cu(OAc)2·H2O in DCE (Scheme 43) [110]. The reaction proceeds regioselectively through ortho-C–H bond activation via the formation of a five-membered ruthenacycle intermediate.
Recently, Ramana and co-workers showed the C–H bond alkenylation of 2-pyridyl benzofurans, leading to C-3 alkylated and alkenylated products [106]. This approach is attractive as it could provide ready access to various biological relevant benzofuran analogues, many of which are the structural units of pharmacophores and natural products. They performed the reaction employing [RuCl2(p-cymene)]2 or [RuCl2(PPh3)3] in the presence of AgOAc as the additive with or without a base. The reaction produced only C-3 alkylated product when [RuCl2(p-cymene)]2 was used without a base,which is comparable to the yield of C-3 alkenylated product in the presence of K2CO3 (Scheme 44).
In 2017, Shome et al. demonstrated that 2-phenylpyrazole or 2-phenyloxazoline could be alkenylated with alkene employing [Ru(MesCO2)2(p-cymene)] in the presence of Cu(OAc)2·H2O at 100 °C under solvent-free condition (Scheme 45a) [111]. Recently, they also demonstrated the alkenylation of 1-phenylpyrazole with acrylate using a ruthenium(II) bipyridine complex, [Ru(MesCO2)(L)(p-cymene)]+, in the presence of Cu(OAc)2·H2O in ethanol at 80 °C, predominantly producing a monoalkenylated product (Scheme 45b) [112].
Recently, the alkenylation of benzimidazoles with acrylates affording various annulated products was performed by Selvaraju et al. using [RuCl2(p-cymene)]2 in the presence of AgSbF6 andCu(OAc)2·H2O in o-xylene (Scheme 46) [113].
They evaluated the efficacy of other oxidants such as CuBr2, AgOAc and AgCO3 as well, but they were found to be less effective. Furthermore, they proposed that the alkenylated product is formed by the protonation of a seven-membered ruthenacycle intermediate [113] (Scheme 47).
Gholamhosseyni and Kianmehr reported aruthenium-catalysed alkenylation‒annulation approach that leads to indazole derivatives [114]. The coupling of N-aryl pyridazinediones and N-aryl phthalazinediones with acrylates proceeded using [RuCl2(p-cymene)]2 in the presence of Cu(OAc)2·H2O and additive in water (Scheme 48). The reaction was most effective when AgSbF6 was used as the additive, giving up to 95% of the desired product. The desired product can be obtained in a competitive yield with KPF6. However, when conducted without an additive,the reaction resulted in lower efficacy, affording only a modest yield of the desired products.
3.2. Alkenylation with Carboxylic Acids and Sulfonic Acids
In 2011, Miura, Satoh and co-workers reported a carboxylic acid directed alkenylation of heteroaromatic sp2C–H bonds with acrylate [102]. The reaction was performed with [RuCl2(p-cymene)]2 in the presence of LiOAc and Cu(OAc)2.H2O in DMF, leading to ortho-monoalkenylated arenes without decarboxylation (Scheme 49).
In the same year, the research group of Ackermann reported the dehydrogenative alkenylation of aromatic carboxylic acids with [RuCl2(p-cymene)]2 in the presence of Cu(OAc)2.H2O in water [115], resulting in annulated lactones (Scheme 50). The optimization of the reaction showed that the reaction proceeded most efficiently without an additive; however, the presence of oxidant is essential as no reaction occurred in the absence of an oxidant.
Recently, Ackermann and co-workers have also demonstrated ruthenium-catalysed meta-selective C–H alkenylation of aromatic carboxylic acid involving decarboxylation [116]. This reaction proceeds efficient with 10 mol% of [Ru(MesCO2)2(p-cymene)] in the presence of an additive, affording meta-alkenylated product involving a decarboxylation under a nitrogen atmosphere (Scheme 51). Although several other additives such as Na2S2O7, tBuC(O)Me and MnO2 could be employed, the best result was obtained when V2O5 was used as the additive in toluene.
Recently, Jin et al. reported an intramolecular C–H bond alkenylation involving a decarboxylation of carboxylic acid with the 5 mol% of [RuCl2(p-cymene)]2 [117]. The reaction was performed in the presence of Cs2CO3 in aqueous media under oxygen, leading to tetrahydropyridoindoles atup to 78% yield (Scheme 52).
Dana et al. reported an orthoC–H bond monoalkenylation of an aromatic carboxylic acid with styrene without a decarboxylation [118]. The reaction proceedsefficiently with [RuCl2(p-cymene)]2 in the presence of CuO and KHPO4 in methanol at 85 °C to give the desired product at 92% yield. The product can be obtained as an ester derivative by further treatment of the acid derivative with methyl iodide (3 eq.), K2CO3 (2 equiv.) in acetonitrile at room temperature (Scheme 53).
They observed that an alteration of the base KHPO4, leads to inferior results, whereas replacing CuO by Cu(OAc)2 drastically reduced the yield to 31%. The reaction gave dialkenylated product when para-substituted benzoic acids were treated with an excess of styrene (Scheme 54).
In 2014, Ackermann and co-workers successfully performed a ruthenium-catalysed alkenylation of aromatic sulfonic acids, sulfonyl chlorides and sulfonamides [119]. This C–H bond alkenylation was achieved using [RuCl2(p-cymene)]2 in the presence of an additive in DMA at 120 °C. They found that the reaction can proceed even without an additive; however, the presence of AgSbF6 as an additive boosts the reaction efficacy considerably. Alkenylation of sulfonic acid or sulfonyl chloride with an alkene afforded mono-alkenylation, whereas oxidative alkenylation of sulphonamides led to C–H/N–H alkene annulation, affording a high yield of annulated products (Scheme 55).
3.3. Alkenylation Involving Weakly Coordinated Directing Group and Dehydrogenative Coupling
Weakly coordinated groups such as ketones, esters, amides and anilides have been successfully used as directing groups in ruthenium-catalysed C–H bond functionalization. The ruthenium-catalysed C–H alkenylation of aromatic ketones with olefins was initiated as early as 1993 with the pioneering work of Murai and co-workers [120], followed by several other groups [121,122,123]. However, in all these attempts, only alkylated products were observed. In 2012, Padala and Jeganmohan described the C–H bond alkenylation of anaromatic ketone with alkene using a [RuCl2(p-cymene)]2/AgSbF6 catalytic system in the presence of Cu(OAc)2·H2O in DCE (Scheme 56) [124]. The reaction proceeds in a highly regio- and stereoselective manner, resulting in the ortho-alkenylated product with acetophenone, but a mixture of mono- and dialkenylated products were observed for benzophenone.
Subsequently, Singh and Dixneuf demonstrated the alkenylation of a ferrocenyl ketone using [RuCl2(p-cymene)]2/AgSbF6 in the presence of Cu(OAc)2·H2O [125]. The reaction leads to the ortho-alkenylation of ferrocene C–H bond when ferrocenyl methyl ketone reacted with acrylate, obtaining a modest yield of the alkenylated product. However, when the reaction was performed with ferrocenyl phenyl ketone, ortho alkenylation occurred at the phenyl ring. The reaction proceeds efficiently under air at room temperature in DCE, leading to the ortho-alkenylated product athigh yield (up to 97%), demonstrating the strong influence of the ferrocenyl group on the arene C–Hfunctionalization.
In 2012, Jeganmohan’s research group described an alkenylation of the aromatic sp2C–H bond directed by an aldehyde [126]. The ortho-alkenylation was achieved using a [RuCl2(p-cymene)]/AgSbF6 catalytic system, which is usually employed for the C–H bond activation, directed by a weakly coordinated group such as ester and ketone. The alkenylation was performed in the presence of Cu(OAc)2·H2O in open air and the reaction proceeded in a highly stereoselective manner. Furthermore, they have shown that the alkenylated product could be converted into four-membered or five-membered cyclic ketones or a polycyclic isochromanone via a photochemical rearrangement (Scheme 57).
In the same year, Jeganmohan’s research group described the ruthenium(II)-catalysed alkenylation of aromatic estersto give ortho-alkenylated products [18]. The alkenylation was performed with a [RuCl2(p-cymene)]2/AgSbF6 catalytic system in the presence of Cu(OAc)2·H2Oin DCE under open air. The reaction was tolerated byvarious aromatic and heteroaromatic esters such as thiophene-3-carboxylate and indole-3-carboxylate, giving an excellent yield of the alkenylated products (Scheme 58).
In the following years, Jeganmohan and co-workers demonstrated the alkenylation of aromatic nitriles with activated alkenes using [RuCl2(p-cymene)]2 in the presence of AgSbF6 and AgOAc in acetic acid providing ortho-alkenylated products [127]. Initially, they reacted nitriles with activated alkenes in the presence of Cu(OAc)2·H2O but failed to produce alkenylated product; instead, the reaction only afforded an amide due to the hydration of nitrile, indicating that copper salts were not suitable for this reaction. They evaluated the alkenylation of nitrile using several Lewis acids such as Mn(OAc)2, AgOAc, Ag(CF3COO) and Ag2CO3, of which AgOAc produced the best results (Scheme 59).
Furthermore, Jeganmohan’s research group have shown that the alkenylation of 2-cyanothiophene gave a monoalkenylated product, whereas 3-cyanothiophene provided a dialkenylated product (Scheme 60).
In 2012, Miura and co-workers demonstrated the alkenylation of N,N-dimethyl benzamide with acrylate using a [RuCl2(p-cymene)]2/AgSbF6 catalytic system in the presence of Cu(OAc)2·H2O in tAmOH affording ortho-alkenylated products [19]. The dehydrogenative alkenylation of benzamide and anilide was described by the research group of Ackermann using a [RuCl2(p-cymene)]2 catalyst. This reaction was performed in water in the presence of an additive and an oxidant [128]. It was found that the most efficient catalysis could be achieved with a cationic ruthenium(II) species. Thus, employing KPF6 as an additive achieved the best catalytic activity, giving 87% yield, whereas AgSbF6 gave a modest yield of 54%. The presence of an additive is essential for this reaction as the absence of it resulted in extremely poor yield, giving only 2% under the same reaction conditions (Scheme 61).
In 2013, Li et al. demonstrated a ruthenium-catalysed C-2 alkenylation of pyrrole and indole derivative directed by an amide group [129]. The reaction proceeded regio- and stereoselectively with [RuCl2(p-cymene)]2 in the presence of an oxidant. In later years, Das and Kapoor demonstrated the ortho-alkenylation of amides including acyclic and cyclic Weinreb amides with alkenes using [RuCl2(p-cymene)]2 in the presence of additive and oxidant in 1,4-dioxane at 100 °C (Scheme 62) [130].
Recently, Ackermann and co-workers demonstrated a ruthenium-catalysed dehydrogenative alkenylation of aromatic tosylamide, aromatic acid and 2-phenoxypyridine with alkene using molecular oxygen as the sole oxidant [131]. The reaction proceeded efficiently with [RuCl2(p-cymene)]2 or [Ru(MesCO2)2(p-cymene)] in the presence of CsOAc or KOAc as additive under an oxygen atmosphere. The reaction of 2-phenoxypyridine and alkene led to ortho-alkenylated products; however, under similar reaction conditions tosylamide and aromatic carboxylic acid gave annulated products (Scheme 63a,b).
3.4. Alkenylation through Hydroarylation with Alkynes
Alkenylarenes could also be obtained efficiently via catalytic hydroarylation of alkynes. In 2008, Zhang and co-workers demonstrated for the first time the alkenylation of arylpyrimidine and aryl pyridine with terminal alkyne using a ruthenium catalyst [132]. This alkenylation of heteroarenes was performed using simple RuCl3 as the catalyst in the presence of potassium carbonate as a base. Subsequently, Wu’s group described the catalytic alkenylation of 2-phenylpyridine with terminal alkynes using ruthenium or zinc compounds containing 12 metallocrown-6 as catalysts in the presence of a base in NMP at 150 °C. In later years, Li et al. have extended the catalytic alkenylation via hydroarylation of alkynes to other arene compounds [133]. They performed the alkenylation of 2-phenyltriazole derivatives with alkynes using [RuCl2(p-cymene)]2/AgSbF6 in the presence of an additive, leading to dialkenylated products (Scheme 64).
The alkenylation via hydroarylation of alkynes was studied with several other arenes as well. Jeganmohan and Manikandan have described the alkenylation of anilide via hydroarylation using [RuCl2(p-cymene)]2 in the presence of AgSbF6 and pivalic acid at 100 °C [134]. They have shown that alkenylated products, on further treatment with 17% HCl:THF (1:1), could lead to the ortho-alkenylated anilines. Recently, Ackermann’s research group demonstrated the hydroarylation of carboxylic acids with diphenylacetylene to give alkenylated products (Scheme 65) [116]. This hydroarylation was performed with [Ru(MesCO2)2(p-cymene)] under nitrogen and the reaction involved a decarboxylation of carboxylic acid.
4. Ruthenium-Catalysed C–H Bond Alkylation and Allylation
The transition metal-catalysed C–H bond alkylation was usually achieved in two ways, namely the hydroarylation of alkene orthe alkylation with the un-reactive alkyl halides. In 1986, Lewis and Smith reported pioneering work on the regioselective ortho-alkylation via hydroarylation of an alkene. In later years, Ackermann and co-workers described the alkylation of 2-phenylpyridine with acrylates via hydroarylation using a ruthenium(II)-catalyst [135]. The reaction proceeded with [RuCl2(p-cymene)]2 in the presence of an additive in toluene or 1,4-dioxane at 80–120 °C (Scheme 66). The reaction achieved the highest efficacy when MesCO2K and AdCO2K were used as additives. However, when the reaction was conducted using anadditive such as PPh3, AgOTf or AgOAc, it resulted in little or no beneficial effect. On the other hand, no reaction occurred without an additive or when employing KPF6 or NaPF6 as the additive.
Subsequently, Ackermann’s research group described an sp3C–H bond alkylation of aryl pyrrolidines with alkenes via hydroarylation using [RuCl2(PPh3)3] in the presence of BINAP and AgOTf [136]. They have shown that the pyridyl group could be removed to generate an N–H-free amine by treatment of the product with Pt/C, followed by acid hydrolysis and fluxing with hydrogen gas (Scheme 67).
In 2010, Cadierno et al. described theC-3 alkylation of indole with a terminal alkyne in the presence of ruthenium catalysts [137]. The reaction has been performed with 2 mol% of ruthenium catalyst in the presence of TFA in water at 100 °C for 24h. They screened the reaction using various ruthenium catalysts such as [RuCl2(η6-C6Me6)]2, [RuCl2(PPh3)3], [RuCl2(p-cymene)]2, [RuCl3·xH2O], etc., of which [RuCl2(p-cymene)]2 and [RuCl2(η6-C10H16)]2 gave the best results (Scheme 68).
The direct alkylation or arenes with alkyl halides is another pathway usually employed to obtain alkylated products. The direct alkylation with alkyl halides was initially studied with palladium catalysts, but the reaction scope was limited to heteroarenes [138,139]. In 2009, Ackermann’s research group demonstrated the direct alkylation of aryl pyridines with alkyl halides using in expensive ruthenium catalysts [140]. The reaction proceeded efficiently with [RuCl2(p-cymene)]2 in the presence of AdCO2H and K2CO3 in NMP at 120 °C. The reaction has a wide substrate scope and could also be performed using pyrazoles and ketimines. In the following years, his research group demonstrated a meta-selective C–H bond alkylation of aromatic C–H bond of ketimine with secondary alkyl halide using [Ru(MesCO2)2(p-cymene)] in the presence of a base in 1,4-dioxane at 100 °C (Scheme 69) [141].
Recently, Li et al. reported a meta-selective C–H bond alkylation of phenol derivative using secondary or tertiary alkyl bromide with [RuCl2(p-cymene)]2 in the presence of AdCO2H and K2CO3 in benzene [142]. Interestingly, the directing group could be removed by treatment of the product with MeOTf followed by dry methanolic solution of sodium to give meta-alkylated phenol derivative (Scheme 70). Notably, this reaction has found application in the synthesis of etoxazole, a marketed pesticide.
Furthermore, Li’s research group have demonstrated a ruthenium-catalysed ortho- and meta-selective C–H bond alkylation of azoarenes using alkyl bromides [143]. The reaction was performed with [RuCl2(p-cymene)]2 in the presence of carboxylic acid and a base in dioxane. The presence of carboxylic acid ligand is essential for this reaction. The most efficient ligand was found to be tBuCO2H, which gave 75% of the alkylated product, whereas employing MesCO2H gave a moderate yield, 35%. Interestingly, the meta-selective alkylated product was obtained with secondary and tertiary alkyl bromides, whereas primary alkyl bromide gave an ortho-alkylated product (Scheme 71).
Recently, Ackermann and co-workers demonstrated a highly efficient ruthenium-catalysed C–H bond alkylation reaction to give a diverse meta-alkylated product [144]. The reaction proceeded profitably with [RuCl2(p-cymene)]2 (5 mol%) in the presence of carboxylic acid and K2CO3 in tert-butylbenzene at 120 °C (Scheme 72). This reaction has a broad substrate scope and thus could provide ready access to meta-alkylated arenes including ketones, alcohols, amines and acids by direct reaction with alkyl bromides.
In 2017, Frost and co-workers described a ruthenium-catalysed para-selective alkylation of aniline withmethyl-α-bromoisobutyrate [145]. The reaction was performed with [RuCl2(p-cymene)]2 in the presence of K2CO3 and a carboxylate ligand in tert-butyl methyl ether (TBME) at 120 °C (Scheme 73). Interestingly, no reaction occurred when the reaction was conducted withouta carboxylate ligand. However, the reaction being performed at a higher temperature, say 140 °C, led to the formation of a minor amount of meta-alkylated product.
In the same year, Frost and co-workers also demonstrated the ruthenium-catalysed remote C-6 alkylation of N-pyrimidinyl indole with ethyl α-bromoisobutyrate [146]. The reaction proceeded with [RuCl2(p-cymene)]2 (5 mol%) in the presence of a base and an additive at 120 °C under air (Scheme 74). The dialkylated product involving a remote C-6 alkylation was obtained in 57% yield along with 39% of the C-3 monoalkylated product when the reaction was performed with 2 eq. of AcOH and KOAc in THF. The reaction conducted in other solvents such as DME or 1, 4-dioxane gavedialkylated and monoalkylated products in quantitative yields. However, the alkylation proceeded selectively, giving 91% monoalkylated and only 5% dialkylated product when the reaction was performed in acetic acid.
Allylic alcohol could also participate as an alkylating agent in the ruthenium-catalysed alkylation reaction. Thus, theruthenium-catalysed alkylation of an aromatic carboxylic acid with allylic alcohol has been reported by Kumar et al. using [RuCl2(p-cymene)]2 in the presence of Cu(OAc)2 and KOAc in dichloroethane to give the desired alkylated product [147]. However, the reaction conducted using AgSbF6 as an additive in place of KOAc in THF did not produce alkylated product; instead, an alkenylated product was formed (Scheme 75).
The dehydrogenative benzylation of 1,2,3,4-tetrahydroquinolines with aryl aldehydes was carried out catalytically by Zhang and co-workers, employing [RuCl2(p-cymene)]2. This reaction requires nitrobenzoic acid as an additive and was performed under an oxygen atmosphere at 120 °C, affording the functionalized quinolines (Scheme 76) [148].
Several ruthenium compounds have been used in the catalytic allylation using allyl halide or allyl acetate. In 2006, Oi et al. reported ortho-allylation of 2-phenylpyridine with allyl acetate using [RuCl2(cod)]n in association with PPh3 in the presence of K2CO3 [149]. Later, Zhang and co-workers performed the ortho-allylation of 2-phenylpyridine with terminal alkynes using RuCl3 as the catalyst in the presence of benzoic acid and K2CO3 [132]. Following this early discovery, Goriya and Ramana described the allylation of 2-phenylpyridine with allyl bromide using [RuCl2(p-cymene]2 in the presence of K2CO3 and AdCO2H in toluene (Scheme 77) [150]. However, this reaction did not give ortho-allylation product but instead afforded a C-2 allylation product on the pyridine ring of the 2-phenylpyridine.
In 2015, Jeganmohan’s research group succeeded in the ortho-allylation of 2-ketoxime with allyl acetate using [RuCl2(p-cymene)]2 in the presence of AgSbF6 in DCE at room temperature (Scheme 78) [151]. They found that the use of a silver salt such as AgSbF6, AgBF4 or AgOTf as an additive is essential for the reaction to occur.
Regio- and stereoselective allylation of indole derivatives with allylic alcohols was reported by Wu and Ji using [RuCl2(p-cymene)]2 in the presence of NaOAc in DCE to give a C-2 allylated product (Scheme 79) [152]. This allylation reaction was performed under mild conditions and was free from oxidant.
5. Ruthenium-Catalysed Annulation Reaction of Arenes with Alkynes
Annulation is one of the most convenient methods for the synthesis of various heterocyclic compounds. Several transition metal catalyst derivatives of Cu [153], Pd [154,155], Ru [26,156], Rh [157] and Ir [158] promote these annulation reactions, providing ready access to various heterocycles. Recently, ruthenium-catalysed C–H/O–H, C–H/N–H bond cleavage reactions leading to pyrroles [159,160], indoles [161], isoquinolines [162], isocoumarins [163], etc. have been reported by several groups.
In 2012, Ackermann’s research group demonstrated annulations of phenylpyrazole and alkyne with a [RuCl2(p-cymene)]2/AgSbF6 catalytic system in the presence of Cu(OAc)2·H2O under air [164]. The reaction did not require a prefunctionalized pyrazole such as the 3-(2-bromophenyl)pyrazole derivative that has been used in similar annulation reactions with copper catalysts [165]. Subsequently, Ackermann’s research group reported oxidative annulation involving C–H/N–H bond functionalization of aniline derivatives containing a removable directing group with [RuCl2(p-cymene)]2 in the presence of KPF6 and Cu(OAc)2·H2O in water at 100 °C, affording diverse indole derivatives [166]. Furthermore, Ackermann and co-workers havedescribed the ruthenium-catalysed annulation of 2-arylsubstituted pyrroles with alkynes, leading to pyrrolo[2,1-a]isoquinolines, a structural unit of biologically active marine lamellarin alkaloids [167]. The reaction involved a C–H/N–H bond cleavage process and proceeded efficiently with [RuCl2(p-cymene)]2 in the presence of Cu(OAc)2·H2O in tAmOH at 100 °C (Scheme 80).
In 2012, Jeganmohan’s research group demonstrated the ruthenium-catalysed regioselective annulation of aromatic carboxylic acids and alkynes, leading to isocoumarins [168]. The reaction was conducted employing a [RuCl2(p-cymene)]2/AgSbF6 catalytic system with Cu(OAc)2·H2O as oxidant and involved a C–H/O–H bond cleavage process (Scheme 81).
In the same year, Ackermann and co-workers demonstrated the annulation of aromatic acids and alkynes with [RuCl2(p-cymene)]2 in the presence of KPF6 and Cu(OAc)2·H2O [169]. The reaction was best performed in tAmOH, giving 87% of the desired isocoumarin for diphenylacetylene. However, the reaction was not productive when performed in water, giving only 52% of the desired isocoumarin (Scheme 82).
Subsequently, Ackermann’s group demonstrated the annulation of aromatic acids and alkynes using molecular oxygen as the sole oxidant instead of Cu(OAc)2·H2O, which is usually employed in oxidative annulation reactions [170]. They performed the annulation with [Ru(MesCO2)2(p-cymene)] in the presence of NaOAc under 1 atmospheric pressure of oxygen or air in methanol, leading to isocoumarin in high yields (Scheme 83).
They proposed a plausible catalytic cycle thatinvolves an initial formation of ruthenium(II) bis-acetate complex (Scheme 84). Thereby, a five-member ruthenacycle is generated along with two equivalent of acetic acid. The next step is the coordination and insertion of alkyne to furnish a seven-membered Ru(II) cycle that rapidly undergoes reductive elimination to yield a ruthenium(0) species, which is then reoxidized to the active ruthenium(II) species by molecular oxygen.
The reaction was evaluated using other ruthenium catalysts as well, such as [RuCl2(p-cymene)]2 and [RuCl2(benzene)]2, which produced the annulated products in 78% and 76% yields, respectively, with diphenylacetylene. However, under similar conditions, the reaction performed in other alcohols was not productive; only traces of isocoumarins were formed.
Recently, Singh and Dixneuf described the ruthenium-catalysed annulation of pyrrole and indole carboxylic acid derivatives with alkynes, leading to pyrrole and indole-fused isocoumarins (Scheme 85) [171]. The reaction was performed using [RuCl2(p-cymene)]2 in the presence of Cu(OAc)2·H2O but without an additive in DMF. The reaction could also be performed profitably in water with competitive yields. The reaction of 1-methylpyrrole-2-carboxylic acid showed a high regiospecificity when performed with an unsymmetrical alkyne, 1-phenyl-1-propyne, giving only one regioisomer in 90% yield. High regioselectivity was observed in water as well for the annulation of 1-methylpyrrole-2-carboxylic and 1-phenyl-1-propyne. However, the annulation of 1-methylindole-3-carboxylic acid and 1-phenyl-1propyne afforded two isomers in 61% and 13% isolated yields.
Subsequently, Singh et al. have reported the annulation of N–Heteroaromatic acid derivatives with alkynes using a ruthenium(II) N^O chelate complex, [RuCl(PySO3)(p-cymene)] (Scheme 86) [172]. They found that the efficiency of [RuCl(PySO3)(p-cymene)] is comparable to that of [RuCl2(p-cymene)]2 for the reaction conducted in DMF, but the reaction performed with [RuCl(PySO3)(p-cymene)] was not as productive as [RuCl2(p-cymene)]2 when performed in water.
The ruthenium-catalysed regioselective cyclization of ketones and alkynes leading to indenols and benzofulvenes was described by Chinnagolla and Jeganmohan using [RuCl2(p-cymene)]2 in association with AgSbF6 in the presence of Cu(OAc)2·H2O in DCE [173]. In the following year, they achieved ruthenium-catalysed cyclization of aromatic nitriles and alkynes employing [RuCl2(p-cymene)]2 in the presence of KPF6 and Cu(OAc)2.H2O in acetic acid under air (Scheme 87) [174].
Jeganmohan co-workers found that the active catalyst was a cationic ruthenium(II) species and the reaction involved deprotonation from aromatic C–H bond, resulting in the formation of a five-membered ruthenacycle as an intermediate. In 2012, the ruthenium(II)-catalysed oxidative coupling of alkyne and naphthol assisted by hydroxyl groups was described by Ackermann and co-workers using [RuCl2(p-cymene)]2 in the presence of Cu(OAc)2·H2O [175]. The presence of Cu(OAc)2·H2O was essential for the reaction, as replacing it with CuBr2 or leaving it out resulted in no reaction. However, CuBr2 in association with NaOAc could be employed for this reaction,which leads to a moderate yield of the desired product. This catalytic system was also effective for the annulation of 4-hydroxycoumarin and alkynes. This annulation reaction was best performed in m-xylene at 80 °C, affording annulated product pyrans in high yields (Scheme 88).
In 2012, Zhao’s research group demonstrated the annulation of N-sulfonyl imines and alkynes leading to indenamines [176]. The reaction was performed using [RuCl2(p-cymene]2 in association with sulphonamides and AgSbF6 and 4 equiv. of AcOH in dioxane. The coupling product, indenamines, were obtained at a modest to good yield in the range of 46–88%. In the following year, Zhao’s research group described a ruthenium-catalysed carbocyclization of aromatic ketimines and alkynes (Scheme 89) [177]. The reaction proceeded efficiently with [Ru(cod)(η3-C4H7)2] in the presence of a NHC ligand (IPr) in toluene at room temperature. Zhao and co-workers also evaluated this reaction with a doubly cyclometalated complex, [Ru(cod)(C,N-ketimine)2], which was produced from the reaction of [Ru(cod)(η3-C4H7)2] with diphenylacetylene. However, this doubly cyclometalated complex alone could not catalyse the coupling of ketimene and alkyne. On the contrary, coupling was promoted in the presence of an NHC ligand affording the annulated product in 45% yield. The reaction was found to be less effective when a saturated NHC ligand (SIPr) was employed. The best reaction condition was achieved when employing [Ru(cod)(η3-C4H7)2] in association with unsaturated NHC ligand (IPr) in hexane at room temperature.
Oxidative coupling of aryl benzimidazole with alkynes has been described employing [RuCl2(p-cymene)]2 as a catalyst in the presence of Cu(OAc)2·H2O in toluene [178]. The reaction could also be conducted using other oxidants such as NaOAc or Ag2CO3 with a lower efficacy (Scheme 90).
Urriolabeitia and co-workers have described an oxidative coupling of primary amines with alkynes through ruthenium catalysis to give isoquinoline derivatives (Scheme 91) [179]. The coupling proceeds with [RuCl2(p-cymene)]2 (10 mol%) in the presence of KPF6 and Cu(OAc)2 in alcohol. The reaction requires 24 h for optimum results in conventional heating but can also be run for only 15 min in a microwave.
In 2015, the ruthenium(II)-catalysed oxidative annulation of 6-aminopurines with alkynes, leading to indole-substituted purines and purine nucleosides,was described by Kumara Swamy and co-workers (Scheme 92) [180]. The annulation was performed employing [RuCl2(p-cymene)]2 (5 mol%) in the presence of an additive in methanol. Among the additives tested, CsOAc was found to be the most effective, followed by Cu(OAc)2·H2O and NaOAc. However, the reaction conducted in DCE gave the desired products only in traces, whereas the reaction performed in water or tBuOH wasfound to be unreactive.
Recently, Sharma et al. described the coupling of alkynes with amine-containing benzothiazole as a removable directing group (Scheme 93) [181]. The ruthenium catalyst employed was the most widely explored, [RuCl2(p-cymene)]2, in association with AgSbF6 in the presence of Cu(OAc)2 in toluene. The coupling involves various types of alkynes, including aryl‒aryl, aryl‒alkyl and alkyl‒alkyl. However, a reaction performed with unsymmetrical alkynes afforded a mixture of regioisomers, whereas dicarboxylated alkynes furnished monodecarboxylated products.
The ruthenium(II)-catalysed decarbonylative annulation of 3-hydroxy-2-phenylchromones with alkynes has been reported by Gogoi and co-workers (Scheme 94) [182]. The reaction was performed with [RuCl2(p-cymene)]2 in the presence of PPh3 and CsOAc in tAmOH, leading to spirobenzofuranones in up to 86% yield. They proposed that the annulation occurred before the extrusion of CO, unlike other transition metal-catalysed decarbonylative annulation reactions. This annulation has proceeded via C–H/C–C activation, alkyne insertion, and decarbonylation to provide spiro-indenebenzofuranones in good yields.
In 2015, a ruthenium-catalysed annulation reaction dictated by a weaker directing group was demonstrated by Patel and co-workers using [RuCl2(p-cymene)]2 in the presence of an oxidant (Scheme 95) [183]. They performed the annulation of 2-arylquinolinone and 2-arylbenzoxazinone, which contained two different directing arenes with alkynes using [RuCl2(p-cymene)]2 in the presence of an oxidant. They observed that the regioselective annulations of 2-arylquinolinone and 2-arylbenzoxazinone containing two different directing arenes with alkynes occurred viathe assistance of weaker carbonyl oxygen over the stronger nitrogen directing site, thus the weaker directing groups dictated the annulation reaction path. Furthermore, the reaction performed in DCE using Cu(OAc)2 as oxidant produced 2-arylquinoline in 68% yield and 2-arylbenzoxazinone in 11%. However, when the reaction was conducted in acetic acid employing AgOAc as an oxidant, 2-arylbenzoxazinone was formed in high yield (78%).
Recently, Xu and co-workers demonstrated the ruthenium-catalysed dehydrogenative annulation of aniline derivative and alkynevia electrochemical-induced hydrogen generation,leading to indole derivatives [184]. The reaction was conducted under constant current electrolysis consisting of an undivided cell equipped with a reticulated vitreous carbon (RVC) anode and a Pt plate cathode. The reaction proceeded efficiently under reflux condition with [RuCl2(p-cymene)]2 in the presence of KPF6 and NaOAc in water or solvent containing a mixture of water:alcohol (1:1) or water:acetonitrile (1:1) (Scheme 96). However, the best result was obtained when it was conducted in water:tAmOH (1:1), affording 92% of the desired indole derivatives.
Haak and co-workers reported a cascade annulation reaction of indole with propargyl alcohols using a bifunctional (cyclopentadienone) ruthenium(0) complex in the presence of trifluoroacetic acid, providing a natural product like scaffolds [185] (Scheme 97). This catalytic process can be conveniently performed with microwave irradiation instead of conventional heating. The reaction proceeded with high selectivity to provide ready access to various annulated products from simple indolestopropargyl alcohols.
6. Conclusions
Catalytic C–H bond functionalization with ruthenium-based catalysts has emerged a powerful method in organic synthesis for ready access to various functional molecules in a greener and time-saving manner. The ruthenium catalysts participating in this C–H bond functionalization reactions are mainly ruthenium(0) and ruthenium(II) species, although a few reactions were also known with the simple RuCl3 species. The C–H bond functionalization reactions with ruthenium complexes such as N^O and O^O chelate ruthenium(II) complexes, as well as ruthenium(II) amine and cationic ruthenium complexes, are presented in this review. In the C–H bond functionalization via ruthenium catalysis, the stable ruthenium(II) catalysts, in particular the [RuCl2(p-cymene)]2 precursor complex, has contributed enormously, and indeed several new reactions and methodologies are being discovered with this ruthenium(II) species. Furthermore, the ruthenium(II) catalysts partnering with carboxylate ligand allowed C–H bond functionalization in a highly attractive manner, providing profitable organic transformations. Several C–H bond functionalization reactions, namely arylation, alkenylation, alkylation and annulation, have been studied, employing ruthenium(II) catalysts partnering with carboxylate ligands in an efficient manner with a better atom economy; sometimes reactions are even conducted in water. This review presents previous reports on recent developments in C–H bond functionalization reactions, covering arylation, alkenylation, alkylation, allylation and annulation reactions. The arylation of nitrogen N–heteroarenes with aryl halides, aryl boronates and aryl silanes as the arylating agents has been discussed. Furthermore, oxidative arylation, decarboxylative arylation and selective monoarylation of 2-phenylpyridine have been presented. Section 3, Section 4 and Section 5 discussed C–H bond functionalization such as alkenylation, alkylation, allylation and annulation of various arenes. In brief, this review has highlighted the various reactions mentioned above with selected reaction mechanisms that appeared over the past eight years. It is hoped that this review might be helpful in further enhancing the research area, which is already a hot topic in the field of catalysis. Furthermore, I believed that the review will be useful to young researchers working on the metal-catalysed C–H bond functionalizationin providing up-to-date information in this field.
Acknowledgments
The author thanks the Director of the CSIR-National Institute of Oceanography for encouragement and Department of Science and Technology (DST) India for financial support.
Conflicts of Interest
The author declares no conflict of interest.
References
- Wang, S.; Fang, K.; Dong, G.; Chen, S.; Liu, N.; Miao, Z.; Yao, J.; Li, J.; Zhang, W.; Sheng, C. Scaffold Diversity Inspired by the Natural Product Evodiamine: Discovery of Highly Potent and Multitargeting Antitumor Agents. J. Med. Chem. 2015, 58, 6678–6696. [Google Scholar] [CrossRef] [PubMed]
- Bathula, C.; Dangi, P.; Hati, S.; Agarwal, R.; Munshi, P.; Singh, A.; Singh, S.; Sen, S. Diverse synthesis of natural product inspired fused and spiro-heterocyclic scaffolds via ring distortion and ring construction strategies. New J. Chem. 2015, 39, 9281–9292. [Google Scholar] [CrossRef]
- Tamao, K.; Sumitani, K.; Kumada, M. Selective carbon-carbon bond formation by cross-coupling of Grignard reagents with organic halides. Catalysis by nickel-phosphine complexes. J. Am. Chem. Soc. 1972, 94, 4374–4376. [Google Scholar] [CrossRef]
- Corriu, R.J.P.; Masse, J.P. Activation of Grignard reagents by transition-metal complexes. A new and simple synthesis of trans-stilbenes and polyphenyls. J. Chem. Soc. Chem. Commun. 1972, 144. [Google Scholar] [CrossRef]
- Ruiz, S.; Villuendas, P.; Urriolabeitia, E.P. Ru-catalysed C–H functionalisations as a tool for selective organic synthesis. Tetrahedron Lett. 2016, 57, 3413–3432. [Google Scholar] [CrossRef]
- Simonetti, M.; Cannas, D.M.; Baringo, X.J.; Vitorica-Yrezabal, I.J.; Larrosa, I. Cyclometallated ruthenium catalyst enables late-stage directed arylation of pharmaceuticals. Nat. Chem. 2018, 10, 724–731. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, J.; Yamaguchi, A.D.; Itami, K. C-H bond functionalization: emerging synthetic tools for natural products and pharmaceuticals. Angew. Chem. Int. Ed. 2012, 51, 8960–9009. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Bulger, P.G.; Sarlah, D. Palladium-catalyzed cross-coupling reactions in total synthesis. Angew. Chem. Int. Ed. 2005, 44, 4442–4489. [Google Scholar] [CrossRef]
- Ackermann, L.; Vicente, R.; Kapdi, A.R. Transition-metal-catalyzed direct arylation of (hetero)arenes by C-H bond cleavage. Angew. Chem. Int. Ed. 2009, 48, 9792–9826. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Bois, J.D.; Yu, J.Q. C-H functionalization in organic synthesis. Chem. Soc. Rev. 2011, 40, 1855–1856. [Google Scholar] [CrossRef]
- Lia, B.J.; Shi, Z.J. From C(sp2)-H to C(sp3)-H: systematic studies on transition metal-catalyzed oxidative C-C formation. Chem. Soc. Rev. 2012, 41, 5588–5598. [Google Scholar] [CrossRef] [PubMed]
- Lyons, T.W.; Sanford, M.S. Palladium-catalyzed ligand-directed C–H functionalization reactions. Chem. Rev. 2010, 110, 1147–1169. [Google Scholar] [CrossRef]
- Lewis, J.C.; Bergman, R.G.; Ellman, J.A. Direct functionalization of nitrogen heterocycles via Rh-catalyzed C–H bond activation. Acc. Chem. Res. 2008, 41, 1013–1025. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.C.; Wiedemann, S.H.; Bergman, R.G.; Ellman, J.A. Arylation of heterocycles via rhodium-catalyzed C-H bond functionalization. Org. Lett. 2004, 6, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Arockiam, P.B.; Bruneau, C.; Dixneuf, P.H. Ruthenium(II)-catalyzed C-H bond activation and functionalization. Chem. Rev. 2012, 112, 5879–5918. [Google Scholar] [CrossRef]
- Manikandan, R.; Jeganmohan, M. Recent advances in the ruthenium(II)-catalyzed chelation-assisted C–H olefination of substituted aromatics, alkenes and heteroaromatics with alkenes via the deprotonation pathway. Chem. Commun. 2017, 53, 8931–8947. [Google Scholar] [CrossRef]
- Kondo, H.; Akiba, N.; Kochi, T.; Kakiuchi, F. Ruthenium-catalyzed monoalkenylation of aromatic ketones by cleavage of carbon–heteroatom bonds with unconventional chemoselectivity. Angew. Chem. Int. Ed. 2015, 54, 9293–9297. [Google Scholar] [CrossRef]
- Padala, K.; Pimparkar, S.; Madasamy, P.; Jeganmohan, M. Ruthenium-catalyzed regioselective oxidative coupling of aromatic and heteroaromatic esters with alkenes underan open atmosphere. Chem Commun. 2012, 48, 7140–7142. [Google Scholar] [CrossRef]
- Hashimoto, T.; Ortloff, Y.; Hirano, K.; Satoh, T.; Bolm, C.; Miura, M. Ru/Ag-catalyzed oxidative alkenylation of benzamides and phenylazoles through regioselective CH bond cleavage. Chem. Lett. 2012, 41, 151–153. [Google Scholar] [CrossRef]
- Mei, R.; Zhang, S.K.; Ackermann, L. Ruthenium(II)-catalyzed C–H alkynylation of weakly coordinating benzoic acids. Org. Lett. 2017, 19, 3171–3174. [Google Scholar] [CrossRef]
- Kishor, P.; Jeganmohan, M. Ruthenium-catalyzed oxidative ortho-benzoxylation of acetanilides with aromatic acids. Chem. Commun. 2013, 49, 9651–9653. [Google Scholar]
- Pimparkar, S.; Padala, K.; Jeganmohan, M. Ruthenium(II)-catalyzed ortho C-O bond formation of substituted aromatics with oxygen nucleophiles throughC-H bond activation. Proc. Indian Nat. Sci. Acad. 2014, 80, 999–1011. [Google Scholar] [CrossRef]
- Sarkar, S.D.; Liu, W.; Kozhushkov, S.I.; Ackermann, L. Weakly coordinating directing groups for ruthenium(II)- catalyzed C-H activation. Adv. Synth. Catal. 2014, 356, 1461–1479. [Google Scholar] [CrossRef]
- Das, R.; Kumar, G.S.; Kapur, M. Amides as weak coordinating groups in proximal C-H bond activation. Eur. J. Org. Chem. 2017, 5439–5459. [Google Scholar] [CrossRef]
- Ackermann, L. Carboxylate-assisted transition-metal-catalyzed C-H bond functionalizations: Mechanism and scope. Chem. Rev. 2011, 111, 1315–1345. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, L. Carboxylate-assisted ruthenium-catalyzed alkyne annulations by C-H/Het-H bond functionalizations. Acc. Chem. Res. 2014, 47, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, L.; Hofmann, N.; Vicente, R. Carboxylate-assisted ruthenium-catalyzed direct alkylations of ketimines. Org. Lett. 2011, 13, 1875–1877. [Google Scholar] [CrossRef] [PubMed]
- Wenbo, L.; Arockiam, P.B.; Fischmeister, C.; Bruneau, C.; Dixneuf, P.H. C–H bond functionalisation with [RuH(codyl)2]BF4 catalyst precursor. Green Chem. 2011, 13, 2315–2319. [Google Scholar]
- Gray, A.; Tsybizova, A.; Roithova, J. Carboxylate-assisted C–H activation of phenylpyridines with copper, palladium and ruthenium: a mass spectrometry and DFT study. Chem. Sci. 2015, 6, 5544–5553. [Google Scholar] [CrossRef]
- Li, B.; Dixneuf, P.H. sp2 C–H bond activation in water and catalytic cross-coupling reactions. Chem. Soc. Rev. 2013, 42, 5744–5767. [Google Scholar] [CrossRef]
- Ackermann, L.; Fenner, S. Ruthenium-catalyzed C–H/N–O bond functionalization: Green isoquinolone syntheses in water. Org. Lett. 2011, 13, 6548–6551. [Google Scholar] [CrossRef] [PubMed]
- Padala, K.; Jeganmohan, M. ortho-benzoxylation of N-alkyl benzamides with aromatic acids catalyzed by ruthenium(II) complex. Chem. Eur. J. 2014, 20, 4092–4097. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Abdukader, A.; Han, J.; Cheng, Y.; Zhu, C. Ruthenium-catalyzed C7 amidation of indoline C-H Bonds with sulfonyl azides. Chem. Eur. J. 2014, 20, 3606–3609. [Google Scholar] [CrossRef] [PubMed]
- Shang, M.; Zeng, S.H.; Sun, S.Z.; Dai, H.X.; Yu, J.Q. Ru(II)-catalyzed ortho-C–H amination of arenes and heteroarenes at room temperature. Org. Lett. 2013, 15, 5286–5289. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Ackermann, L. Versatile ruthenium(II)-catalyzed C–H cyanations of benzamides. Chem. Commun. 2014, 50, 1878–1881. [Google Scholar] [CrossRef] [PubMed]
- Teskey, C.J.; Lui, A.Y.W.; Greaney, M.F. Ruthenium-catalyzed meta-selective C-H bromination. Angew. Chem. Int. Ed. 2015, 54, 11677–11680. [Google Scholar] [CrossRef]
- Shu, S.; Fan, Z.; Yao, Q.; Zhang, A. Ru(II)-catalyzed direct C(sp2)–H activation/selenylation of arenes with selenyl chlorides. J. Org. Chem. 2016, 81, 5263–5269. [Google Scholar] [CrossRef]
- Li, B.; Dixneuf, P.H. Ruthenium(II)-catalysed sp2 C-H bond functionalization by C–C bond formation. Top. Organomet. Chem. 2014, 48, 119–194. [Google Scholar]
- Thirunavukkarasu, V.S.; Kozhushkov, S.I.; Ackermann, L. C–H nitrogenation and oxygenation by ruthenium catalysis. Chem. Commun. 2014, 50, 29–39. [Google Scholar] [CrossRef]
- Wang, Z.; Xie, P.; Xia, Y. Recent progress in Ru(II)-catalyzed C–H activations with oxidizing directing groups. Chin. Chem. Lett. 2018, 29, 47–53. [Google Scholar] [CrossRef]
- Shan, C.; Zhu, L.; Qu, L.B.; Bai, R.; Lan, Y. Mechanistic view of Ru-catalyzed C–H bond activation and functionalization: Computational advances. Chem. Soc. Rev. 2018, 47, 7552–7576. [Google Scholar] [CrossRef] [PubMed]
- Mcintyre, J.; Mayoral-Soler, I.; Salvador, P.; Poater, A.; Nelson, D.J. Insights into mechanism and selectivity in ruthenium(II)-catalysed ortho-arylation reactions directed by Lewis basic groups. Catal. Sci. Technol. 2018, 8, 3174–3182. [Google Scholar] [CrossRef]
- Oi, S.; Fukita, S.; Hirata, N.; Watanuki, N.; Miyano, S.; Inoue, Y. Ruthenium complex-catalyzed direct ortho arylation and alkenylation of 2-arylpyridines with organic halides. Org. Lett. 2001, 3, 2579–2581. [Google Scholar] [CrossRef]
- Oi, S.; Sakai, K.; Inoue, Y. Ruthenium-catalyzed arylation of 2-alkenylpyridines with aryl bromides: Alternative E, Z-selectivity to Mizoroki−Heck reaction. Org. Lett. 2005, 7, 4009–4011. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, L.; Vicente, R.; Althammer, A. Assisted ruthenium-catalyzed C–H bond activation: Carboxylic acids as cocatalysts for generally applicable direct arylations in apolar solvents. Org. Lett. 2008, 10, 2299–2302. [Google Scholar] [CrossRef] [PubMed]
- Christakakou, M.; Schön, M.; Schnürch, M.; Mihovilovic, M.D. Arylation of pyridines via Suzuki–Miyaura cross-coupling and pyridine- directed C–H activation using a continuous-flow approach. Synlett 2013, 24, 2411–2418. [Google Scholar] [CrossRef]
- Drev, M.; Grošelj, U.; Ledinek, B.; Perdih, F.; Svete, J.; Štefane, B.; Požgan, F. Ruthenium(II)-catalyzed microwave-promoted multiple C–H activation in synthesis of hexa(heteroaryl)benzenes in water. Org. Lett. 2018, 20, 5268–5273. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Devaraj, K.; Darcel, C.; Dixneuf, P.H. Catalytic C-H bond arylation of aryl imines and oxazolines in water with ruthenium(II)-acetate catalyst. Tetrahedron 2012, 68, 5179–5184. [Google Scholar] [CrossRef]
- Ackermann, L.; Vicente, R.; Potukuchi, H.K.; Pirovano, V. Mechanistic insight into direct arylations with ruthenium(II) carboxylate catalysts. Org. Lett. 2010, 12, 5032–5035. [Google Scholar] [CrossRef]
- Požgan, F.; Dixneuf, P.H. Ruthenium(II) acetate catalyst for direct functionalisation of sp2-C-H bonds with aryl chlorides and access to tris-heterocyclic molecules. Adv. Synth. Catal. 2009, 351, 1737–1743. [Google Scholar] [CrossRef]
- Arockiam, P.B.; Fischmeister, C.; Bruneau, C.; Dixneuf, P.H. C-H bond functionalization in water catalyzed by carboxylato ruthenium(II) systems. Angew. Chem. Int. Ed. 2010, 49, 6629–6632. [Google Scholar] [CrossRef] [PubMed]
- Arockiam, P.; Poirier, V.; Fischmeister, C.; Bruneau, C.; Dixneuf, P.H. Diethyl carbonate as a solvent for ruthenium catalysed C–H bond functionalisation. Green Chem. 2009, 11, 1871–1875. [Google Scholar] [CrossRef]
- Singh, K.S.; Dixneuf, P.H. Direct C-H bond arylation in water promoted by (O,O)-and (O,N)-chelate ruthenium(II) catalysts. ChemCatChem 2013, 5, 1313–1316. [Google Scholar] [CrossRef]
- Binnani, C.; Rai, R.K.; Tyagi, D.; Mobin, S.M.; Singh, S.K. Ligand-tuned C–H bond activation/arylation of 2-arylpyridines over pyridine-based N,O/N,N ligated ruthenium–arene complexes. Eur. J. Inorg. Chem. 2018, 1435–1445. [Google Scholar] [CrossRef]
- Doherty, S.; Knight, J.G.; Addyman, C.R.; Smyth, C.H.; Ward, N.A.B.; Harrington, R.W. Ruthenium complexes of κ(P)- and κ(P)-η6-coordinated KITPHOS monophosphines: Efficient catalysts for the direct ortho arylation of 2-phenylpyridine and N-phenylpyrazole with aryl chlorides. Organometallics 2011, 30, 6010–6016. [Google Scholar] [CrossRef]
- Arockiam, P.B.; Fischmeister, C.; Bruneau, C.; Dixneuf, P.H. Ruthenium(II)-catalyzed selective monoarylation in water and sequential functionalisations of C–H bonds. Green Chem. 2013, 15, 67–71. [Google Scholar] [CrossRef]
- Ho, J.S.; Castro, L.C.M.; Aihara, Y.; Tobisu, M.; Chatani, N. Ruthenium(II)-catalyzed chelation-assisted arylation of C-H bonds with diaryliodonium salts. Asian J. Org. Chem. 2014, 3, 48–51. [Google Scholar] [CrossRef]
- Reddy, G.M.; Rao, N.S.S.; Satyanarayana, P.; Maheswaran, H. PhI(OCOCF3)2-mediated ruthenium catalyzed highly site-selective direct ortho-C–H monoarylation of 2-phenylpyridine and 1-phenyl-1H-pyrazole and their derivatives by arylboronic acids. RSC Adv. 2015, 5, 105347–105352. [Google Scholar] [CrossRef]
- Binnani, C.; Tyagi, D.; Rai, R.K.; Mobin, S.M.; Singh, S.K. C-H bond activation/arylation catalyzed by arene-ruthenium-aniline complexes in water. Chem. Asian J. 2016, 11, 3022–3031. [Google Scholar] [CrossRef]
- Seki, M. Highly efficient catalytic system for C–H activation: A practical approach to angiotensin II receptor blockers. ACS Catal. 2011, 1, 607–610. [Google Scholar] [CrossRef]
- Wexler, R.R.; Greenlee, W.J.; Irvin, J.D.; Goldberg, M.R.; Prendergast, K.; Smith, R.D.; Timmermans, P.B.M.W.M. Nonpeptide angiotensin II receptor antagonists: The next generation in antihypertensive therapy. J. Med. Chem. 1996, 39, 625–656. [Google Scholar] [CrossRef] [PubMed]
- Hubrich, J.; Ackermann, L. Amino Acid Ligands for Ruthenium(II)-Catalyzed C–H Arylation of Aryltetrazoles with Chlorides: Expedient Access to Antihypertension Drugs. Eur. J. Org. Chem. 2016, 3700–3704. [Google Scholar] [CrossRef]
- Ackermann, L.; Diers, E.; Manvar, A. Ruthenium-catalyzed C–H bond arylations of arenes bearing removable directing groups via six-membered ruthenacycles. Org. Lett. 2012, 14, 1154–1157. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Darcel, C.; Dixneuf, P.H. Ruthenium(II)-catalysed functionalisation of C-H bonds via a six-membered cyclometallate: monoarylation of aryl 2-pyridyl ketones. ChemCatChem. 2014, 6, 127–130. [Google Scholar] [CrossRef]
- Kaloğlu, N.; Özdemir, I.; Gürbüz, I.N.; Arslan, H.; Dixneuf, P.H. Ruthenium(η6,η1-arene-CH2-NHC) catalysts for direct arylation of 2-phenylpyridine with (hetero)aryl chlorides in water. Molecules 2018, 23, 647. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, L.; Vicente, R. Catalytic direct arylations in polyethylene glycol (PEG): Recyclable Palladium(0) catalyst for C-H bond cleavages in the presence of air. Org. Lett. 2009, 11, 4922–4925. [Google Scholar] [CrossRef] [PubMed]
- Luo, N.; Yu, Z. RuCl3⋅x H2O-Catalyzed direct arylation of arenes with aryl chlorides in the presence of triphenylphosphine. Chem. Eur. J. 2010, 16, 787–791. [Google Scholar] [CrossRef]
- Adrio, L.A.; Gimeno, J.; Vicent, C. One-pot direct C–H arylation of arenes in water catalysed by RuCl3·nH2O–NaOAc in the presence of Zn. Chem. Commun. 2013, 49, 8320–8322. [Google Scholar] [CrossRef]
- Doria, R.G.; Achelle, S.; Bruneau, C.; Guen, F.R.; Dorcet, V.; Roisnel, T. Ruthenium(II)-catalyzed C–H (hetero)arylation of alkenylic 1,n-diazines (n = 2, 3, and 4): Scope, mechanism, and application in tandem hydrogenations. J. Org. Chem. 2018, 83, 1462–1477. [Google Scholar] [CrossRef]
- Siopa, F.; Cladera, V.A.R.; Afonso, C.A.M.; Oble, J.; Poli, G. Ruthenium-catalyzed C-H arylation and alkenylation of furfural imines with boronates. Eur. J. Org. Chem. 2018, 6101–6106. [Google Scholar] [CrossRef]
- Ackermann, L.; Mulzer, M. Dehydrative direct arylations of arenes with phenols via ruthenium-catalyzed C–H and C−OH bond functionalizations. Org. Lett. 2008, 10, 5043–5045. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, L.; Pospech, J.; Potukuchi, H.K. Well-defined ruthenium(II) carboxylate as catalyst for direct C–H/C–O bond arylations with phenols in water. Org. Lett. 2012, 14, 2146–2149. [Google Scholar] [CrossRef] [PubMed]
- Roger, J.; Hierso, J.C. Phenol derivatives in ruthenium-catalyzed C–H arylation: A general synthetic access to azole-based congested polyaromatics. Eur. J. Inorg. Chem. 2018, 4953–4958. [Google Scholar] [CrossRef]
- Ackermann, L.; Lygin, A.V. Ruthenium-catalyzed direct C-H bond arylations of heteroarenes. Org. Lett. 2011, 13, 3331–3335. [Google Scholar] [CrossRef]
- Sollert, C.; Devaraj, K.; Orthaber, A.; Gates, P.J.; Pilarski, L.T. Ru-catalysed C-H arylation of indoles and pyrroles with boronic acids: Scope and mechanistic Studies. Chem. Eur. J. 2015, 21, 5380–5386. [Google Scholar] [CrossRef]
- Chinnagolla, R.K.; Jeganmohan, M. Regioselective ortho-arylation and alkenylation of N-alkyl benzamides with boronic acids via ruthenium-catalyzed C–H bond activation: An easy route to fluorenones synthesis. Org. Lett. 2012, 14, 5246–5249. [Google Scholar] [CrossRef]
- Nareddy, P.; Jordan, F.; Szostak, M. Ruthenium(II)-catalyzed ortho-C–H arylation of diverse N-heterocycles with aryl silanes by exploiting solvent-controlled N-coordination. Org. Biomol. Chem. 2017, 15, 4783–4788. [Google Scholar] [CrossRef]
- Nareddy, P.; Jordan, F.; Brenner-Moyer, S.E.; Szostak, M. Ruthenium(II)-catalyzed regioselective C–H arylation of cyclic and N,N-dialkyl benzamides with boronic acids by weak coordination. ACS Catal. 2016, 6, 4755–4759. [Google Scholar] [CrossRef]
- Nareddy, P.; Jordan, F.; Szostak, M. Highly chemoselective ruthenium(II)-catalyzed direct arylation of cyclic and N,N-dialkyl benzamides with aryl silanes. Chem. Sci. 2017, 8, 3204–3210. [Google Scholar] [CrossRef]
- Nareddy, P.; Jordan, F.; Szostak, M. Ruthenium(II)-catalyzed direct C–H arylation of indoles with arylsilanes in water. Org. Lett. 2018, 20, 341–344. [Google Scholar] [CrossRef]
- Norinder, J.; Matsumoto, A.; Yoshikai, N.; Nakamura, E. Iron-catalyzed direct arylation through directed C–H bond activation. J. Am. Chem. Soc. 2008, 130, 5858–5859. [Google Scholar] [CrossRef]
- Gu, S.; Chen, C.; Chen, W. Ortho-functionalization of 2-phenoxypyrimidines via Palladium-catalyzed C–H bond activation. J. Org. Chem. 2009, 74, 7203–7206. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Zhang, Y.; Zhao, J.; Xie, C. Peroxide-promoted regioselective arylation of 2-phenylpyridines and related substrates with aryl iodides. Synlett 2008, 1325–1330. [Google Scholar] [CrossRef]
- Stefane, B.; Fabris, J.; Pozgan, F. C–H bond functionalization of arylpyrimidines catalyzed by an in situ generated ruthenium(II) carboxylate system and the construction of tris(heteroaryl)-substituted benzenes. Eur. J. Org. Chem. 2011, 3474–3481. [Google Scholar] [CrossRef]
- Štefane, B.; Žugelj, H.B.; Grošelj, U.; Kuzman, P.; Svete, J.; Požgan, F. Quinazoline-Directed C–H Bond Functionalization Catalyzed by Ruthenium(II) Carboxylate—Construction of Polyconjugated Aryl-Heteroaryl Systems. Eur. J. Org. Chem. 2017, 1855–1864. [Google Scholar]
- Zhao, Y.; Snieckus, V. Quinazoline-directed C–H bond functionalization catalyzed by ruthenium(II) carboxylate – construction of polyconjugated aryl-heteroaryl systems. Adv. Synth. Catal. 2014, 356, 1527–1532. [Google Scholar] [CrossRef]
- Al Mamari, H.H.; Diers, E.; Ackermann, L. Triazole-assisted ruthenium-catalyzed C-H arylation of aromatic amides. Chem. Eur. J. 2014, 20, 9739–9743. [Google Scholar] [CrossRef]
- Yamamoto, T.; Yamakawa, T. Ruthenium/base-catalyzed ortho-selective C–H arylation of acylarenes with halogenated arylboronates. RSC Adv. 2015, 5, 105829–105836. [Google Scholar] [CrossRef]
- Koseki, Y.; Kitazawa, K.; Miyake, M.; Kochi, T.; Kakiuchi, F. Ruthenium-catalyzed ortho C–H arylation of aromatic nitriles with arylboronates and observation of partial para arylation. J. Org. Chem. 2017, 82, 6503–6510. [Google Scholar] [CrossRef]
- Huang, L.; Weix, D.J. Ruthenium-catalyzed C–H arylation of diverse aryl carboxylic acids with aryl and heteroaryl halides. Org. Lett. 2016, 18, 5432–5435. [Google Scholar] [CrossRef]
- Simonetti, M.; Perry, G.J.P.; Cambeiro, X.C.; Hernández, F.J.; Arokianathar, J.N.; Larrosa, I. Ru-catalyzed C–H arylation of fluoroarenes with aryl halides. J. Am. Chem. Soc. 2016, 138, 3596–3606. [Google Scholar] [CrossRef] [PubMed]
- Flegeau, E.F.; Bruneau, C.; Dixneuf, P.H.; Jutand, A. Autocatalysis for C–H bond activation by ruthenium(II) complexes in catalytic arylation of functional arenes. J. Am. Chem. Soc. 2011, 133, 10161–10170. [Google Scholar] [CrossRef] [PubMed]
- Simonetti, M.; Cannas, D.M.; Panigrahi, A.; Kujawa, S.; Kryjewski, M.; Xie, P.; Larrosa, I. Ruthenium-catalyzed C–H arylation of benzoic acids and indole carboxylic acids with aryl halides. Chem. Eur. J. 2017, 23, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Pastine, S.J.; Gribkov, D.V.; Sames, D. sp3 C–H bond arylation directed by amidine protecting group: α-arylation of pyrrolidines and piperidines. J. Am. Chem. Soc. 2006, 128, 14220–14221. [Google Scholar] [CrossRef] [PubMed]
- Peschiulli, A.; Smout, V.; Storr, T.E.; Mitchell, E.A.; Eliáš, Z.; Herrebout, W.; Berthelot, D.; Meerpoel, L.; Maes, B.U.W. Ruthenium-catalyzed a-(hetero)arylation of saturated cyclic amines: Reaction scope and mechanism. Chem. Eur. J. 2013, 19, 10378–10387. [Google Scholar] [CrossRef]
- Dastbaravardeh, N.; Schnürch, M.; Mihovilovic, M.D. Ruthenium(II)-catalyzed sp3 C–H bond arylation of benzylic amines using aryl halides. Org. Lett. 2012, 14, 3792–3795. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.Y.P.; Jeyachandran, R.; Ackermann, L. C(sp3)–H bond arylations catalyzed by well-defined [Ru(O2CMes)2(p-cymene)]. J. Org. Chem. 2013, 78, 4145–4152. [Google Scholar] [CrossRef] [PubMed]
- Gribkov, D.V.; Pastine, S.J.; Schnürch, M.; Sames, D. Ruthenium catalyzed decarbonylative arylation at sp3 carbon centers in pyrrolidine and piperidine heterocycles. J. Am. Chem. Soc. 2007, 129, 11750–11755. [Google Scholar] [CrossRef]
- Moselage, M.; Li, J.; Kramm, F.; Ackermann, L. Ruthenium(II)-catalyzed C-C arylations and alkylations: Decarbamoylative C-C functionalizations. Angew. Chem. Int. Ed. 2017, 56, 5341–5344. [Google Scholar] [CrossRef]
- Weissman, H.; Song, X.; Milstein, D. Ru-catalyzed oxidative coupling of arenes with olefins using O2. J. Am. Chem. Soc. 2001, 123, 337–338. [Google Scholar] [CrossRef]
- Yi, C.S.; Lee, D.W. Intermolecular dehydrative coupling reaction of aryl ketones with cyclic alkenes catalyzed by a well-defined cationic ruthenium−hydride complex: A novel ketone olefination method via vinyl C–H bond activation. Organometallics 2010, 29, 1883–1885. [Google Scholar] [CrossRef] [PubMed]
- Ueyama, T.; Mochida, S.; Fukutani, T.; Hirano, K.; Satoh, T.; Miura, M. Ruthenium-Catalyzed Oxidative Vinylation of Heteroarene Carboxylic Acids with Alkenes via Regioselective C–H Bond Cleavage. Org. Lett. 2011, 13, 706–708. [Google Scholar] [CrossRef] [PubMed]
- Kozhushkov, S.I.; Ackermann, L. Ruthenium-catalyzed direct oxidative alkenylation of arenes through twofold C–H bond functionalization. Chem. Sci. 2013, 4, 886–896. [Google Scholar] [CrossRef]
- Arockiam, P.B.; Fischmeister, C.; Bruneau, C.; Dixneuf, P.H. Ruthenium diacetate-catalysed oxidative alkenylation of C–H bonds in air: Synthesis of alkenyl N-arylpyrazoles. Green Chem. 2011, 13, 3075–3078. [Google Scholar] [CrossRef]
- Ogiwara, Y.; Kochi, T.; Kakiuchi, F. Ruthenium-catalyzed ortho-selective aromatic CH alkenylation with alkenyl carbonates. Chem. Lett. 2014, 43, 667–669. [Google Scholar] [CrossRef]
- Kommagalla, Y.; Mullapudi, V.B.; Francis, F.; Ramana, C.V. Ruthenium(II)-catalyzed switchable C3-alkylation versus alkenylation with acrylates of 2-pyridylbenzofurans via C–H bond activation. Catal. Sci. Technol. 2015, 5, 114–117. [Google Scholar] [CrossRef]
- Bin, L.; Devaraj, K.; Darcel, C.; Dixneuf, P.H. Ruthenium(II) catalysed synthesis of unsaturated oxazolines via arene C–H bond alkenylation. Green Chem. 2012, 14, 2706–2709. [Google Scholar]
- Ogiwara, Y.; Tamura, M.; Kochi, T.; Matsuura, Y.; Chatani, N.; Kakiuchi, F. Ruthenium-catalyzed ortho-selective C–H alkenylation of aromatic compounds with alkenyl esters and ethers. Organometallics 2014, 33, 402–420. [Google Scholar] [CrossRef]
- Ma, W.; Ackermann, L. Ruthenium(II)-catalyzed C-H alkenylations of phenols with removable directing groups. Chem. Eur. J. 2013, 19, 13925–13928. [Google Scholar] [CrossRef]
- Sawant, D.; Singh, I.; Tulsyan, G.; Abbagani, K.; Pardasani, R.T. Ruthenium-catalyzed oxidative C–H bond alkenylation of 2-phenylimidazo[1,2-a]pyridine. Synlett 2015, 26, 1671–1676. [Google Scholar] [CrossRef]
- Shome, S.; Singh, S.P. Solvent-free ruthenium(II)-catalyzed C–H activation: Synthesis of alkenylarylpyrazole derivatives. Eur. J. Org. Chem. 2015, 6025–6032. [Google Scholar] [CrossRef]
- Shome, S.; Singh, S.P. Ruthenium-bipyridine complex catalyzed C–H alkenylation of arylpyrazole derivatives. J. Chem. Sci. 2018, 130, 87. [Google Scholar] [CrossRef]
- Selvaraju, M.; Wang, Y.L.; Sun, C.M. Ruthenium(II)-catalyzed C–H alkenylation/annulation cascade for the rapid synthesis of benzoimidazoisoindoles. Org. Chem. Front. 2017, 4, 1358–1362. [Google Scholar] [CrossRef]
- Gholamhosseyni, M.; Kianmehr, E. A ruthenium-catalyzed alkenylation–annulation approach for the synthesis of indazole derivatives via C–H bond activation. Org. Biomol. Chem. 2018, 16, 5973–5978. [Google Scholar] [CrossRef]
- Ackermann, L.; Pospech, J. Ruthenium-catalyzed oxidative C–H bond alkenylations in water: Expedient synthesis of annulated lactones. Org. Lett. 2011, 13, 4153–4155. [Google Scholar] [CrossRef]
- Kumar, N.Y.P.; Bechtoldt, A.; Raghuvanshi, K.; Ackermann, L. Ruthenium(II)-catalyzed decarboxylative C–H activation: Versatile routes to meta-alkenylated arenes. Angew. Chem. Int. Ed. 2016, 55, 6929–6932. [Google Scholar] [CrossRef]
- Jin, X.Y.; Xie, L.J.; Cheng, H.P.; Liu, A.D.; Li, X.D.; Wang, D.; Cheng, L.; Liu, L. Ruthenium-catalyzed decarboxylative C–H alkenylation in aqueous media: Synthesis of tetrahydropyridoindoles. J. Org. Chem. 2018, 83, 7514–7522. [Google Scholar] [CrossRef]
- Dana, S.; Mandal, A.; Sahoo, H.; Mallik, S.; Grandhi, G.S.; Baidya, M. Ru(II)-catalyzed oxidative Heck-type olefination of aromatic carboxylic acids with styrenes through carboxylate-assisted C–H bond activation. Org. Lett. 2018, 20, 716–719. [Google Scholar] [CrossRef]
- Ma, W.; Mei, R.; Tenti, G.; Ackermann, L. Ruthenium(II)-catalyzed oxidative C-H alkenylations of sulfonic acids, sulfonyl chlorides and sulfonamides. Chem. Eur. J. 2014, 20, 15248–15251. [Google Scholar] [CrossRef]
- Murai, S.; Kakiuchi, F.; Sekine, S.; Tanaka, Y.; Kamatani, A.; Sonoda, M.; Chatani, N. Efficient catalytic addition of aromatic carbon-hydrogen bonds to olefins. Nature 1993, 366, 529–531. [Google Scholar] [CrossRef]
- Martinez, R.; Genet, J.P.; Darses, S. Anti-Markovnikov hydroarylation of styrenes catalyzed by an in situ generated ruthenium complex. Chem. Commun. 2008, 3855–3857. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.O.; Genet, J.-P.; Darses, S. Ruthenium chloride as an efficient catalytic precursor for hydroarylation reactions via C-H bond activation. Org. Lett. 2010, 12, 3038–3041. [Google Scholar] [CrossRef] [PubMed]
- Kakiuchi, F.; Kochi, T.; Mizushima, E.; Murai, S. Room-Temperature regioselective C-H/olefin coupling of aromatic ketones using an activated ruthenium catalyst with a carbonyl ligand and structural elucidation of key intermediates. J. Am. Chem. Soc. 2010, 132, 17741–17750. [Google Scholar] [CrossRef] [PubMed]
- Padala, K.; Jeganmohan, M. Ruthenium-catalyzed ortho-alkenylation of aromatic ketones with alkenes by C -H bond activation. Org. Lett. 2011, 13, 6144–6147. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.S.; Dixneuf, P.H. Ruthenium(II)-catalyzed alkenylation of ferrocenyl ketones via C–H bond activation. Organometallics 2012, 31, 7320–7323. [Google Scholar] [CrossRef]
- Padala, K.; Jeganmohan, M. Highly regio- and stereoselective ruthenium(II)-catalyzed direct ortho-alkenylation of aromatic and heteroaromatic aldehydes with activated alkenes under open atmosphere. Org. Lett. 2012, 14, 1134–1137. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.C.; Jeganmohan, M. Ruthenium-catalyzed ortho alkenylation of aromatic nitriles with activated alkenes via C–H bond activation. Chem. Commun. 2015, 51, 10738–10741. [Google Scholar] [CrossRef]
- Ackermann, L.; Wang, L.; Wolfram, R.; Lygin, A.V. Ruthenium-catalyzed oxidative C–H alkenylations of anilides and benzamides in water. Org. Lett. 2012, 14, 728–731. [Google Scholar] [CrossRef]
- Li, B.; Ma, J.; Xie, W.; Song, H.; Xu, S.; Wang, B. Ruthenium-catalyzed regioselective C2 alkenylation of indoles and pyrroles via C–H bond functionalization. J. Org. Chem. 2013, 78, 9345–9353. [Google Scholar] [CrossRef]
- Das, R.; Kapur, M. Fujiwara–Moritani reaction of Weinreb amides using a ruthenium-catalyzed C–H functionalization reaction. Chem. Asian J. 2015, 10, 1505–1512. [Google Scholar] [CrossRef]
- Bechtoldt, A.; Tirler, C.; Raghuvanshi, K.; Warratz, S.; Kornhaaß, C.; Ackermann, L. Ruthenium oxidase catalysis for site-selective C-H alkenylations with ambient O2 as the sole oxidant. Angew. Chem. Int. Ed. 2016, 55, 264–267. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Yao, B.B.; Zhao, J.L.; Zhang, Y.H. RuCl3-catalyzed alkenylation of aromatic C–H bonds with terminal alkynes. Org. Lett. 2008, 10, 5309–5312. [Google Scholar] [CrossRef] [PubMed]
- Li, X.G.; Liu, K.; Zou, G.; Liu, P.N. Ruthenium-catalyzed alkenylation of arenes with alkynes or alkenes by 1,2,3-triazole-directed C–H activation. Eur. J. Org. Chem. 2014, 7878–7888. [Google Scholar] [CrossRef]
- Manikandan, R.; Jeganmohan, M. Ruthenium-catalyzed hydroarylation of anilides with alkynes: An efficient route to ortho-alkenylated anilines. Org. Lett. 2014, 16, 912–915. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, M.; Marek, I.; Ackermann, L. Carboxylate-assisted ruthenium(II)-catalyzed hydroarylations of unactivated alkenes through C-H cleavage. Angew. Chem. Int. Ed. 2013, 52, 3977–3980. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, M.; Wang, L.; Bielefeld, K.; Ackermann, L. Ruthenium(II)-catalyzed C(sp3)–H α-alkylation of pyrrolidines. Org. Lett. 2014, 16, 1876–1879. [Google Scholar] [CrossRef] [PubMed]
- Cadierno, V.; Francos, J.; Gimeno, J. Ruthenium/TFA-catalyzed regioselective C-3-alkylation of indoles with terminal alkynes in water: Efficient and unprecedented access to 3-(1-methylalkyl)-1H-indoles. Chem. Commun. 2010, 46, 4175–4177. [Google Scholar] [CrossRef]
- Yao, T.; Hirano, K.; Satoh, T.; Miura, M. Palladium- and Nickel-catalyzed direct alkylation of azoles with unactivated alkyl bromides and chlorides. Chem. Eur. J. 2010, 16, 12307–12311. [Google Scholar] [CrossRef]
- Ackermann, L.; Punji, B.; Song, W. User-Friendly [(Diglyme)NiBr2]-catalyzed direct alkylations of heteroarenes with unactivated alkyl halides through C-H bond cleavages. Adv. Synth. Catal. 2011, 353, 3325–3329. [Google Scholar] [CrossRef]
- Ackermann, L.; Novák, P.; Vicente, R.; Hofmann, N. Ruthenium-catalyzed regioselective direct alkylation of arenes with unactivated alkyl halides through C-H bond cleavage. Angew. Chem. Int. Ed. 2009, 48, 6045–6048. [Google Scholar] [CrossRef]
- Hofmann, N.; Ackermann, L. meta-Selective C–H Bond Alkylation with Secondary Alkyl Halides. J. Am. Chem. Soc. 2013, 135, 5877–5884. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Gao, P.; Lv, X.; Qu, C.; Yan, Q.; Wang, Y.; Yang, S.; Wang, J. Synthesis of m-alkylphenols via a ruthenium-catalyzed C–H bond functionalization of phenol derivatives. Org. Lett. 2017, 19, 2682–2685. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Ma, X.; Jia, C.; Han, Q.; Wang, Y.; Wang, J.; Yu, L.; Yang, S. Ruthenium-catalyzed meta/ortho-selective C–H alkylation of azoarenes using alkyl bromides. Chem. Commun. 2017, 53, 1261–1264. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Korvorapun, K.; Sarkar, S.D.; Rogge, T.; Burns, D.J.; Warratz, S.; Ackermann, L. Ruthenium(II)-catalysed remote C–H alkylations as a versatile platform to meta-decorated arenes. Nat. Commun. 2017, 8, 15430. [Google Scholar] [CrossRef] [PubMed]
- Leitch, J.A.; McMullin, C.L.; Paterson, A.J.; Mahon, M.F.; Bhonoah, Y.; Frost, C.G. Ruthenium-catalyzed para-selective C-H alkylation of aniline derivatives. Angew. Chem. Int. Ed. 2017, 56, 15131–15135. [Google Scholar] [CrossRef] [PubMed]
- Leitch, J.A.; McMullin, C.L.; Mahon, M.F.; Bhonoah, Y.; Frost, C.G. Remote C6-selective ruthenium-catalyzed C–H alkylation of indole derivatives via σ-activation. ACS Catal. 2017, 7, 2616–2623. [Google Scholar] [CrossRef]
- Kumar, G.S.; Chand, T.; Singh, D.; Kapur, M. Ruthenium-catalyzed C–H functionalization of benzoic acids with allyl alcohols: A controlled reactivity switch between C–H alkenylation and C–H alkylation pathways. Org. Lett. 2018, 20, 4934–4937. [Google Scholar] [CrossRef]
- Tan, Z.; Jiang, H.; Zhang, M. Ruthenium-catalyzed dehydrogenative β-benzylation of 1,2,3,4-tetrahydroquinolines with aryl aldehydes: Access to functionalized quinolines. Org. Lett. 2016, 18, 3174–3177. [Google Scholar] [CrossRef]
- Oi, S.; Tanaka, Y.; Inoue, Y. Ortho-selective allylation of 2-pyridylarenes with allyl acetates catalyzed by ruthenium complexes. Organometallics 2006, 25, 4773–4778. [Google Scholar] [CrossRef]
- Goriya, Y.; Ramana, C.V. Ruthenium-catalyzed C6-propenylation reactions of substituted pyridine derivatives: Directed and direct C-H activation. Chem. Eur. J. 2012, 18, 13288–13292. [Google Scholar] [CrossRef]
- Manikandan, R.; Madasamy, P.; Jeganmohan, M. Ruthenium-catalyzed oxidant-free allylation of aromatic ketoximes with allylic acetates at room temperature. Chem. Eur. J. 2015, 21, 13934–13938. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Ji, H. Ruthenium(II)-catalyzed rgio- and stereoselective C–H allylation of indoles with allyl alcohols. Org. Lett. 2018, 20, 2224–2227. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.X. Synthesis of isocoumarin derivatives by copper-catalyzed addition of o-halobenzoic acids to active internal alkynes. J. Org. Chem. 2013, 78, 1660–1664. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Huang, L.; Wu, W.; Jiang, H. Palladium-catalyzed oxidative annulation of acrylic acid and amide with alkynes: A practical route to synthesize α-pyrones and pyridones. Org. Lett. 2014, 16, 2146–2149. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Chen, D.; Song, G.; Han, K.; Li, X. Palladium-catalyzed cascade cyclization–Oxidative olefination of tert-butyl 2-alkynylbenozates. J. Org. Chem. 2012, 77, 1579–1584. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; John, M.; Ackermann, L. Amidines for versatile ruthenium(II)-catalyzed oxidative C-H activations with internal alkynes and acrylates. Chem. Eur. J. 2014, 20, 5403–5408. [Google Scholar] [CrossRef] [PubMed]
- Midya, S.P.; Sahoo, M.K.; Landge, V.G.; Rajamohanan, P.R.; Balaraman, E. Reversed reactivity of anilines with alkynes in the rhodium-catalysed C–H activation/carbonylation tandem. Nat. Commun. 2015, 6, 8591. [Google Scholar] [CrossRef] [PubMed]
- Nagamoto, M.; Yamauchi, D.; Nishimura, T. Iridium-catalyzed asymmetric [3+2] annulation of aromatic ketimines with alkynes via C–H activation: Unexpected inversion of the enantioselectivity induced by protic acids. Chem. Commun. 2016, 52, 5876–5879. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ackermann, L. Versatile pyrrole synthesis through ruthenium(II)-catalyzed alkene C–H bond functionalization on enamines. Org. Lett. 2013, 15, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wang, N.; Liang, Y.; Xu, S.; Wang, B. Ruthenium-catalyzed pyrrole synthesis via oxidative annulation of enamides and alkynes. Org. Lett. 2013, 15, 136–139. [Google Scholar] [CrossRef]
- Lin, H.; Li, S.S.; Dong, L. Synthesis of indoles and polycyclic amides via ruthenium(II)-catalyzed C–H activation and annulations. Org. Biomol. Chem. 2015, 13, 11228–11234. [Google Scholar] [CrossRef] [PubMed]
- Villuendas, P.; Urriolabeitia, E.P. Primary amines as directing groups in the Ru-catalyzed synthesis of isoquinolines, benzoisoquinolines, and thienopyridines. J. Org. Chem. 2013, 78, 5254–5263. [Google Scholar] [CrossRef] [PubMed]
- Kaishap, P.P.; Sarma, B.; Gogoi, S. The amide C–N bond of isatins as the directing group and the internal oxidant in Ru-catalyzed C–H activation and annulation reactions: Access to 8-amido isocoumarins. Chem. Commun. 2016, 52, 9809–9812. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Graczyk, K.; Ackermann, L. Ruthenium-catalyzed alkyne annulations with substituted 1H-pyrazoles by C–H/N–H bond functionalizations. Org. Lett. 2012, 14, 6318–6321. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ren, H.; Wang, D.; Shi, F.; Wu, C. Cu-Catalyzed alkynylation–cyclization cascade for the construction of the pyrazolo[5,1-a]isoquinoline skeleton. RSC Adv. 2013, 3, 10434–10441. [Google Scholar] [CrossRef]
- Ackermann, L.; Lygin, A.V. Cationic Ruthenium(II) catalysts for oxidative C–H/N–H bond functionalizations of anilines with removable directing group: Synthesis of indoles in water. Org. Lett. 2012, 14, 764–767. [Google Scholar] [CrossRef]
- Ackermann, L.; Wang, L.; Lygin, A.V. Ruthenium-catalyzed aerobic oxidative coupling of alkynes with 2-aryl-substituted pyrroles. Chem. Sci. 2012, 3, 177–180. [Google Scholar] [CrossRef]
- Chinnagolla, R.K.; Jeganmohan, M. Regioselective synthesis of isocoumarins by ruthenium-catalyzed aerobic oxidative cyclization of aromatic acids with alkynes. Chem. Commun. 2012, 48, 2030–2032. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, L.; Pospech, J.; Graczyk, K.; Rauch, K. Versatile synthesis of isocoumarins and α-pyrones by ruthenium-catalyzed oxidative C–H/O–H bond cleavages. Org. Lett. 2012, 14, 930–933. [Google Scholar] [CrossRef] [PubMed]
- Warratz, S.; Kornhaaß, C.; Cajaraville, A.; Niepçtter, B.; Stalke, D.; Ackermann, L. Ruthenium(II)-catalyzed C-H activation/alkyne annulation by weak coordination with O2 as the sole oxidant. Angew. Chem. Int. Ed. 2015, 54, 5513–5517. [Google Scholar] [CrossRef]
- Singh, K.S.; Sawant, S.G.; Dixneuf, P.H. Ruthenium(II)-catalyzed synthesis of pyrrole- and indole fused isocoumarins by C-H bond activation in DMF and water. ChemCatChem 2016, 8, 1046–1050. [Google Scholar] [CrossRef]
- Singh, K.S.; Sawant, S.G.; Kaminsky, W. Regioselective synthesis of pyrrole and indole-fused isocoumarins catalysed by N∧O chelate ruthenium(II) complex. J. Chem. Sci. 2018, 130, 120. [Google Scholar] [CrossRef]
- Chinnagolla, R.K.; Jeganmohan, M. Ruthenium-catalyzed regioselective cyclization of aromatic ketones with alkynes: An efficient route to indenols and benzofulvenes. Eur. J. Org. Chem. 2012, 417–423. [Google Scholar] [CrossRef]
- Reddy, M.C.; Manikandan, R.; Jeganmohan, M. Ruthenium-catalyzed aerobic oxidative cyclization of aromatic and heteroaromatic nitriles with alkynes: a new route to isoquinolones. Chem. Commun. 2013, 49, 6060–6062. [Google Scholar] [CrossRef] [PubMed]
- Thirunavukkarasu, V.S.; Donati, M.; Ackermann, L. Hydroxyl-directed ruthenium-catalyzed C–H bond functionalization: Versatile access to fluorescent pyrans. Org. Lett. 2012, 14, 3416–3419. [Google Scholar] [CrossRef]
- Zhao, P.; Wang, F.; Han, K.; Li, X. Ruthenium- and sulfonamide-catalyzed cyclization between N-sulfonyl imines and alkynes. Org. Lett. 2012, 14, 5506–5509. [Google Scholar] [CrossRef]
- Zhang, J.; Ugrinov, A.; Zhao, P. Ruthenium(II)/N-heterocyclic carbene catalyzed [3+2] carbocyclization with aromatic N-H ketimines and internal alkynes. Angew. Chem. Int. Ed. 2013, 52, 6681–6684. [Google Scholar] [CrossRef] [PubMed]
- Kavitha, N.; Sukumar, G.; Kumar, V.P.; Mainkar, P.S.; Chandrasekhar, S. Ruthenium-catalyzed benzimidazoisoquinoline synthesis via oxidative coupling of 2-arylbenzimidazoles with alkynes. Tetrahedron Lett. 2013, 54, 4198–4201. [Google Scholar] [CrossRef]
- Ruiz, S.; Villuendas, P.; Ortuño, M.A.; Lledós, A.; Urriolabeitia, E.P. Ruthenium-catalyzed oxidative coupling of primary amines with internal alkynes through C-H bond activation: Scope and mechanistic studies. Chem. Eur. J. 2015, 21, 8626–8636. [Google Scholar] [CrossRef]
- Allu, S.; Kumara Swamy, K.C. Ruthenium-catalyzed oxidative annulation of 6-anilinopurines with alkynes via C-H activation: Synthesis of indole-substituted purines/purine nucleosides. Adv. Synth. Catal. 2015, 357, 2665–2680. [Google Scholar] [CrossRef]
- Sharma, S.; Singh, S.; Kumari, A.; Sawant, D.M.; Pardasani, R.T. Ruthenium-catalyzed oxidative annulation of anilines using benzothiazole as a removable directing group. Asian J. Org. Chem. 2017, 6, 728–736. [Google Scholar] [CrossRef]
- Kaishap, P.P.; Duarah, G.; Sarma, B.; Chetia, D.; Gogoi, S. Ruthenium(II)-catalyzed synthesis of spirobenzofuranones by a decarbonylative annulation reaction. Angew. Chem. Int. Ed. 2018, 57, 456–460. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Santra, S.K.; Mohanta, P.R.; Patel, B.K. Ruthenium(II) catalyzed regiospecific C–H/O−H annulations of directing arenes via weak coordination. Org. Lett. 2015, 17, 5678–5681. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Li, Y.J.; Huang, C.; Xu, H.C. Ruthenium-catalyzed electrochemical dehydrogenative alkyne annulation. ACS Catal. 2018, 8, 3820–3824. [Google Scholar] [CrossRef]
- Kaufmann, J.; Jäckel, E.; Haak, E. Ruthenium-catalyzed cascade annulation of indole with propargyl alcohols. Angew. Chem. Int. Ed. 2018, 57, 5908–5911. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Classical cross-coupling (a); various strategies for C–H bond functionalization (b–d).
Scheme 2.
Arylation of (a) 2-phenylpyridine with aryl halides; (b) alkenyl pyridine with aryl bromides.
Scheme 2.
Arylation of (a) 2-phenylpyridine with aryl halides; (b) alkenyl pyridine with aryl bromides.

Scheme 3.
Ruthenium-catalysed direct arylation of N-heteroarenes with aryl halides.

Scheme 4.
Ruthenium(II)-catalysed direct arylation of 2-phenylpyridine with aryl bromides.
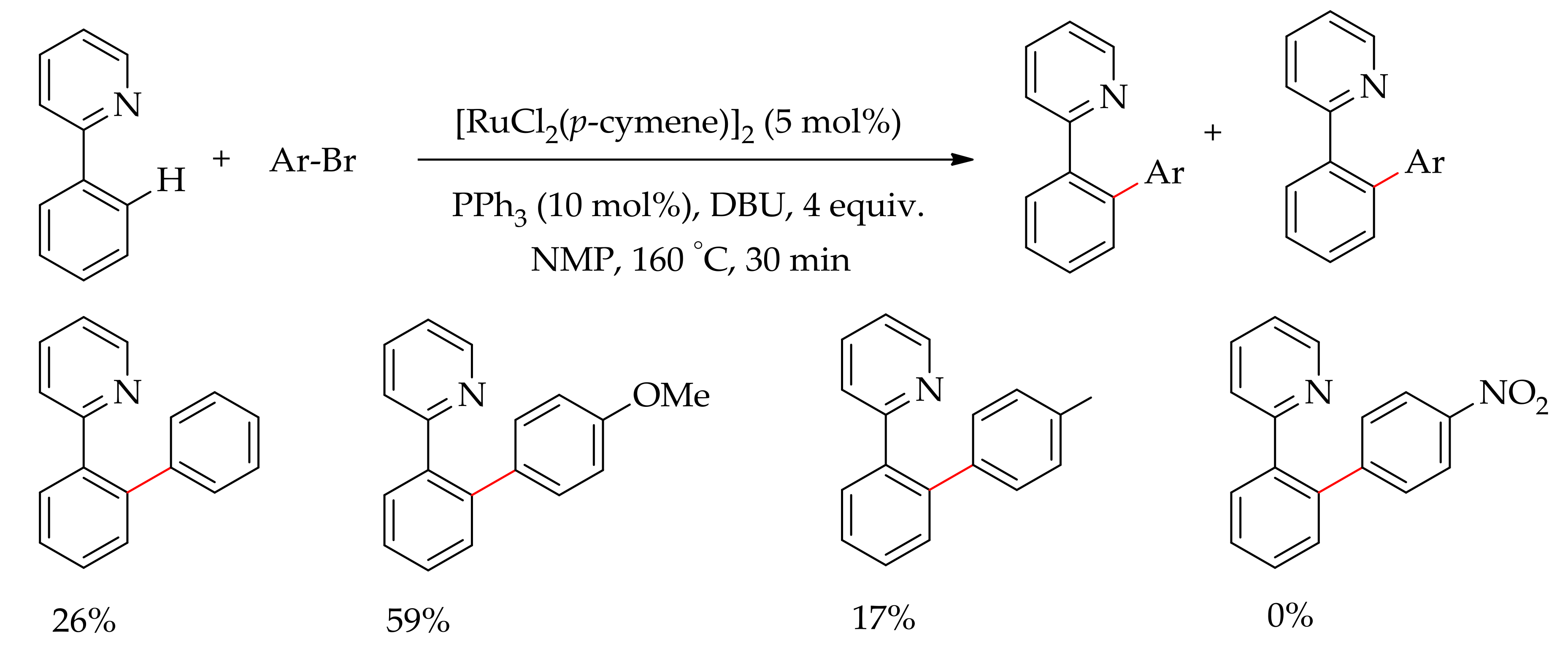
Scheme 5.
Ruthenium-catalysed microwave assisted multiple C–H activation of 2-phenylpyridine.
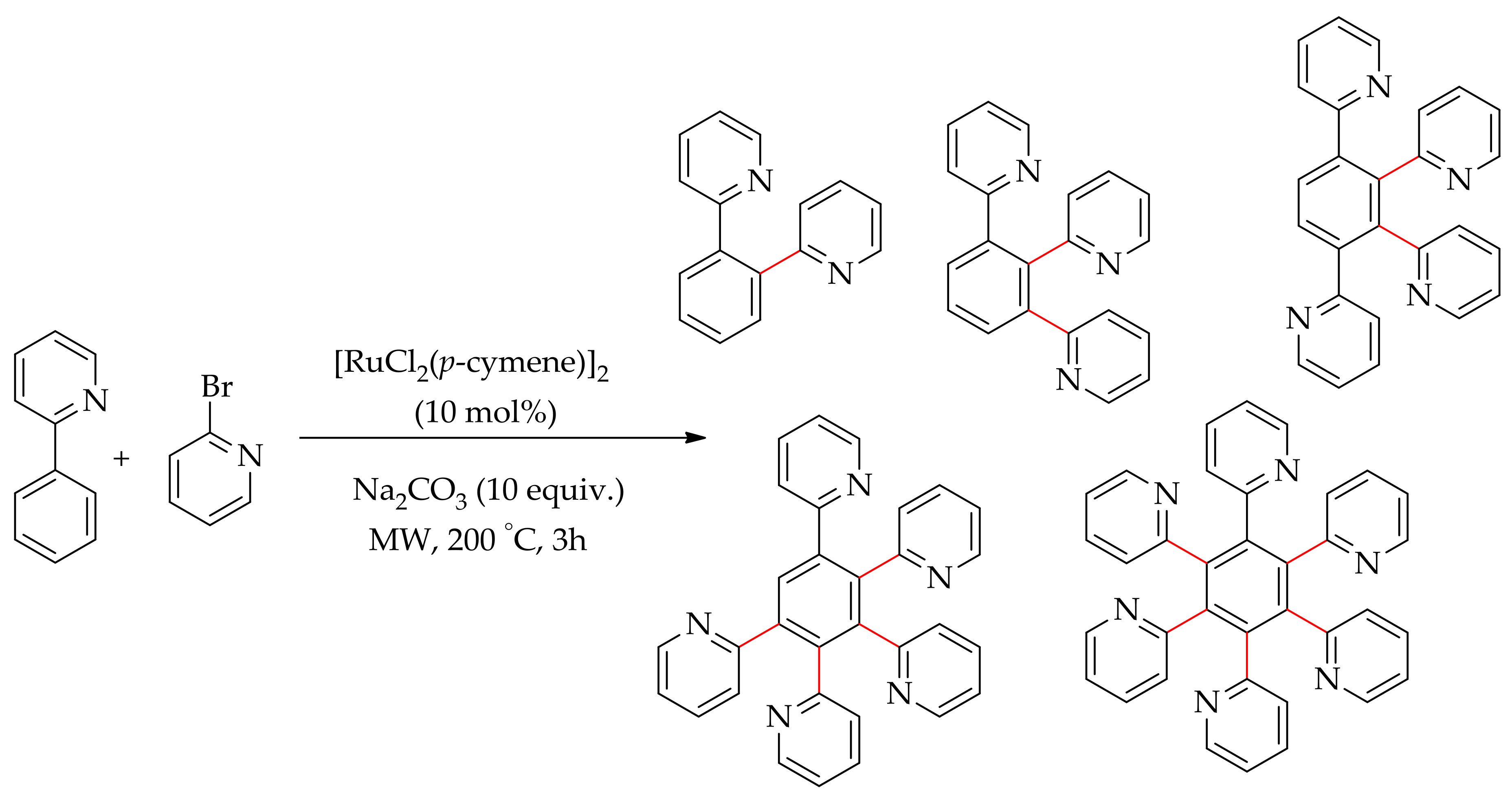
Scheme 6.
Ruthenium-catalysed synthesis of diphenylated benzaldehyde.

Scheme 7.
Arylation of heteroarne with aryl halides by O^O or N^O chelate ruthenium(II)-catalysts.
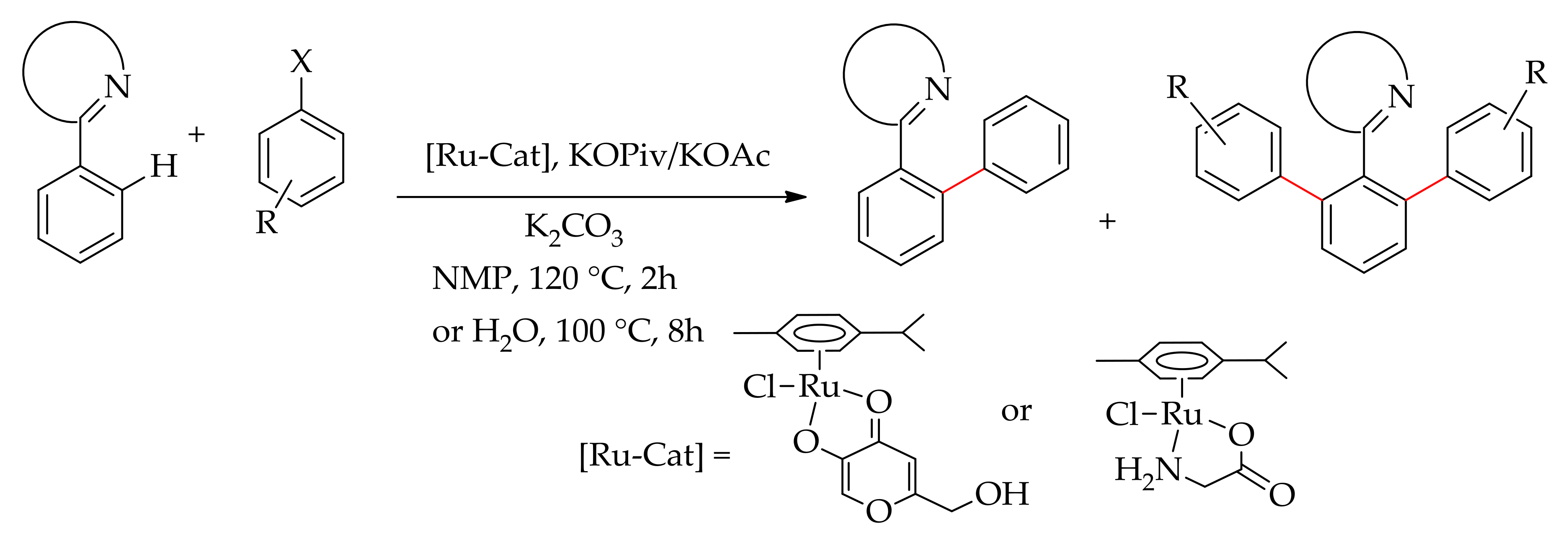
Scheme 8.
Arylation of 2-phenylpyridine with N^O andN^N chelate ruthenium-catalysts.

Scheme 9.
Selective monoarylation of 2-phenylpyridine with ruthenium(II)-catalysts.

Scheme 10.
Ruthenium(II)-catalysed selective monoarylation in water.
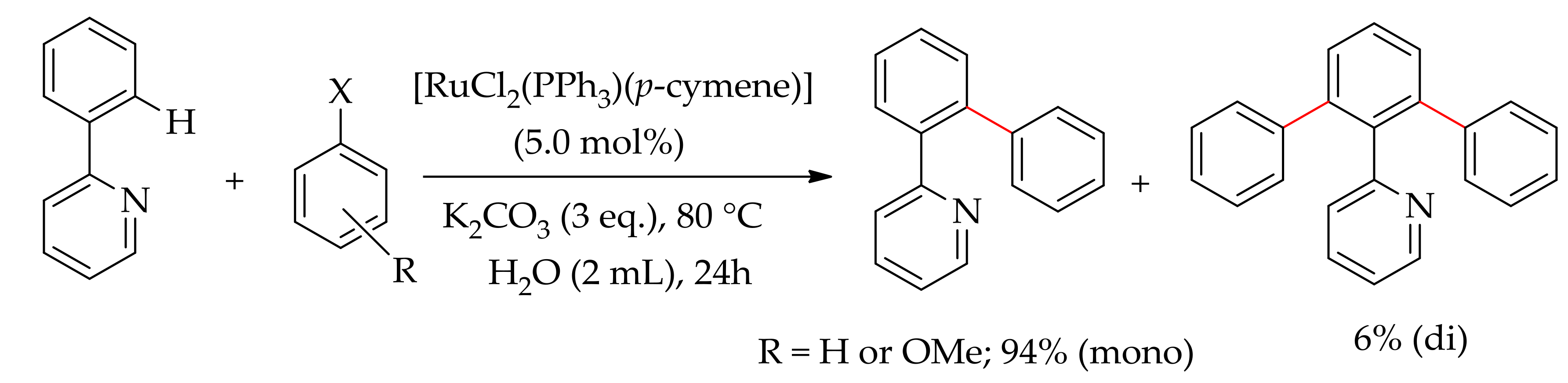
Scheme 11.
Selective monoarylation of 2-phenylpyridine with Ph2IOTf.

Scheme 12.
Proposed mechanisms for the ortho-phenylation of C–H bonds [57].
Scheme 12.
Proposed mechanisms for the ortho-phenylation of C–H bonds [57].
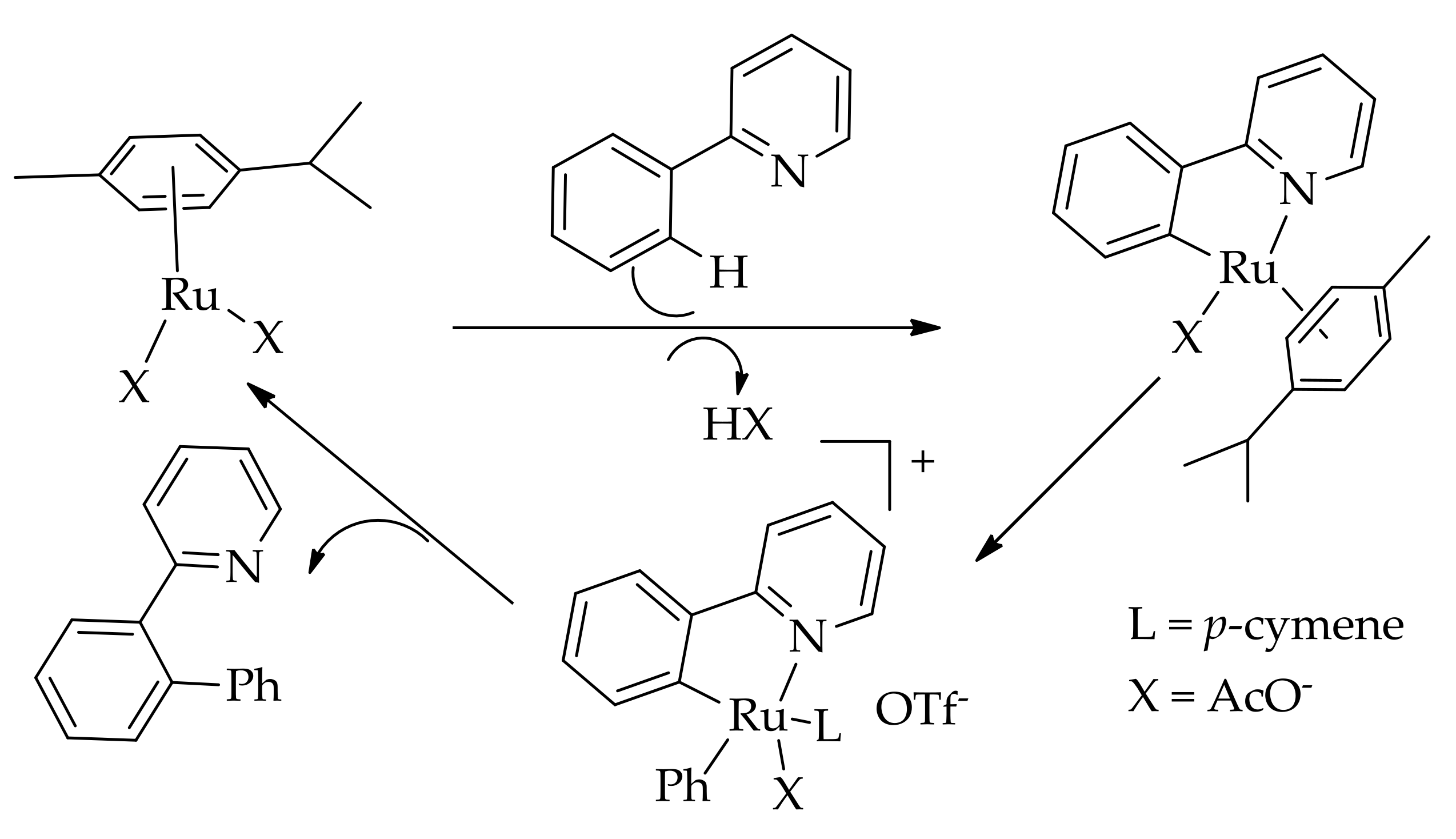
Scheme 13.
Ruthenium-catalysed monoarylation of arenes with phenyl boronic acids.
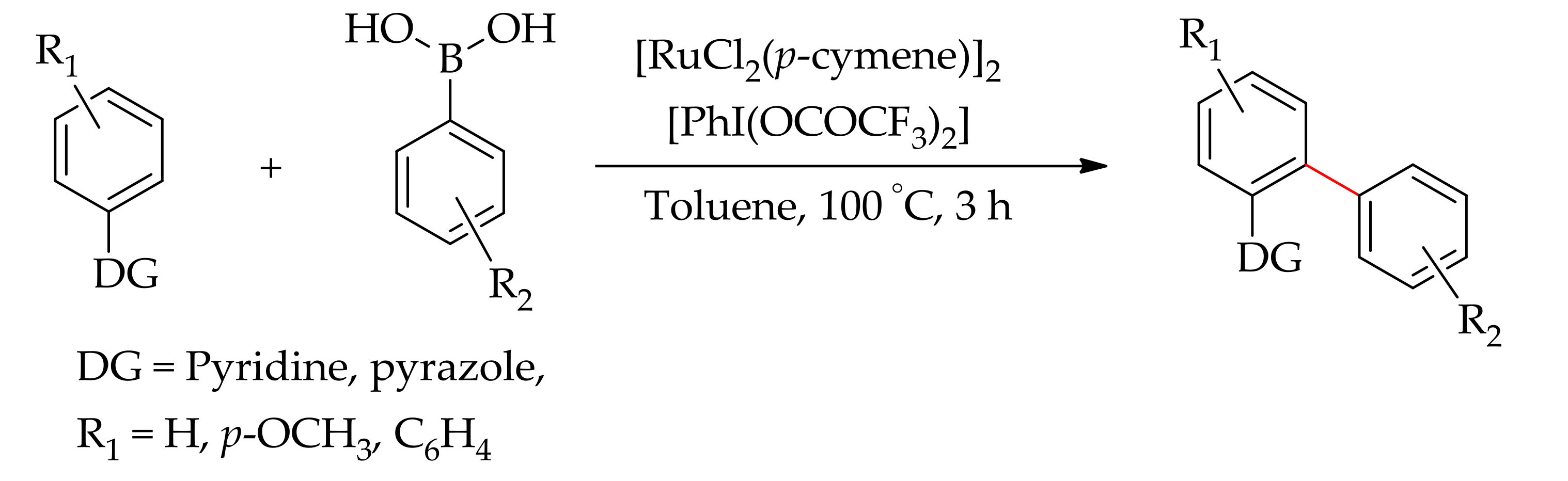
Scheme 14.
Selective monoarylation of 2-phenylpyridine with arene-ruthenium(II) amine catalysts.

Scheme 15.
Arylation of aryltetrazole with aryl chloride via rutheniumcatalyst promoted by amino acid ligands.
Scheme 15.
Arylation of aryltetrazole with aryl chloride via rutheniumcatalyst promoted by amino acid ligands.
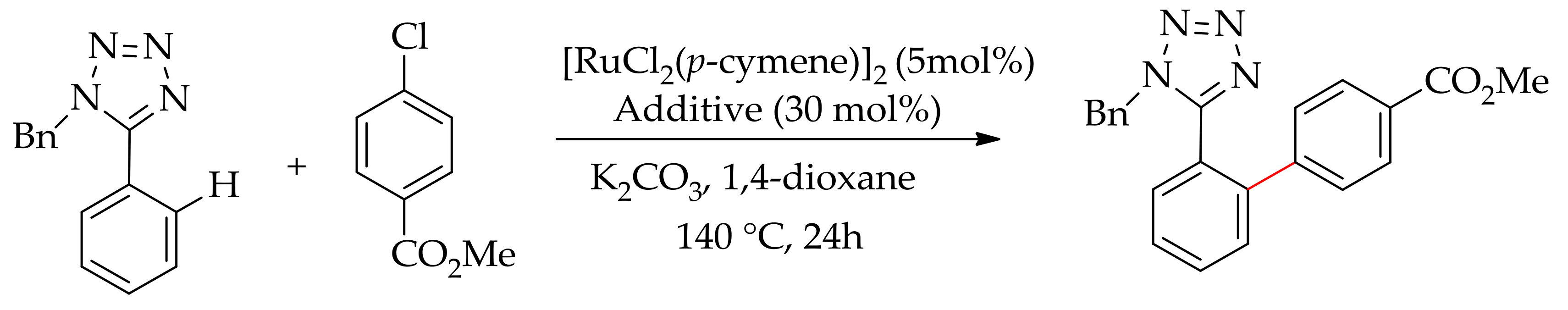
Scheme 16.
Arylation of 2-phenoxypyridines with aryl chlorides and synthesis of arylated phenols.
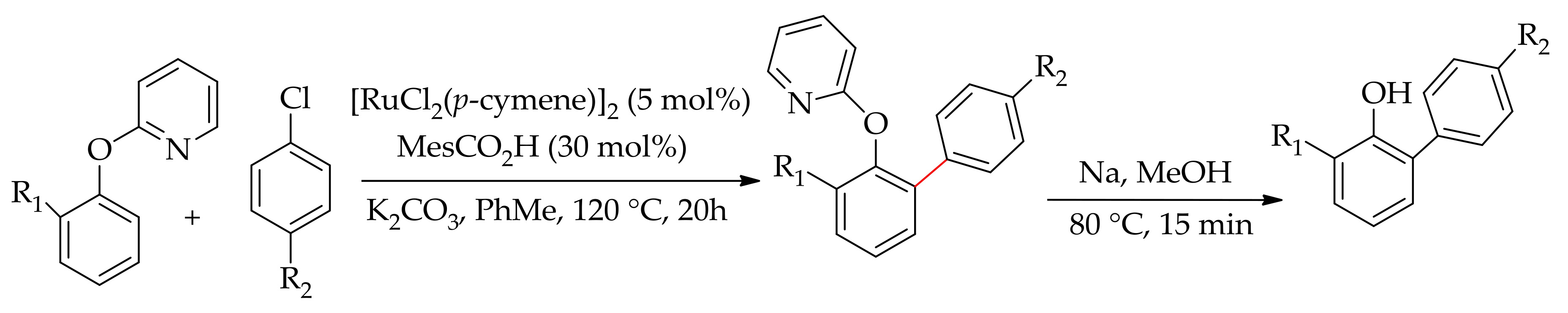
Scheme 17.
Ruthenium-catalysed selective arylation of 2-pyridyl aryl ketone with aryl bromide.
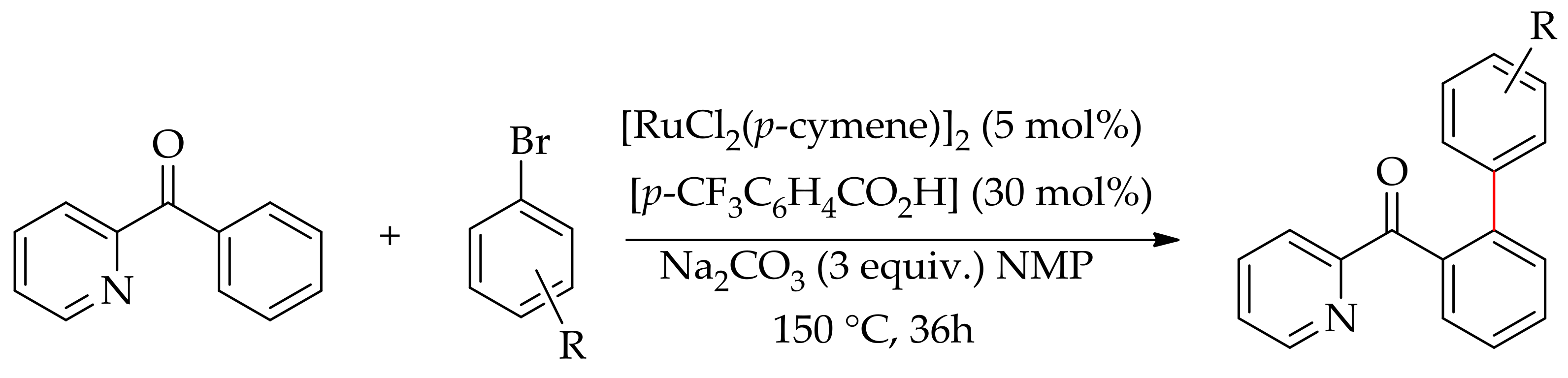
Scheme 18.
Selective mono arylation of 2-phenylpyridine with arene-rutehnium(II)-NHC catalysts.
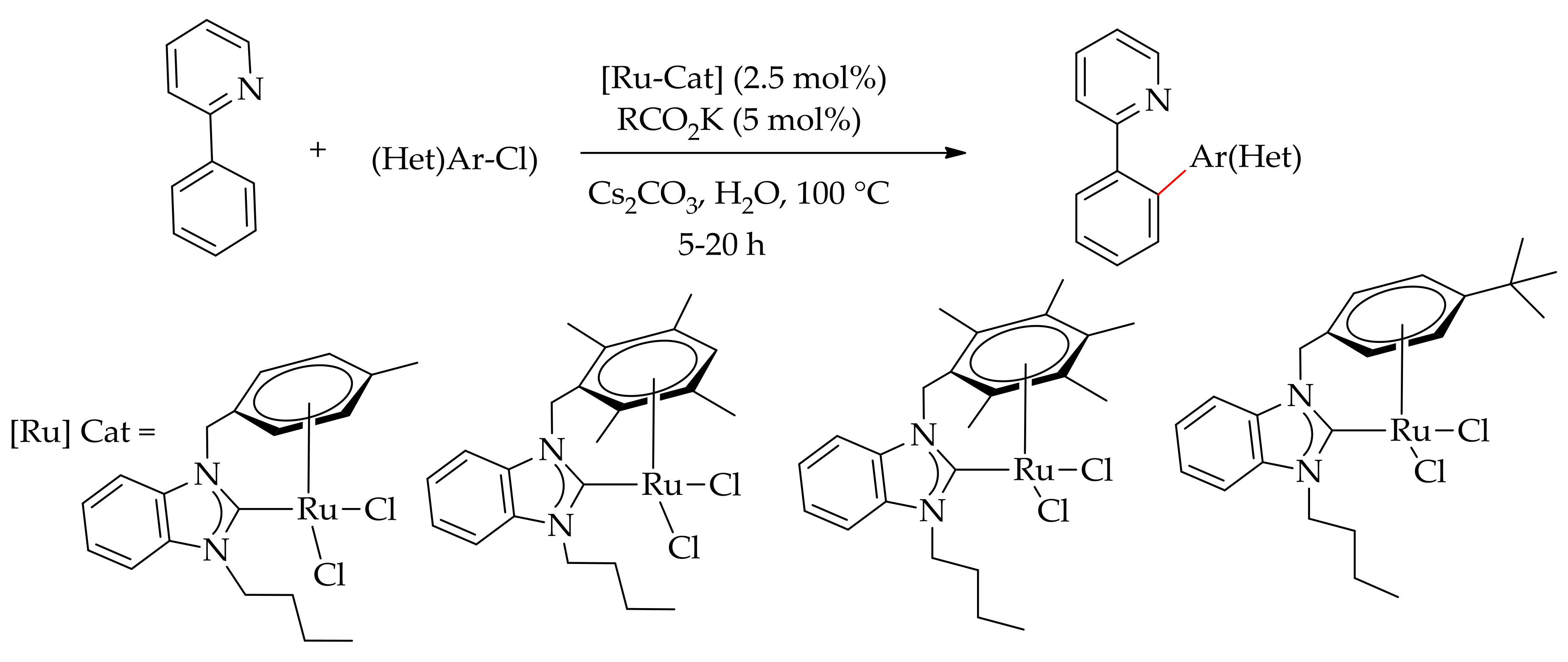
Scheme 19.
Ruthenium-catalysed direct arylation of arenes with aryl bromides in PEG-2000.
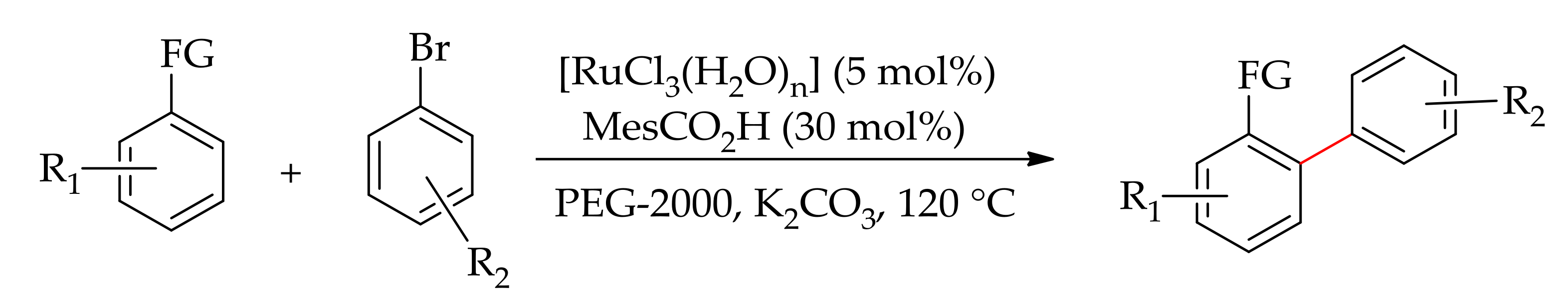
Scheme 20.
Ruthenium-catalysed C-H arylation of alkenylic diazines.

Scheme 21.
Ruthenium-catalysed C-3 arylation of fufural imines with aryl boronate.

Scheme 22.
Ruthenium-catalysed dehydrative arylation of pyrazole with phenols.
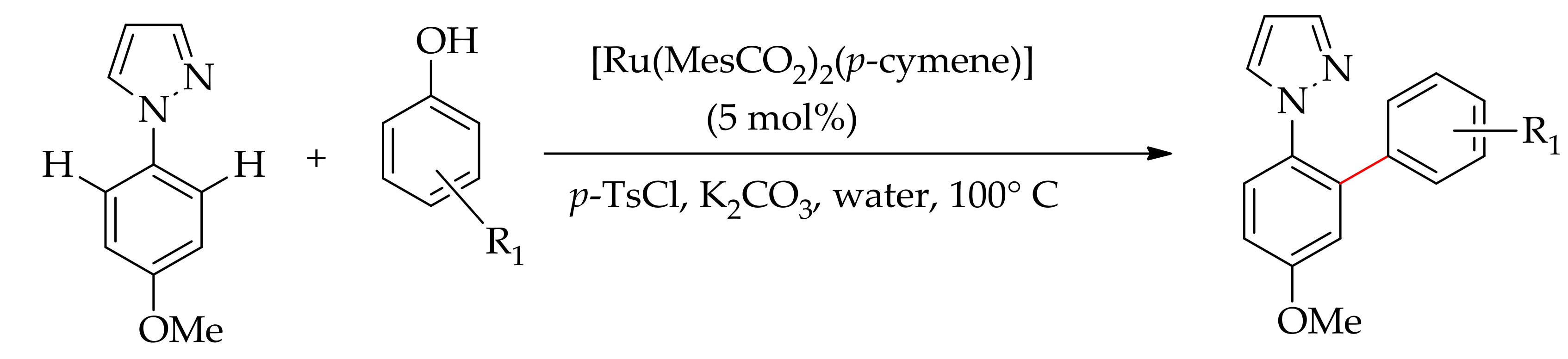
Scheme 23.
Ruthenium-catalysed diarylation of phenyl-1H-pyrazole from functional aryl triflates.

Scheme 24.
Ruthenium-catalysed oxidative arylation of pyrrole or indole derivatives.
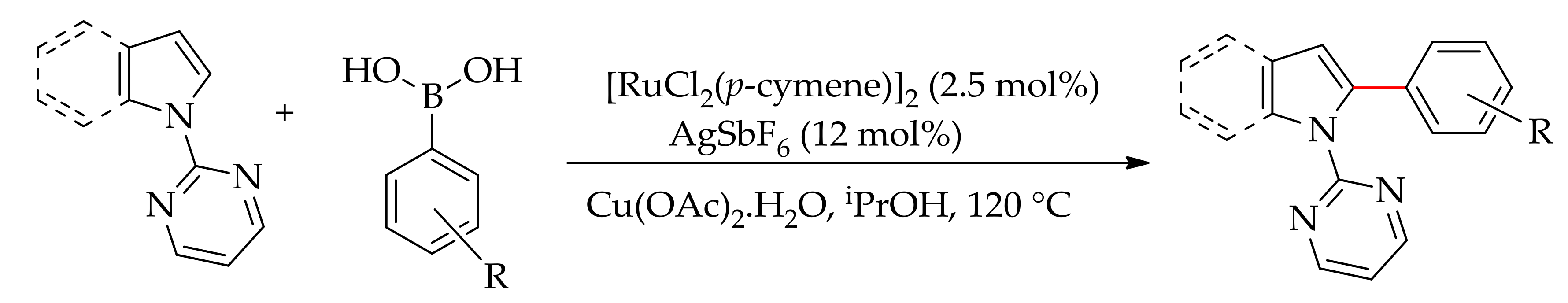
Scheme 25.
Oxidative arylation of alkylbenzamide with aryl boronic acid and synthesis of fluorenones.
Scheme 25.
Oxidative arylation of alkylbenzamide with aryl boronic acid and synthesis of fluorenones.

Scheme 26.
Ruthenium- heterocycle with catalysed oxidative arylation of N- aryl silane.
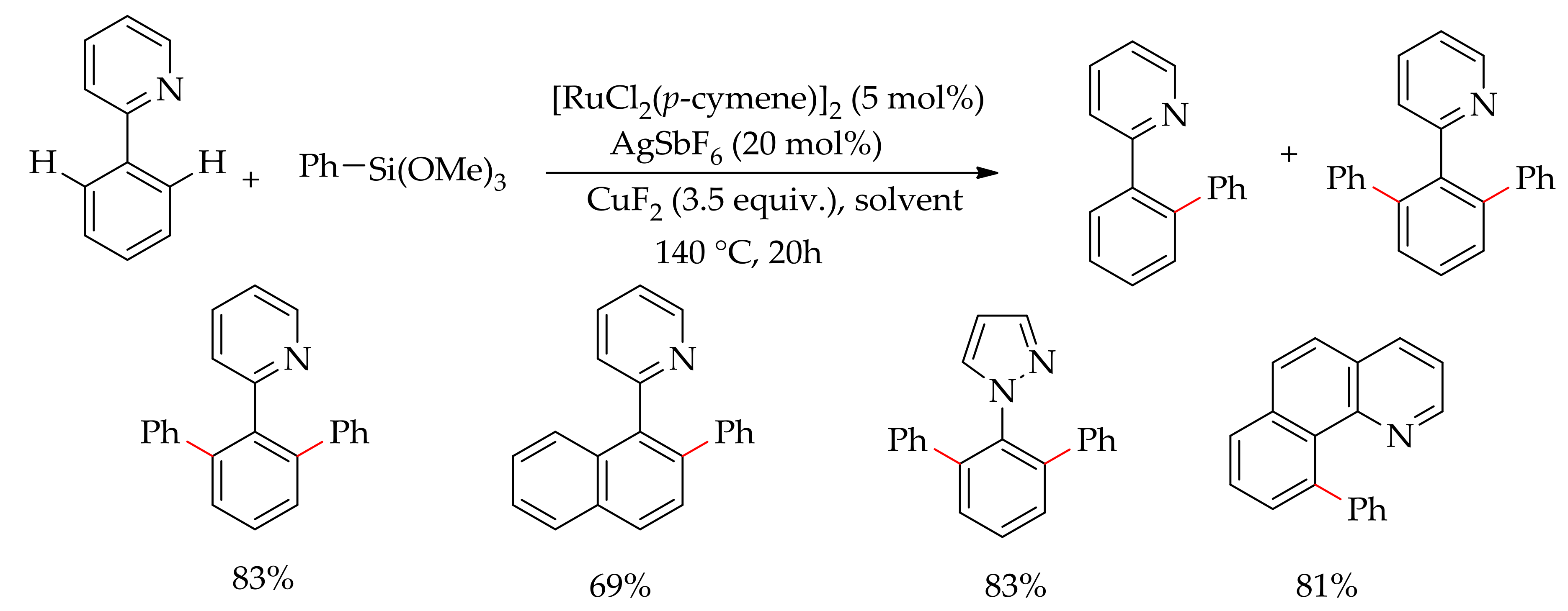
Scheme 27.
Ruthenium-catalysed oxidative arylation of N,N-dialkylbenzamides with phenylboronic acid.
Scheme 27.
Ruthenium-catalysed oxidative arylation of N,N-dialkylbenzamides with phenylboronic acid.
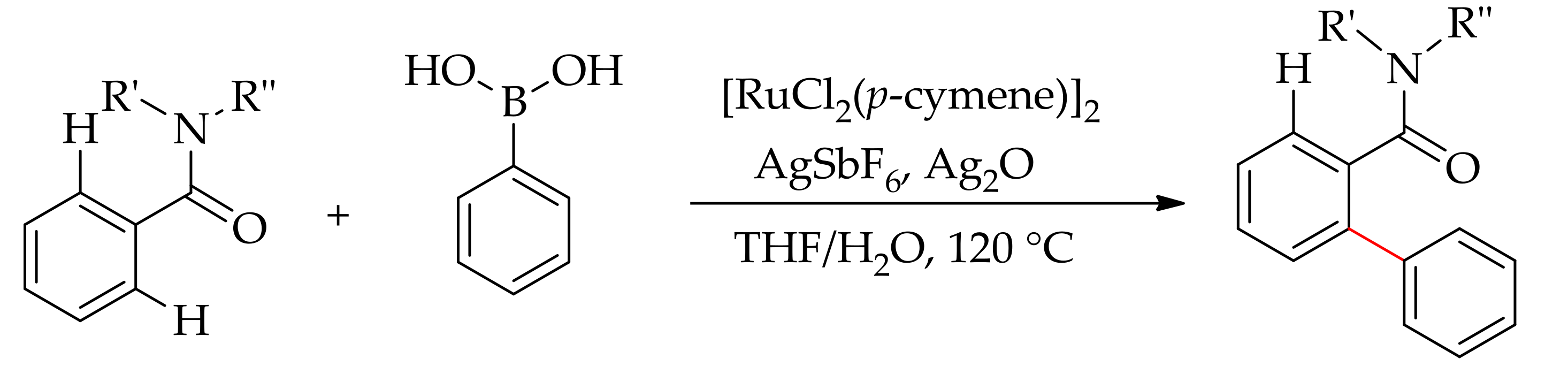
Scheme 28.
Ruthenium-catalysed oxidative arylation of N,N’-dialkylbenzamides with phenylsilane.
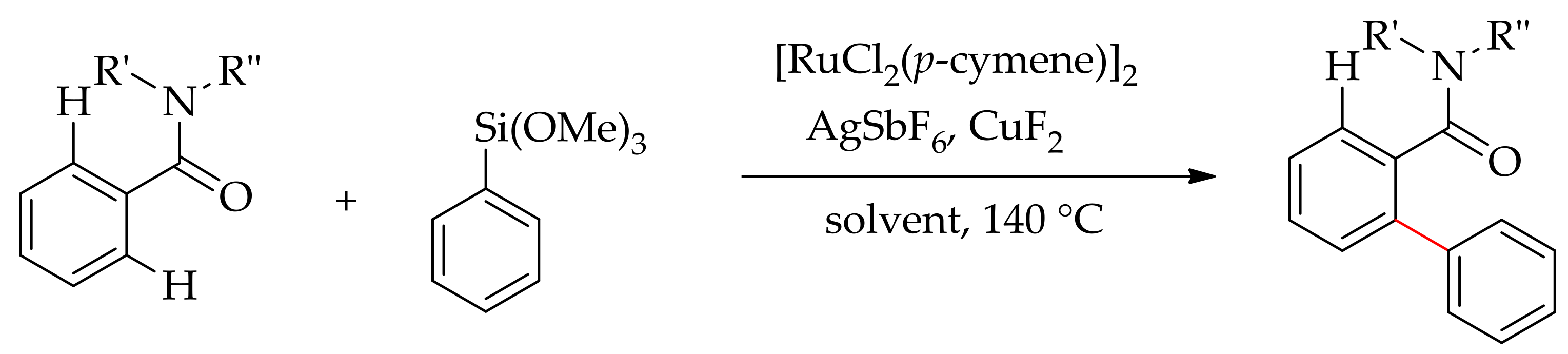
Scheme 29.
Ruthenium-catalysed direct C-2 arylation of indoles with arylsilanes in water.

Scheme 30.
Ruthenium(II)-catalysed C-H arylation of arene with aryl bromide directed by quinazoline.
Scheme 30.
Ruthenium(II)-catalysed C-H arylation of arene with aryl bromide directed by quinazoline.
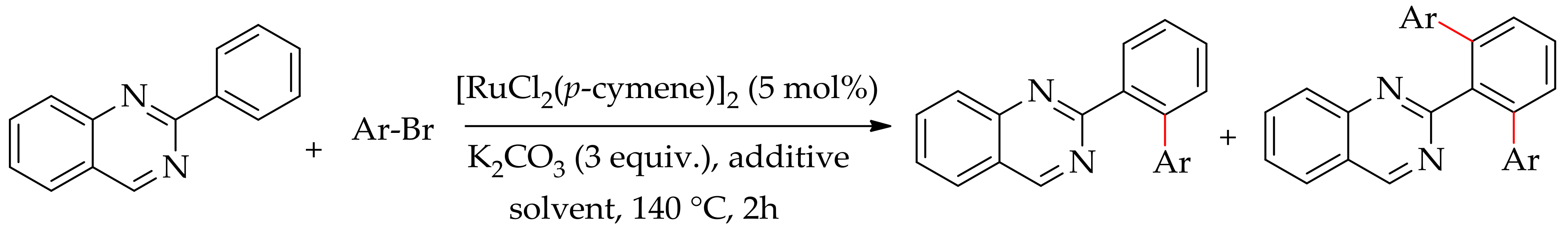
Scheme 31.
Triazole assitsted ruthenium-catalysed C-H arylation of aryl amide.
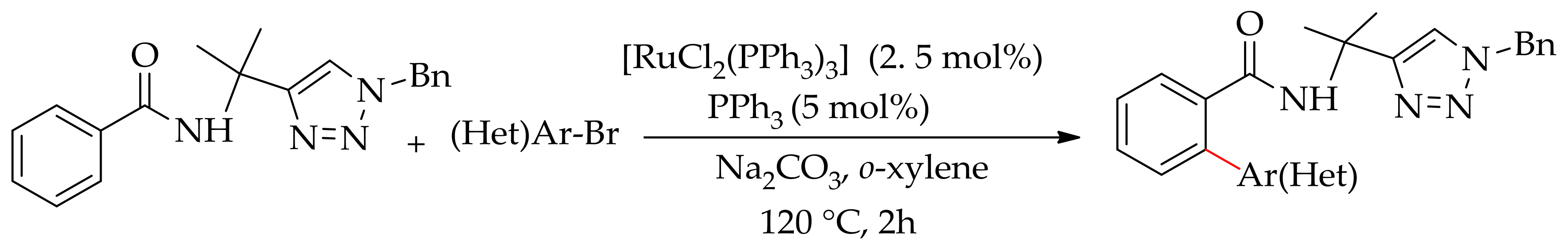
Scheme 32.
Ruthenium-catalysed arylation of acylarenes with halogenated arylboronates.
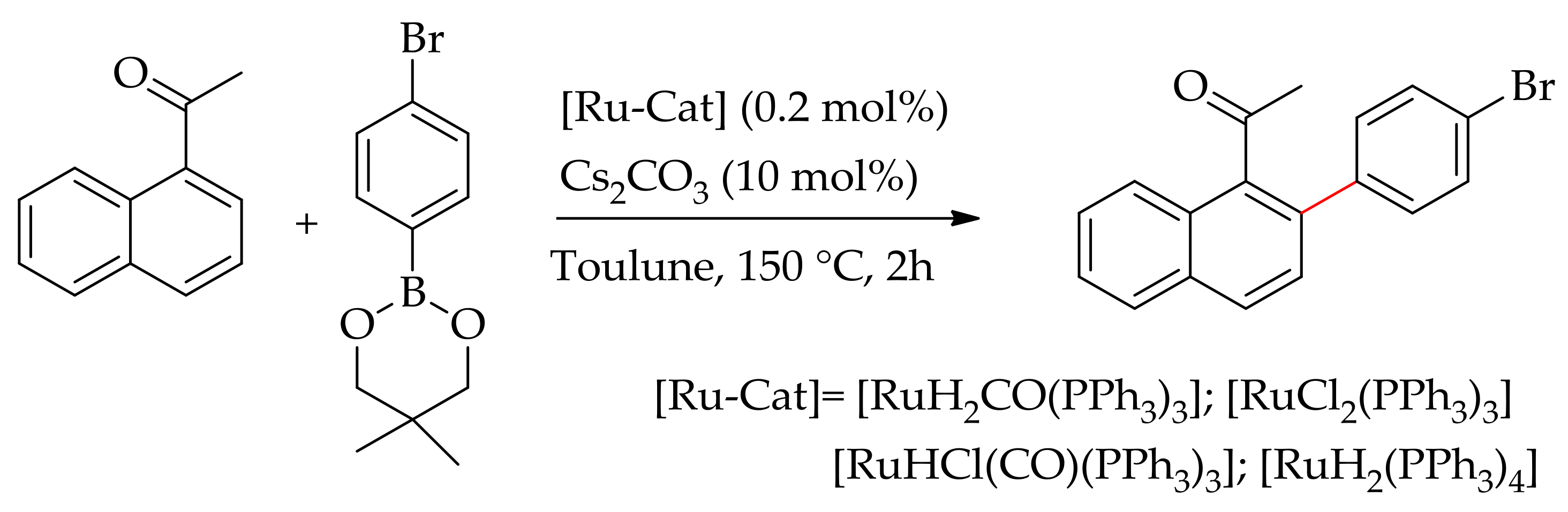
Scheme 33.
Ruthenium-catalysed arylation of aromatic nitriles with arylboronates.

Scheme 34.
Ruthenium-catalysed arylation of with aryl or heteroaryl halides.

Scheme 35.
Ruthenium-catalysed C-H arylation of fluoroarnes with aryl halides.
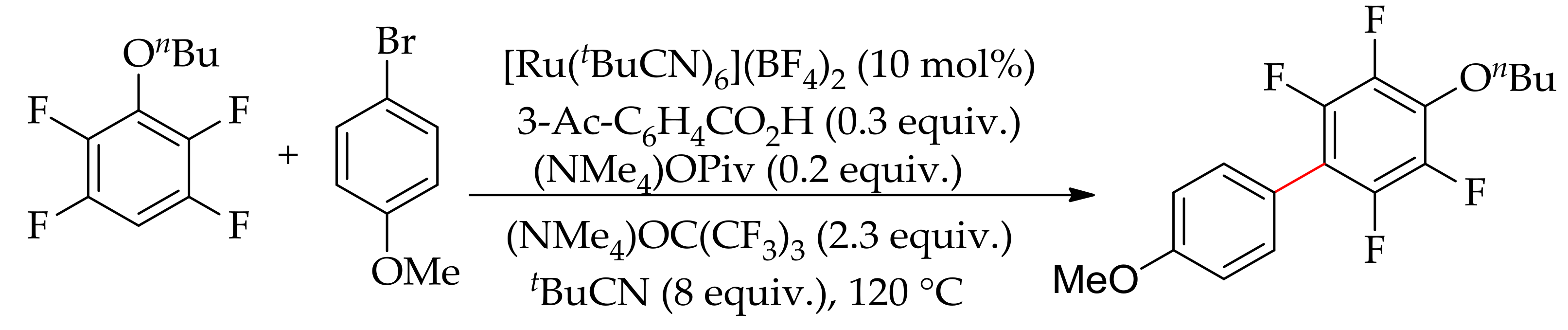
Scheme 36.
Ruthenium-catalysed arylation of carboxylic acids with aryl iodides.
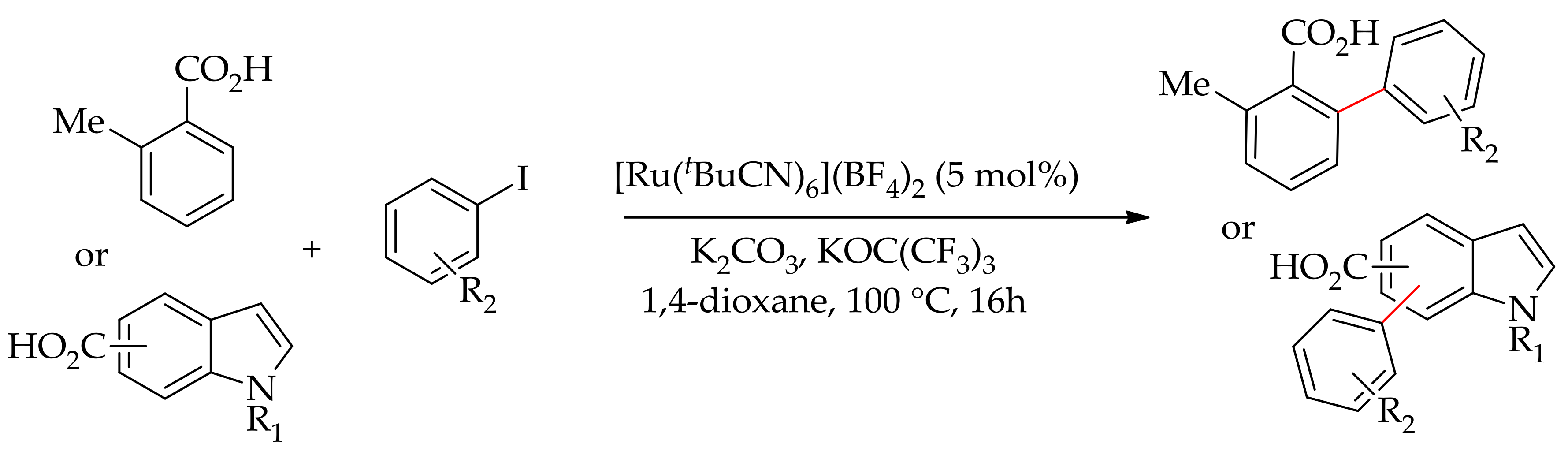
Scheme 37.
Ruthenium(II) -catalysed sp3 C-H bond arylation of benzylic amines with aryl halides.
Scheme 38.
Ruthenium(II)-catalysed decarbonylative arylation of pyrazole and indole with aryl halides.
Scheme 38.
Ruthenium(II)-catalysed decarbonylative arylation of pyrazole and indole with aryl halides.
Scheme 39.
Proposed mechnism for the ruthenium-catalysed decarbonylative arylation of pyrazole [99].
Scheme 39.
Proposed mechnism for the ruthenium-catalysed decarbonylative arylation of pyrazole [99].

Scheme 40.
Ruthenium(II)-catalysed alkenylation of N-aryl pyrazoles.
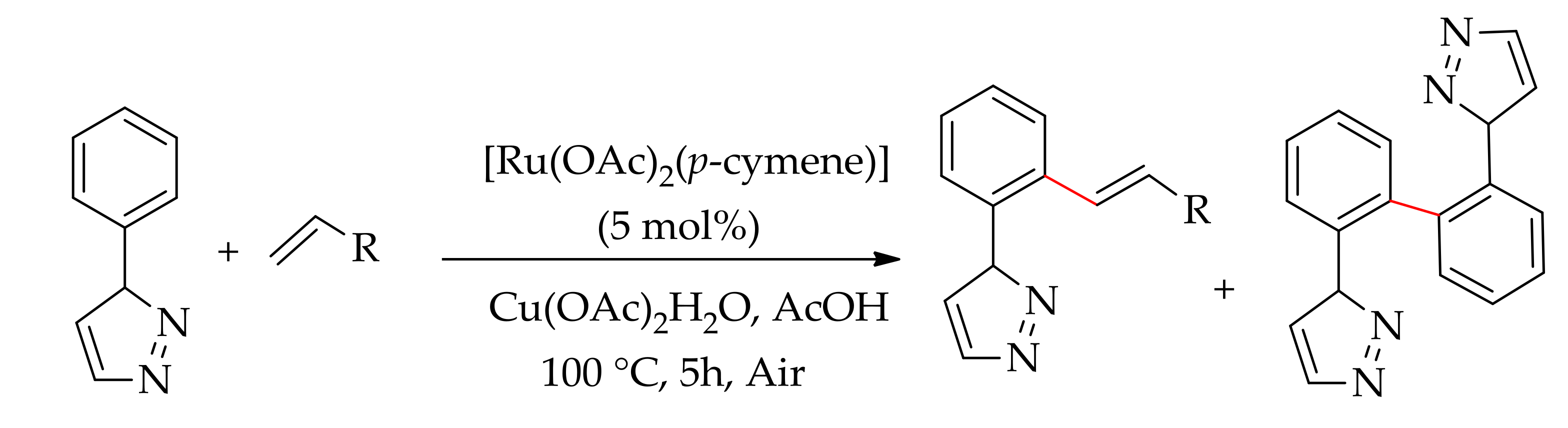
Scheme 41.
Ruthenium-catalysed alkenylation of phenyloxazolines with alkenes.
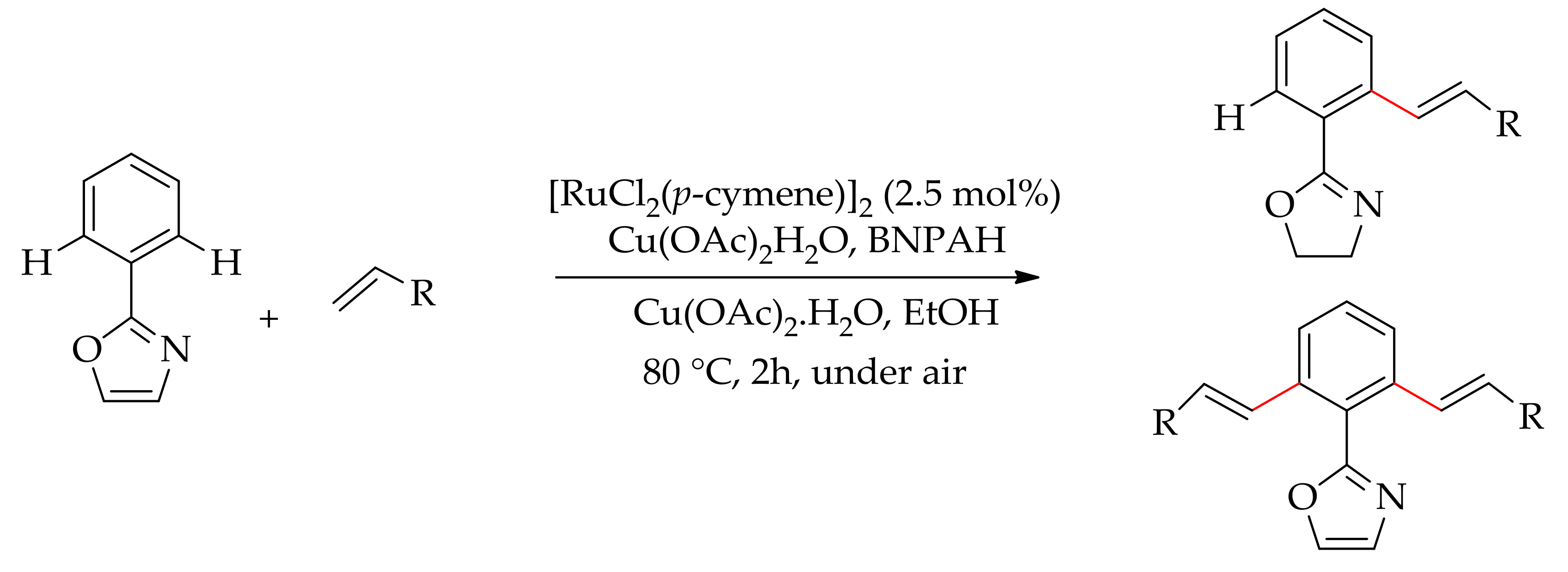
Scheme 42.
Alkenylation of 2-phenoxypyridines with acrylates.
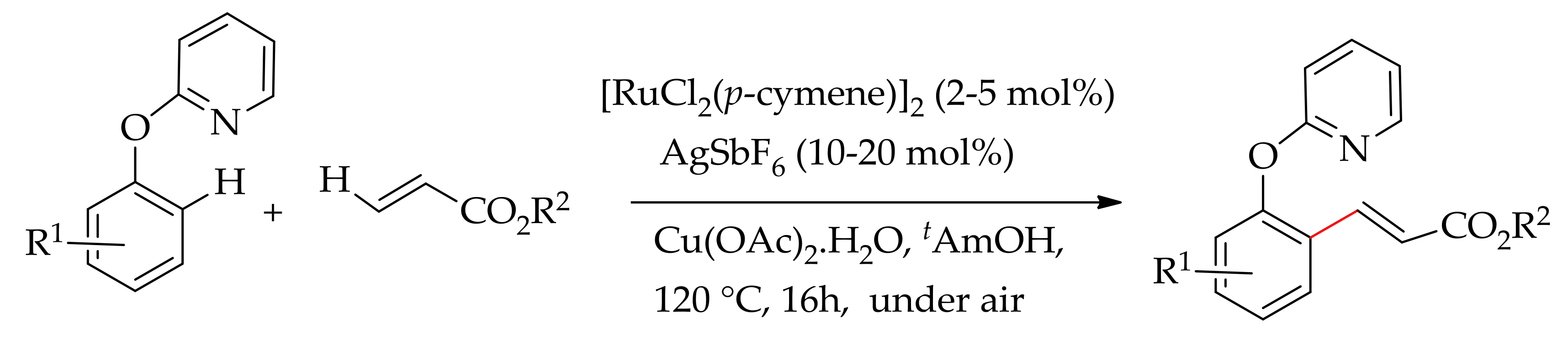
Scheme 43.
Ruthenium-catalysed alkenylation of 2-phenylimidazo[1,2-a] pyridine.
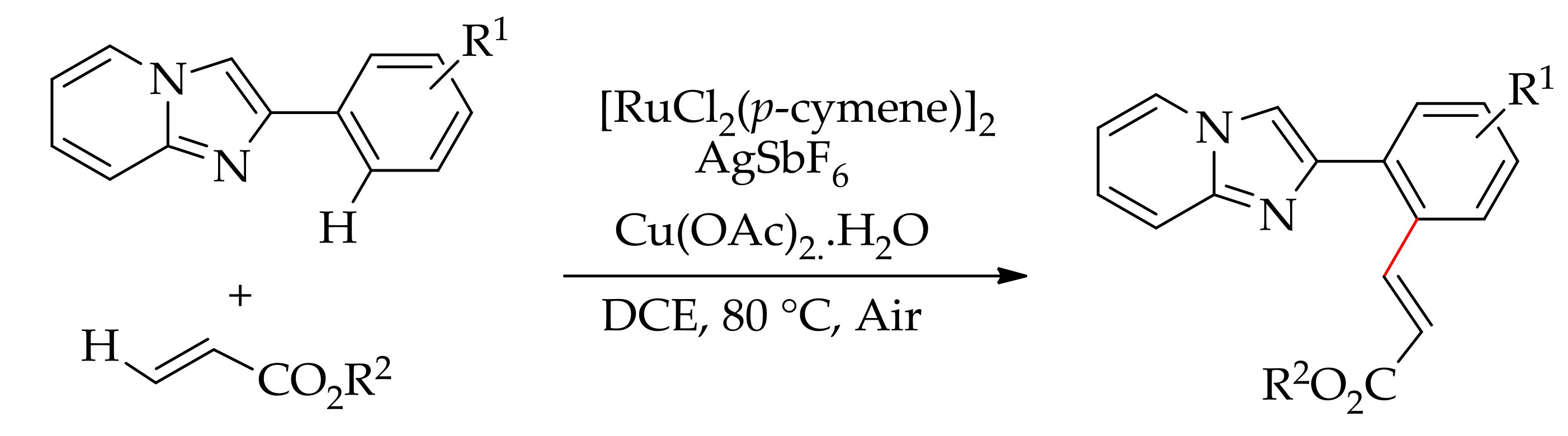
Scheme 44.
Ruthenium-catalysed switchable C-3 alkylation and alkenylation of 2-pyridinebenzofurans.
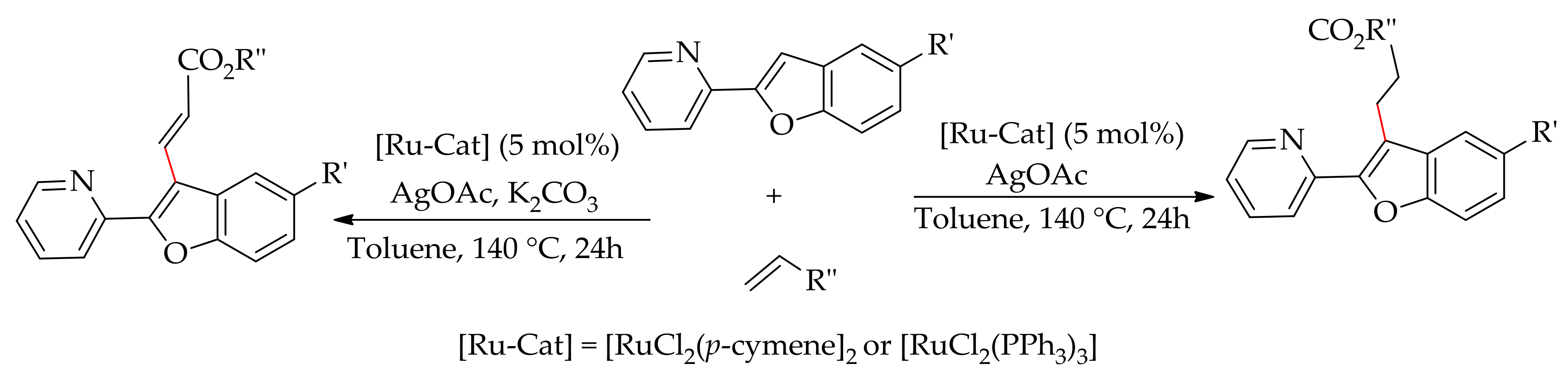
Scheme 45.
Alkenylation of pyraozle/oxazole with alkenes under neat condition (a); Alkenylation of pyrazoles with acrylates (b).
Scheme 45.
Alkenylation of pyraozle/oxazole with alkenes under neat condition (a); Alkenylation of pyrazoles with acrylates (b).
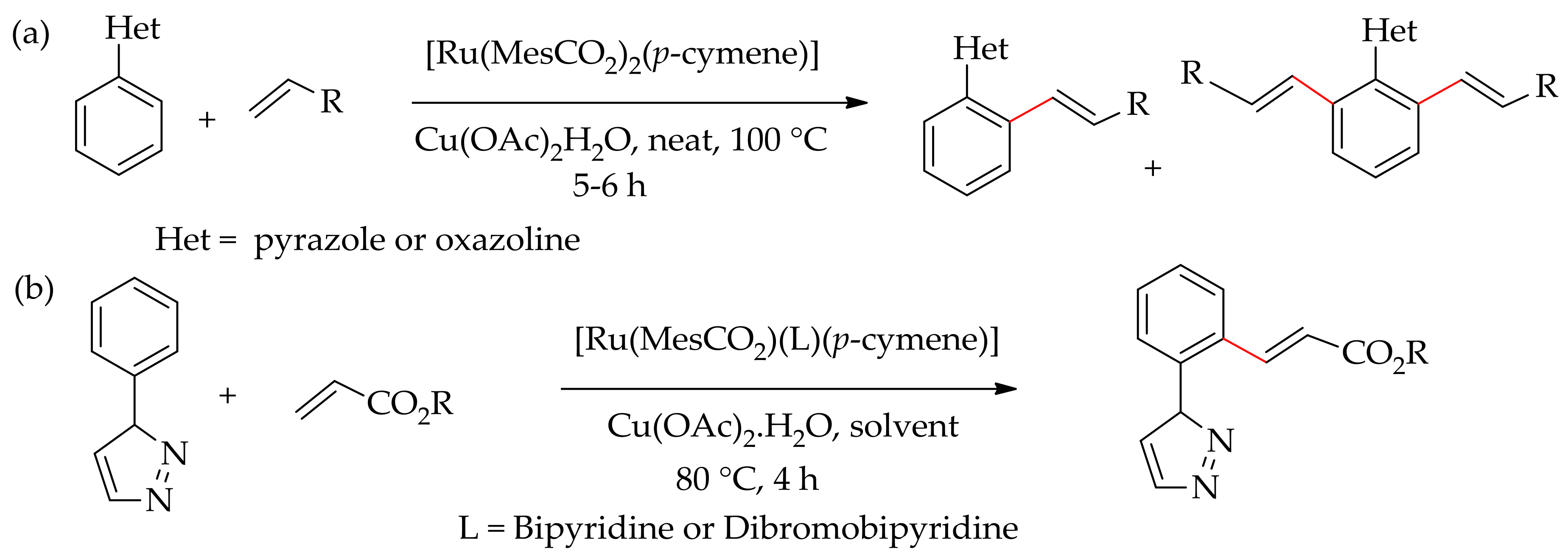
Scheme 46.
Alkenylation/annulation of benzimidazoles with acrylates to benzoimidazoisoindoles.
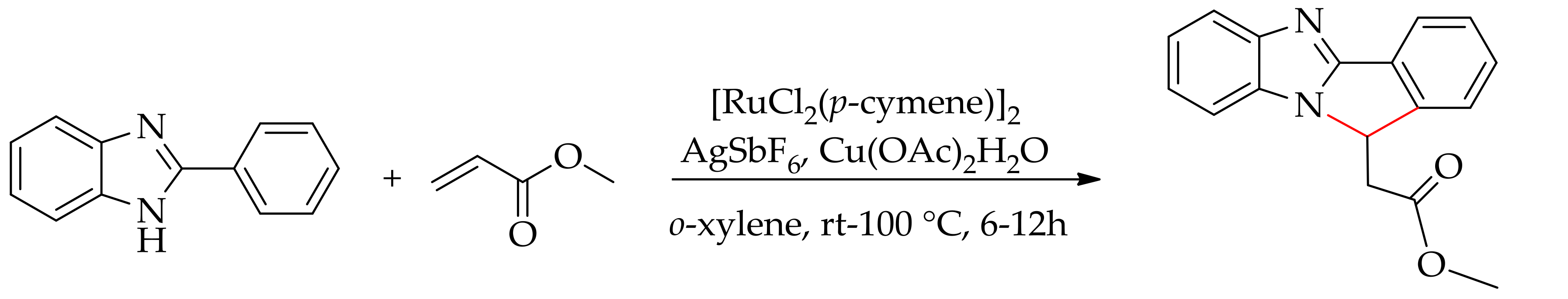
Scheme 47.
Proposed mechanism for the formation of benzoimidazoisoindoles [113].
Scheme 47.
Proposed mechanism for the formation of benzoimidazoisoindoles [113].
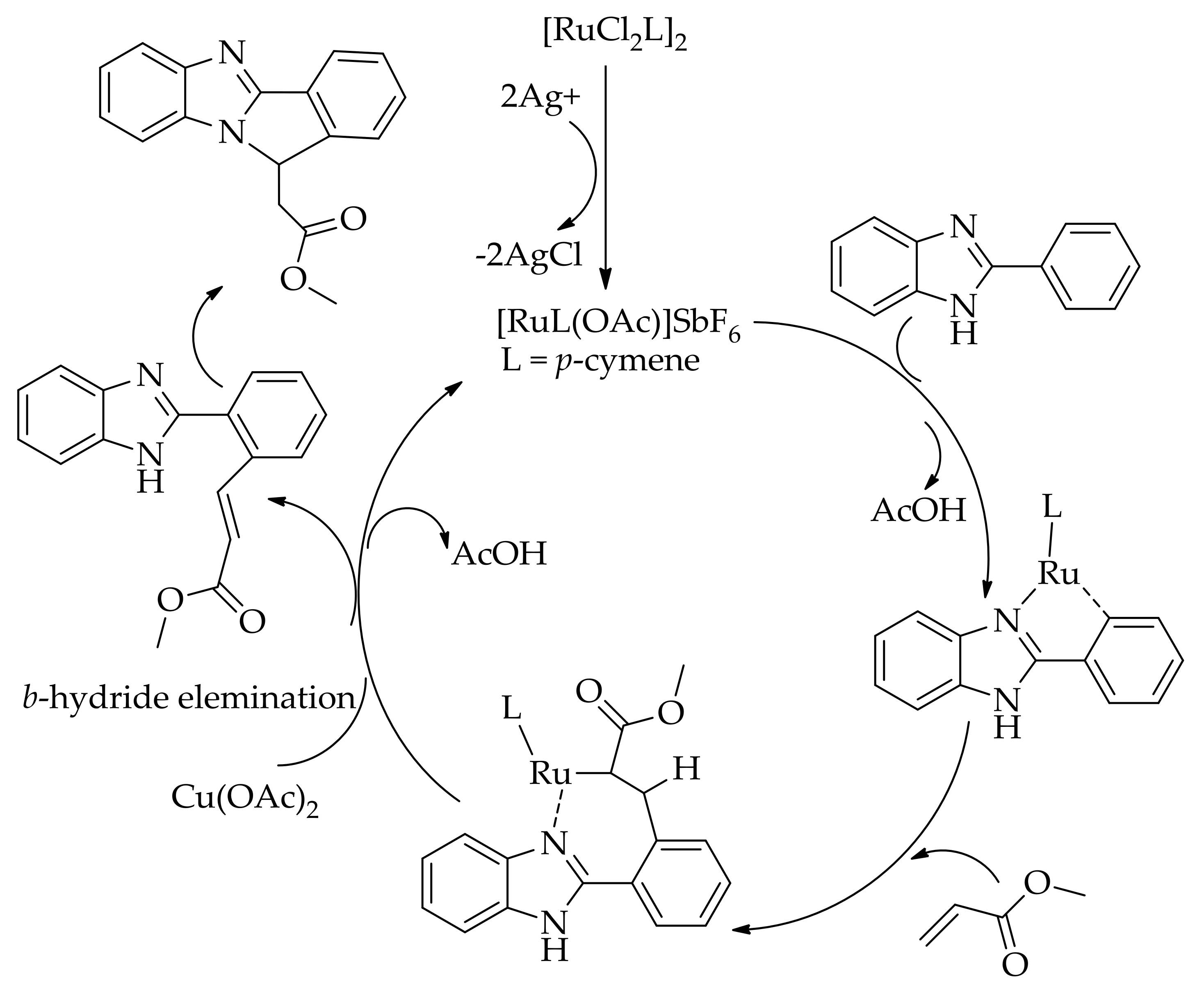
Scheme 48.
Ruthenium-catalysed synthesis of indazole derivatives via alkenylation-annulation approach.
Scheme 48.
Ruthenium-catalysed synthesis of indazole derivatives via alkenylation-annulation approach.

Scheme 49.
Ruthenium-catalysed alkenylation of heteroaromatic carboxylic acids with alkenes.
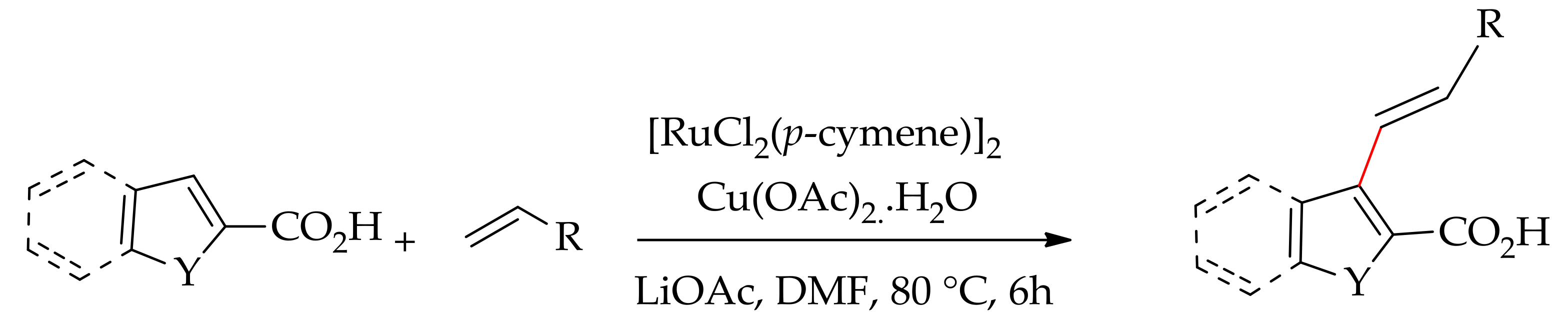
Scheme 50.
Ruthenium-catalysed akenylation of carboxylic acids leading to annulated lactones.

Scheme 51.
Ruthenium-catalysed decarboxylative meta-alkenylation of aromatic carboxylic acids.
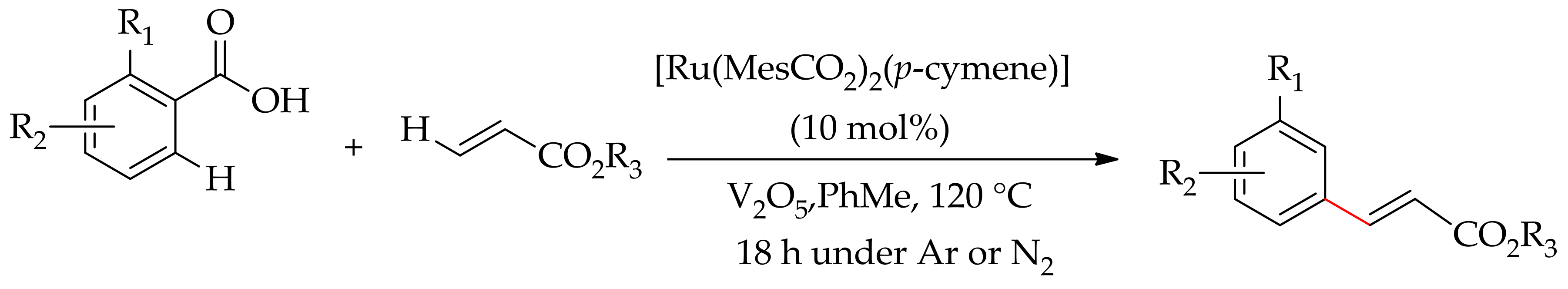
Scheme 52.
Ruthenium-catalysed decarboxylative intramolecular alkenylation of indole derivatives.
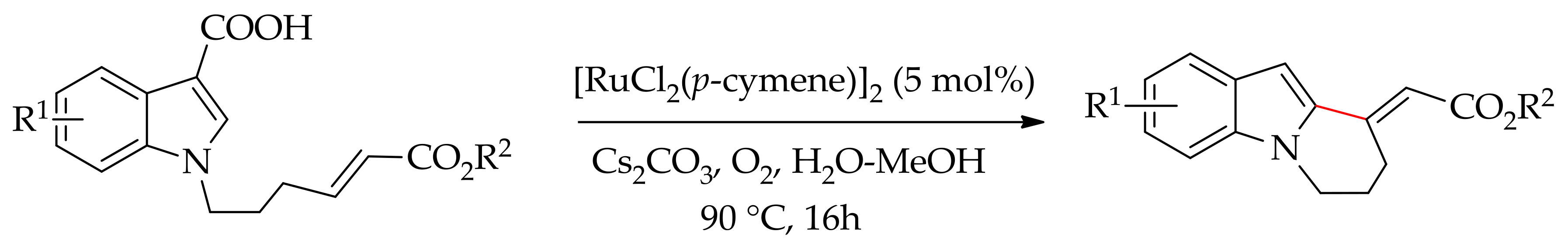
Scheme 53.
Ruthenium catalysed mono alkenylation of aromatic acids without decarboxylation.
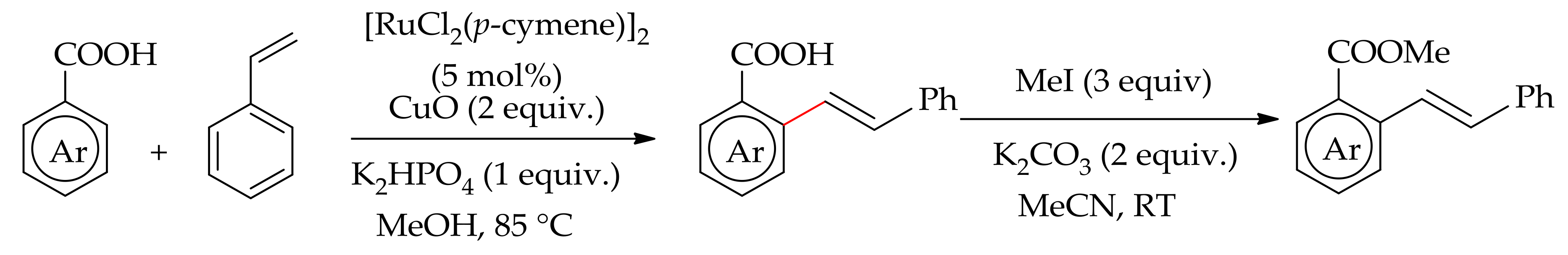
Scheme 54.
Ruthenium catalysed dialkenylation of para-substituted aromatic acids without decarboxylation.
Scheme 54.
Ruthenium catalysed dialkenylation of para-substituted aromatic acids without decarboxylation.
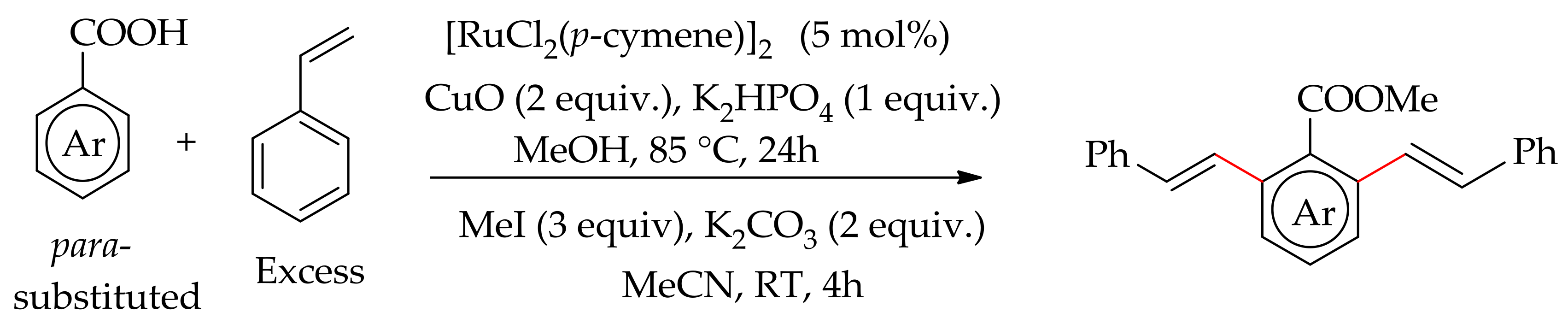
Scheme 55.
Ruthenium-catalysed (a) ortho-alkenylation of sulfonic acids or sulfonyl chlorides; (b) Oxidative alkenylation of sulfonamides.
Scheme 55.
Ruthenium-catalysed (a) ortho-alkenylation of sulfonic acids or sulfonyl chlorides; (b) Oxidative alkenylation of sulfonamides.
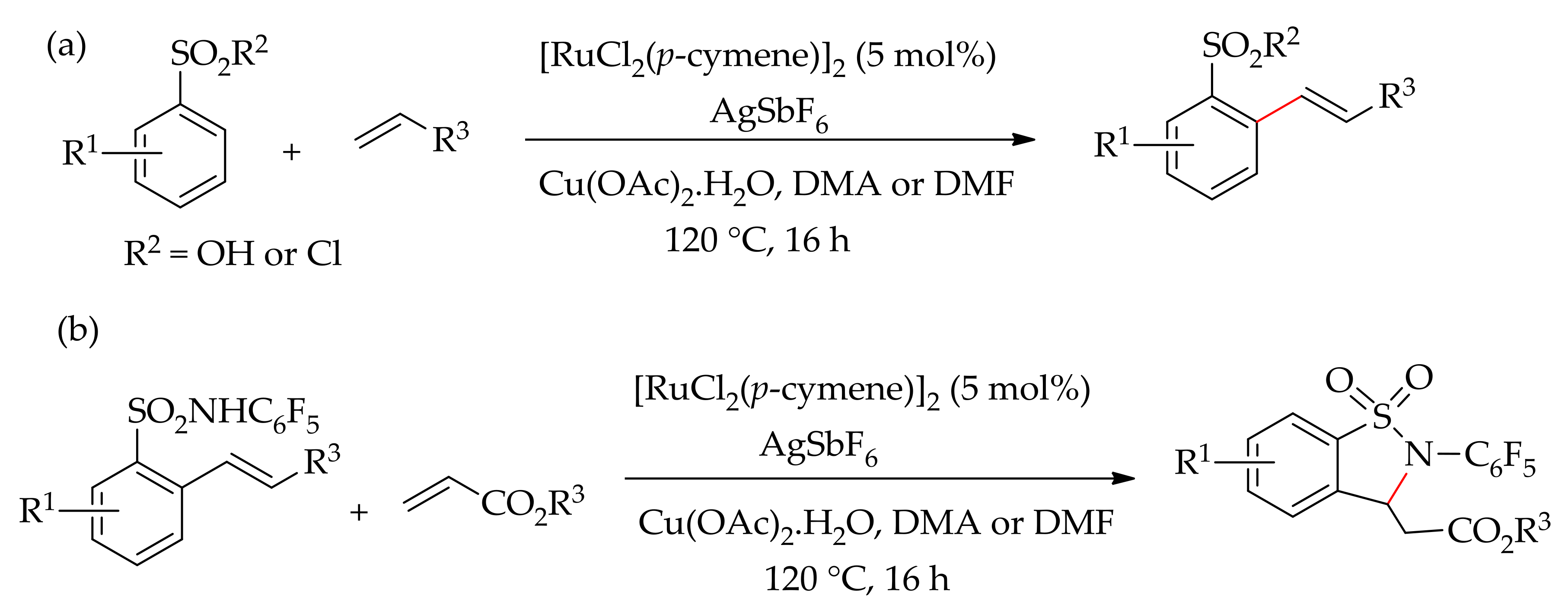
Scheme 56.
Ruthenium(II)-catalysed alkenylation of aromatic ketones.

Scheme 57.
Alkenylation of aromatic aldehydes and photochemical rearrangement to cyclic ketones or polycyclic isochromanone.
Scheme 57.
Alkenylation of aromatic aldehydes and photochemical rearrangement to cyclic ketones or polycyclic isochromanone.
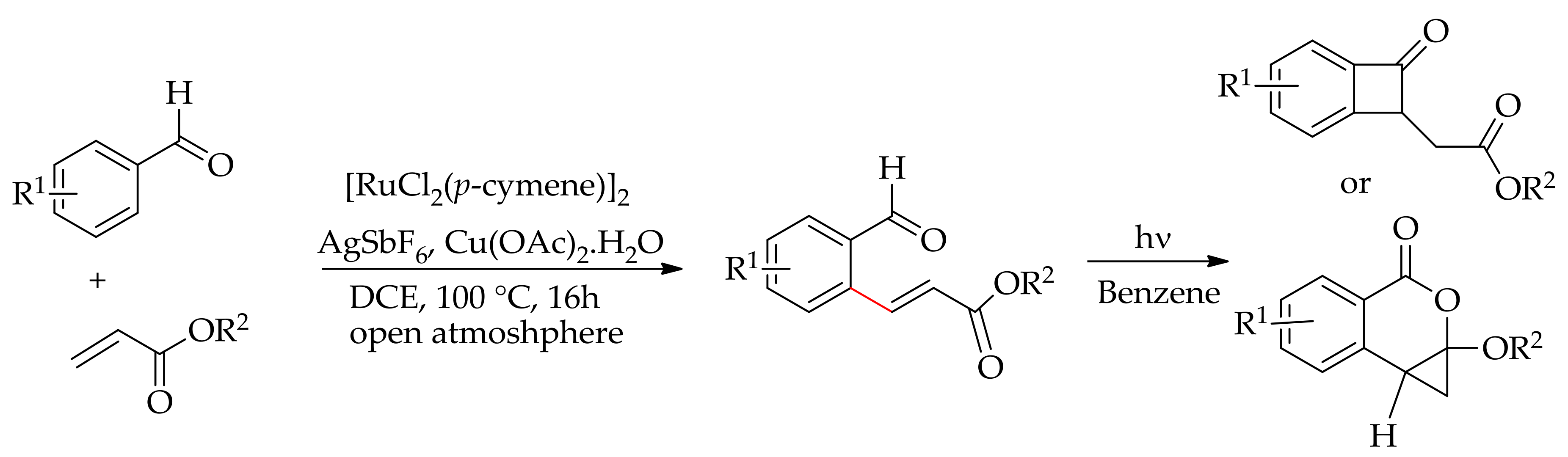
Scheme 58.
Ruthenium-catalysed oxidative alkenylation of aromatic esters.

Scheme 59.
Ruthenium-catalysed alkenylation of aromatic nitriles with activated alkynes.

Scheme 60.
Alkenylated products of 2-cyanothiophene and 3-cyanothiophene with acrylates.

Scheme 61.
Ruthenium-catalysed oxidative alkenylation of benzamides and anilides.
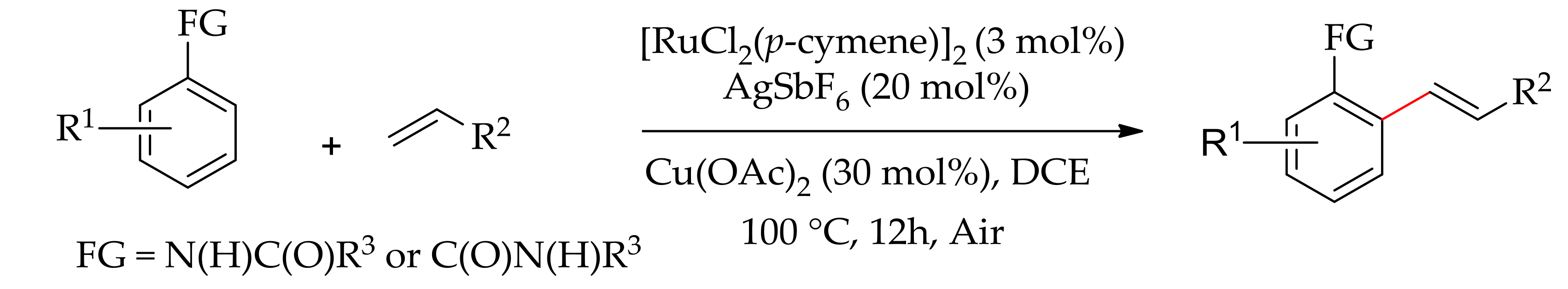
Scheme 62.
Ruthenium-catalysed alkenylation of Weinreb’s amides.

Scheme 63.
Ruthenium catalysed alkenylation of removable phenol derivatives (a); alkenylation of amide or acid with acrylates (b).
Scheme 63.
Ruthenium catalysed alkenylation of removable phenol derivatives (a); alkenylation of amide or acid with acrylates (b).
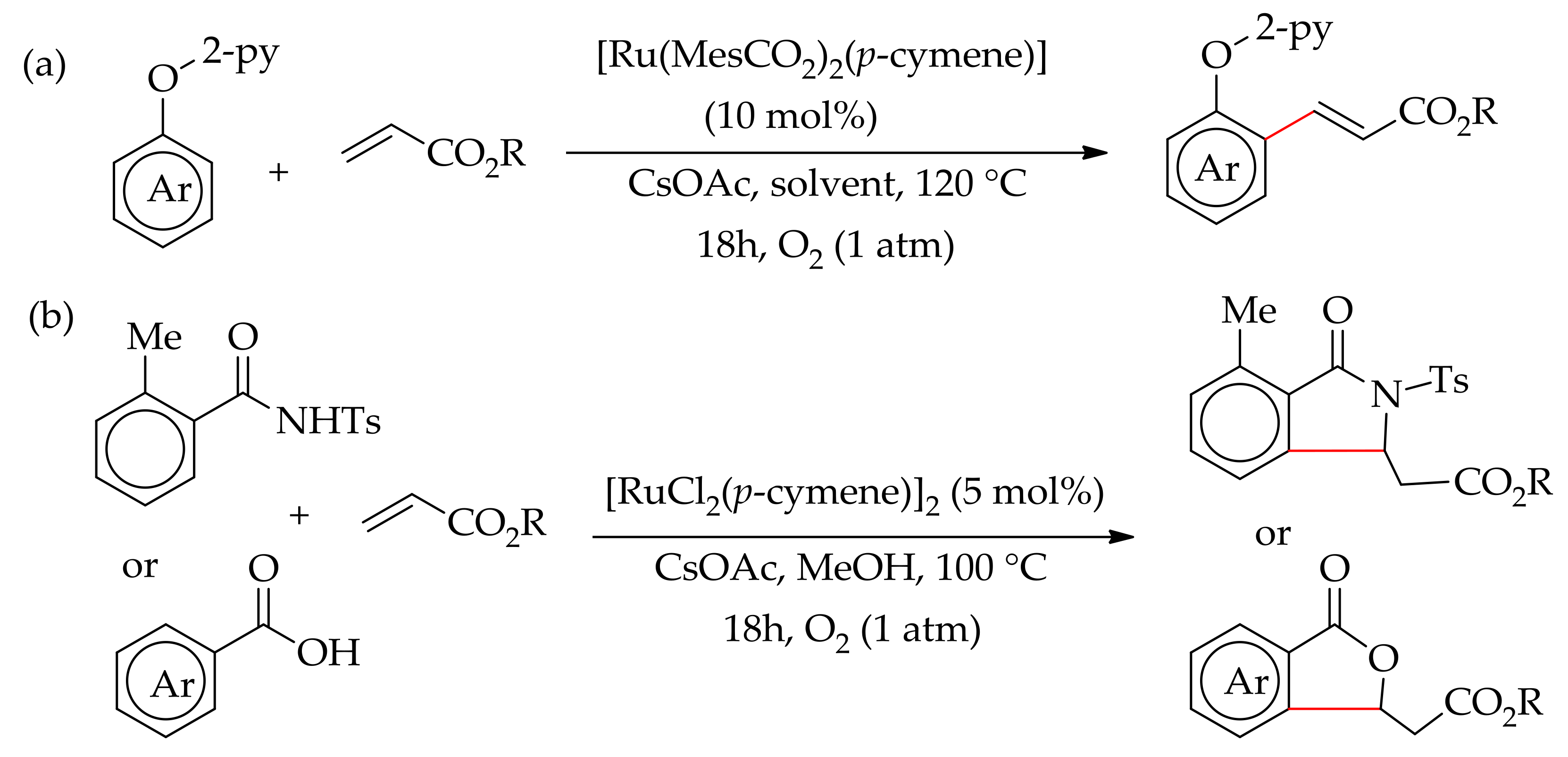
Scheme 64.
Ruthenium-catalysed hydroarylation of phenyltriazoles with alkynes.

Scheme 65.
Ruthenium-catalysed hydroarylation of aromatic acids with decarboxylation.
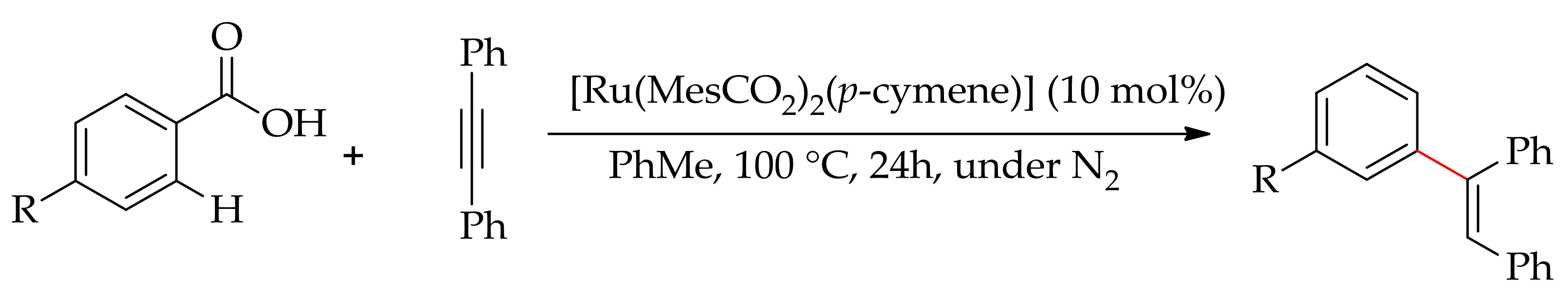
Scheme 66.
Ruthenium-catalysed alkylation of 2-phenylpyridine with alkene via hydroaylation.
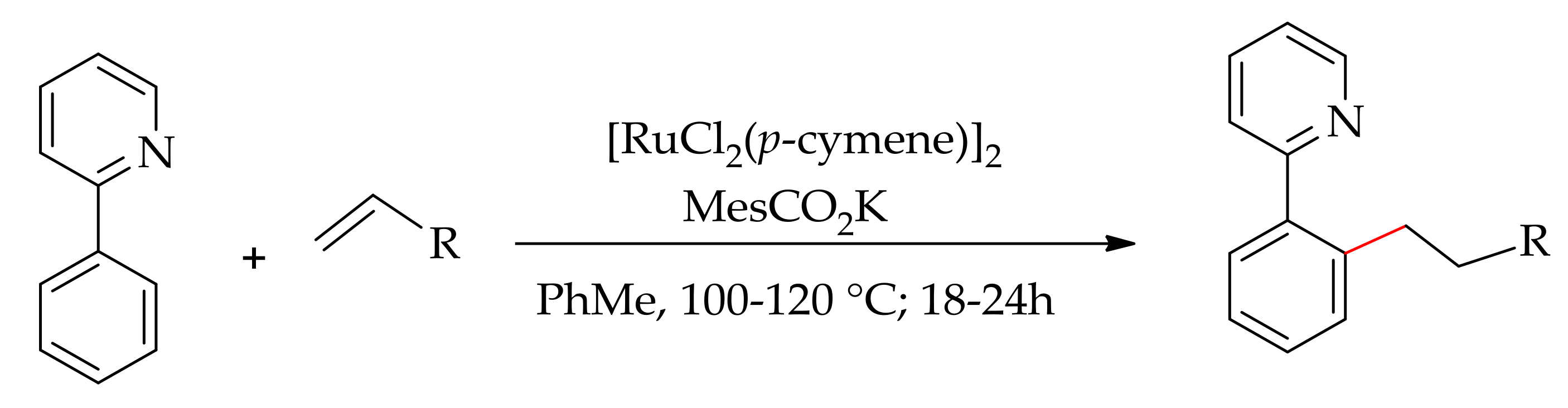
Scheme 67.
Ruthenium-catalysed sp3 C-H alkylation via hydroaylation (A) and removal of directing group leading to free pyrolidines (B).
Scheme 67.
Ruthenium-catalysed sp3 C-H alkylation via hydroaylation (A) and removal of directing group leading to free pyrolidines (B).
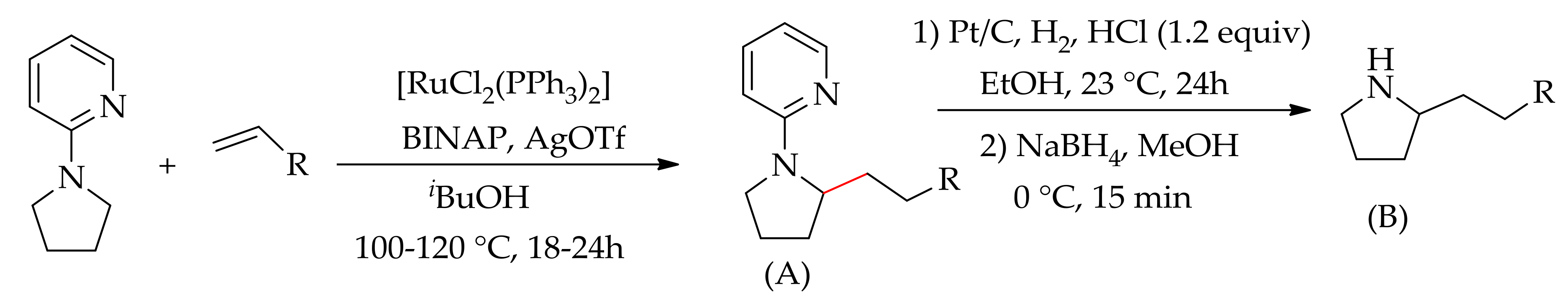
Scheme 68.
Ruthenium-catalysed C-3 alkylation of indoles with alkynes.
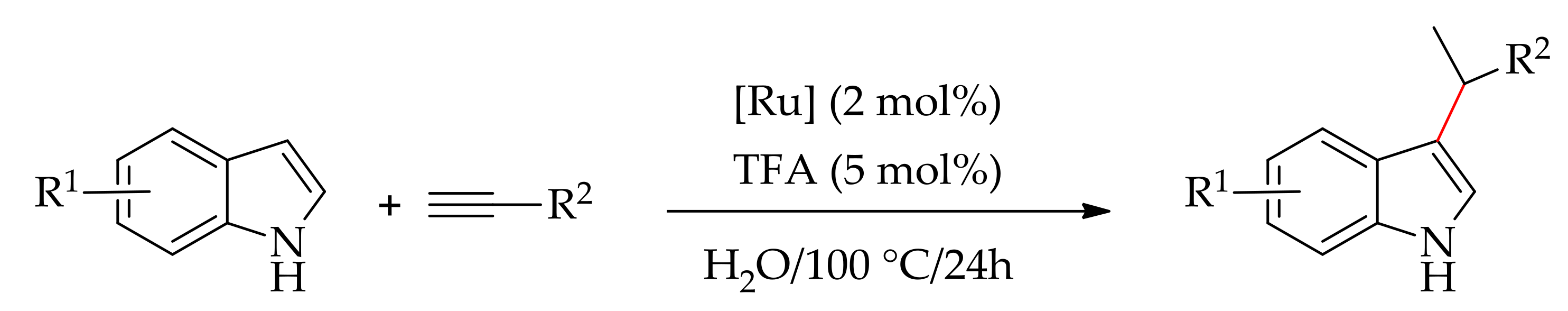
Scheme 69.
Ruthenium-catalysed meta-selective alkylation of 2-phenylpyridine derivative with alkyl bromides.
Scheme 69.
Ruthenium-catalysed meta-selective alkylation of 2-phenylpyridine derivative with alkyl bromides.

Scheme 70.
Ruthenium-catalysed meta-selective C-H alkylation of phenol derivatives.
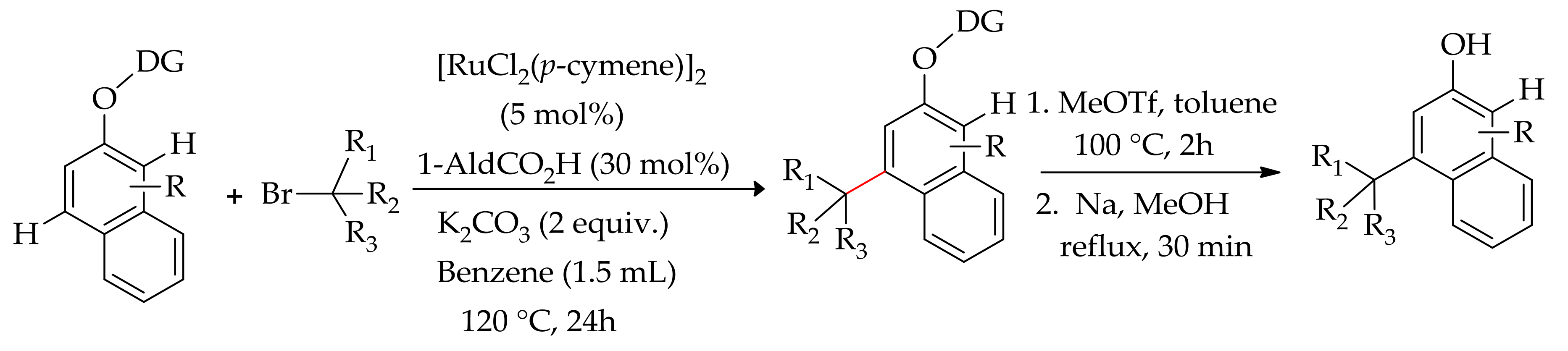
Scheme 71.
Ruthenium-catalysed ortho-and meta-selective alkylation of azoarenes.
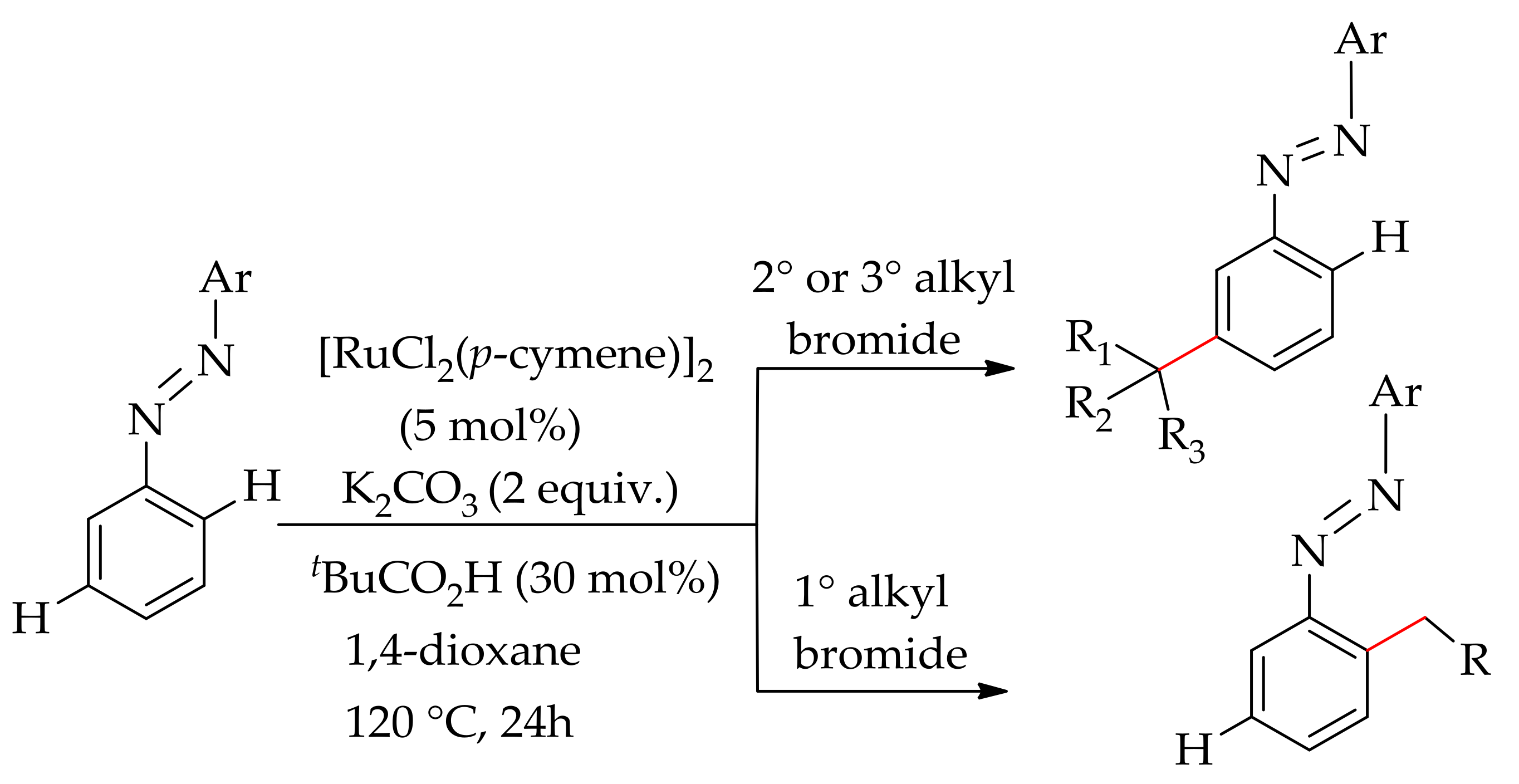
Scheme 72.
Ruthenium-catalysed direct meta-alkylation of arene with alkyl bromide.
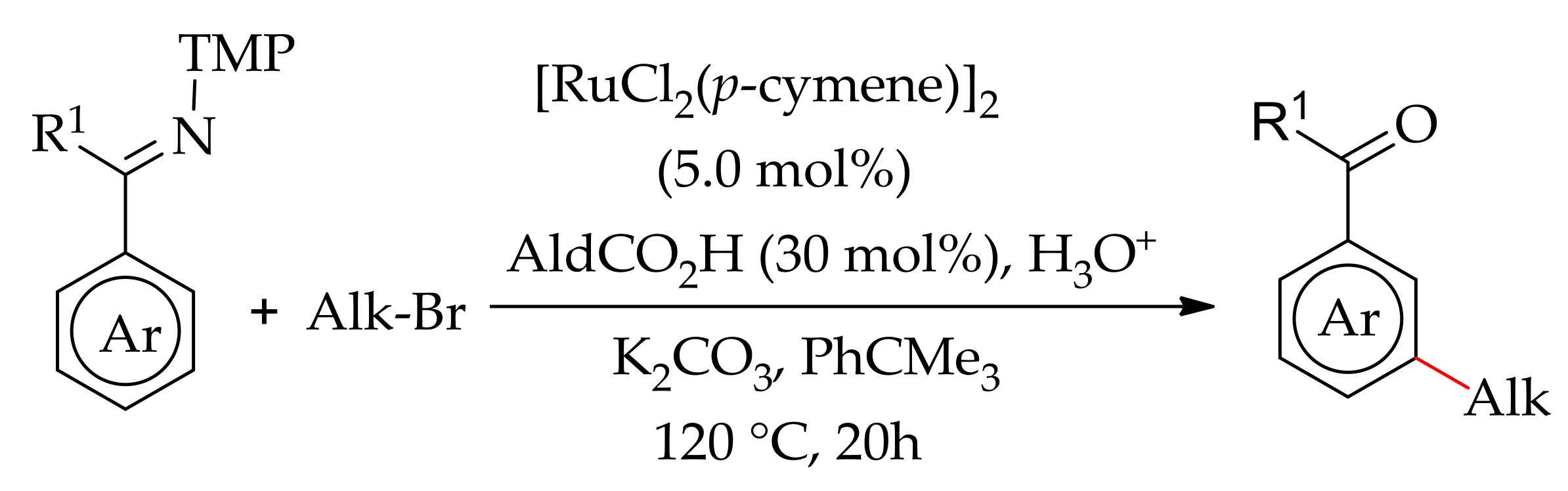
Scheme 73.
Ruthenium-catalysed para-selective C-H alkylation of aniline derivatives.
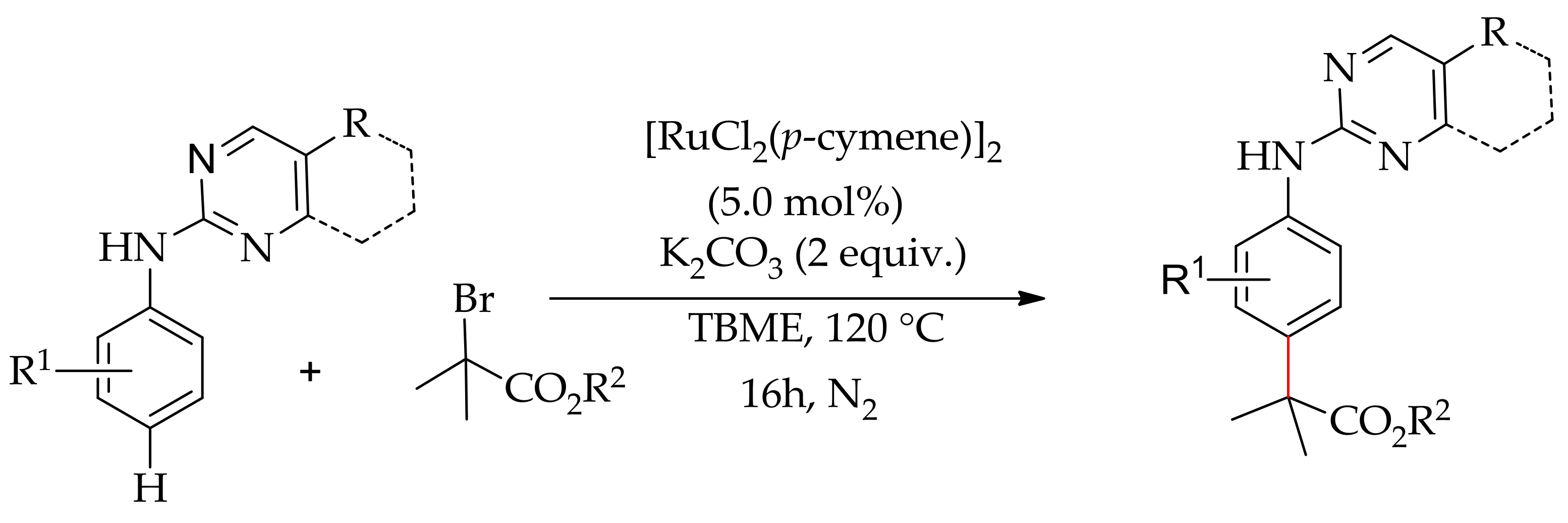
Scheme 74.
Ruthenium-catalysed C-6 remote alkylation of indole derivative.

Scheme 75.
Ruthenium-catalysed C-H bond functionalization with allylic alcohols.
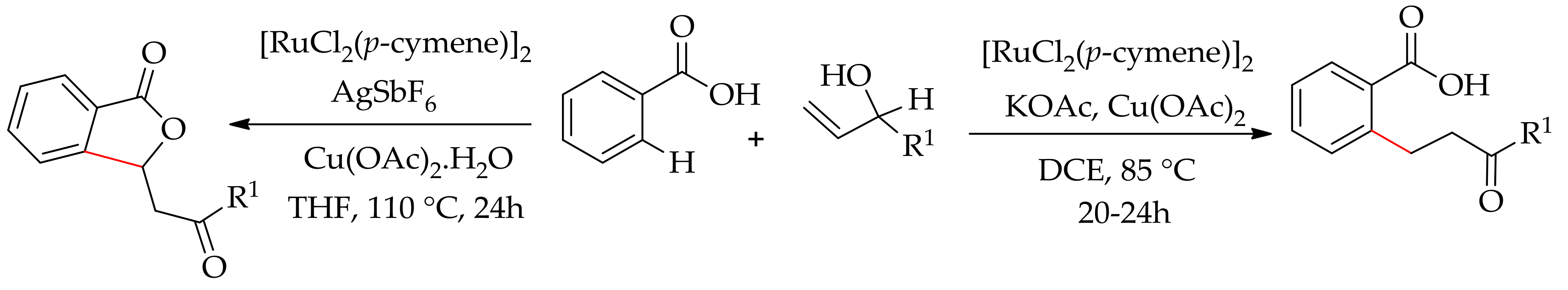
Scheme 76.
Ruthenium-catalysed benzylation of 1,2,3,4,-tetrahydroquinolines with aryl aldehydes.
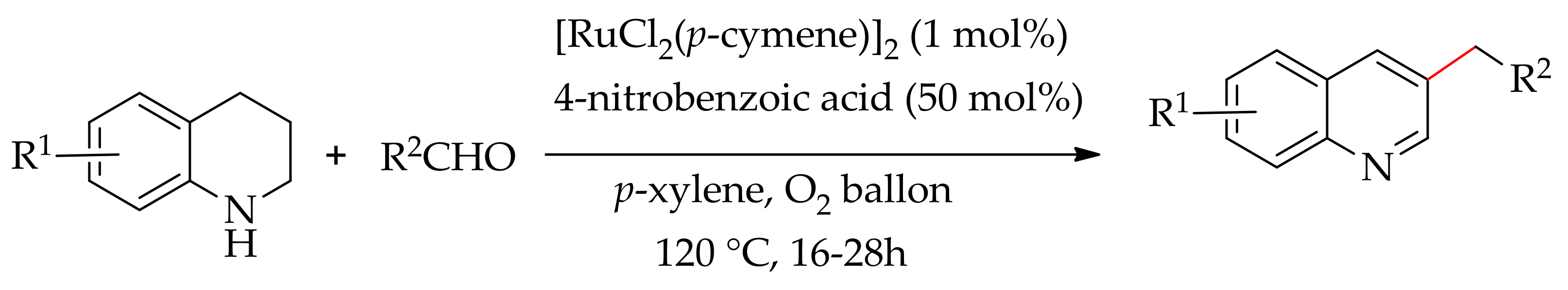
Scheme 77.
Rutheium-catalysed allylation of 2-phenylpyridine with allyl halide.

Scheme 78.
Ruthenium-catalysed allylation of ketoximes with allyl acetate.
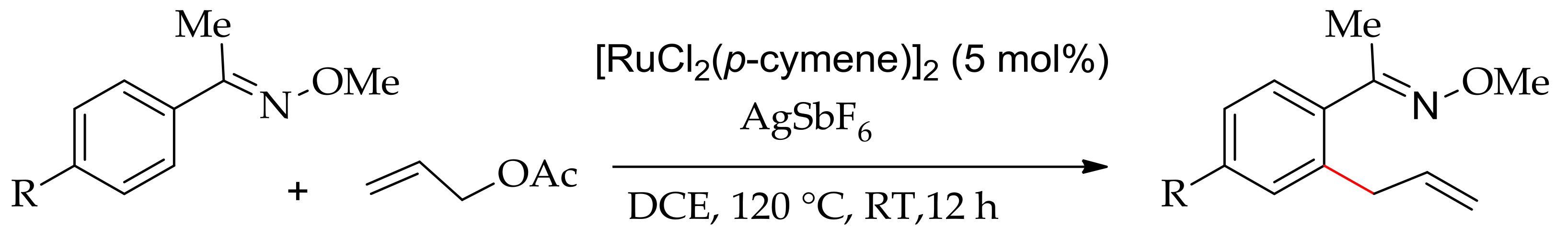
Scheme 79.
Ruthenium-catalysed stereoselective C-2 allylation of indole derivatives with allyl alcohols.
Scheme 79.
Ruthenium-catalysed stereoselective C-2 allylation of indole derivatives with allyl alcohols.

Scheme 80.
Ruthenium-catalysed aerobic annulation of 2-substituted pyrroles with alkynes.
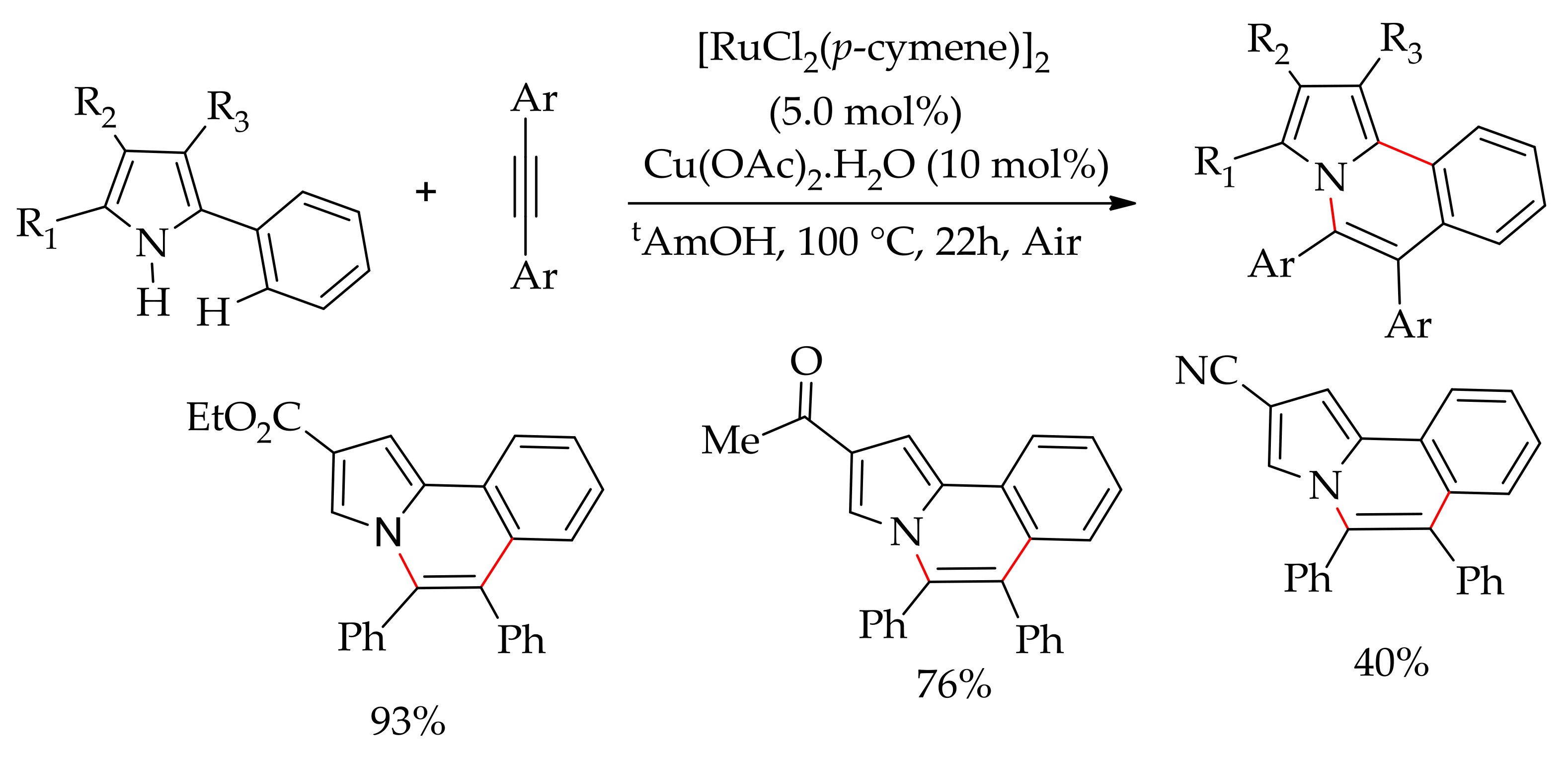
Scheme 81.
Ruthenium-catalysed regioselective annulation of aromatic acids with alkynes.
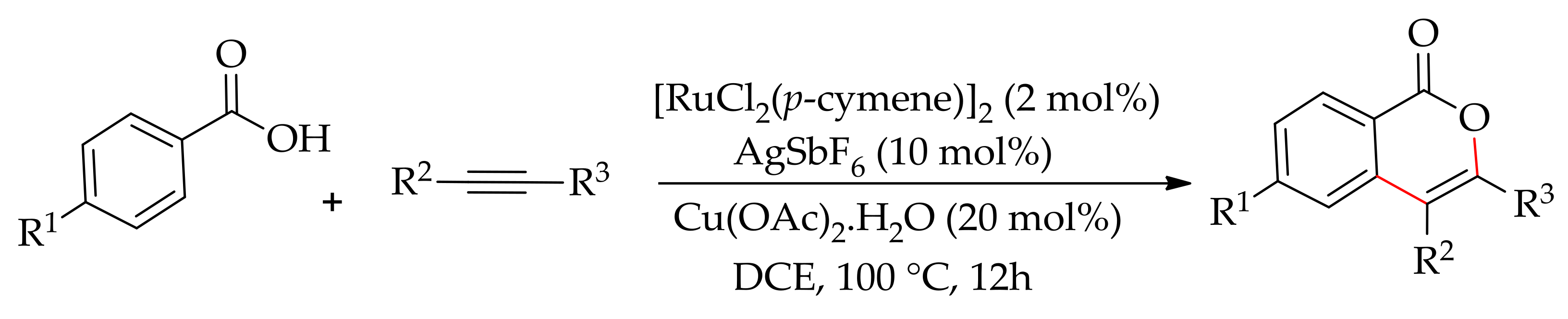
Scheme 82.
Ruthenium-catalysed annulation via C-H/O-H bond cleavage of aromatic acids with alkynes.
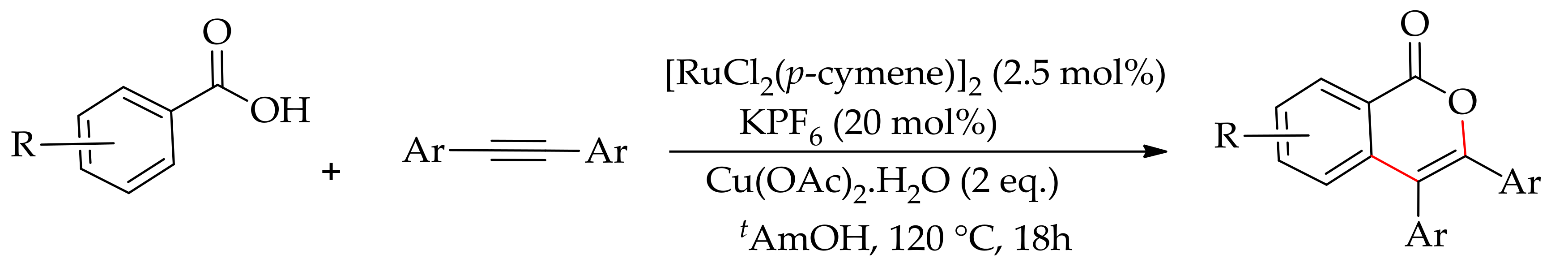
Scheme 83.
Ruthenium-catalysed annulation of aromatic acids and alkynes with O2 as sole oxidant.
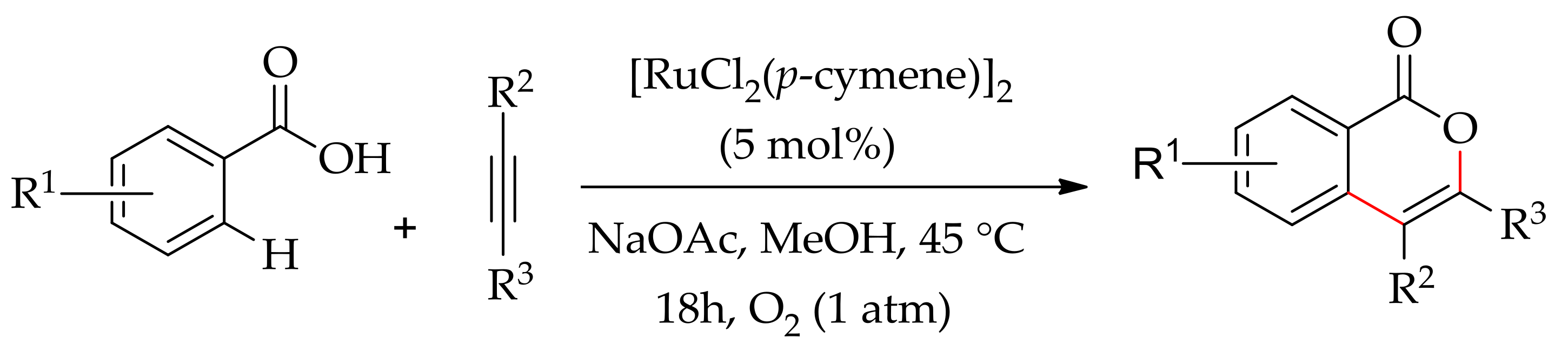
Scheme 84.
Plausible mechanism for annulation of aromatic acids and alkynes with O2 as oxidant [170].
Scheme 84.
Plausible mechanism for annulation of aromatic acids and alkynes with O2 as oxidant [170].

Scheme 85.
Ruthenium-catalysed regioselective annulation of N-heteroaromatic acids with alkynes.
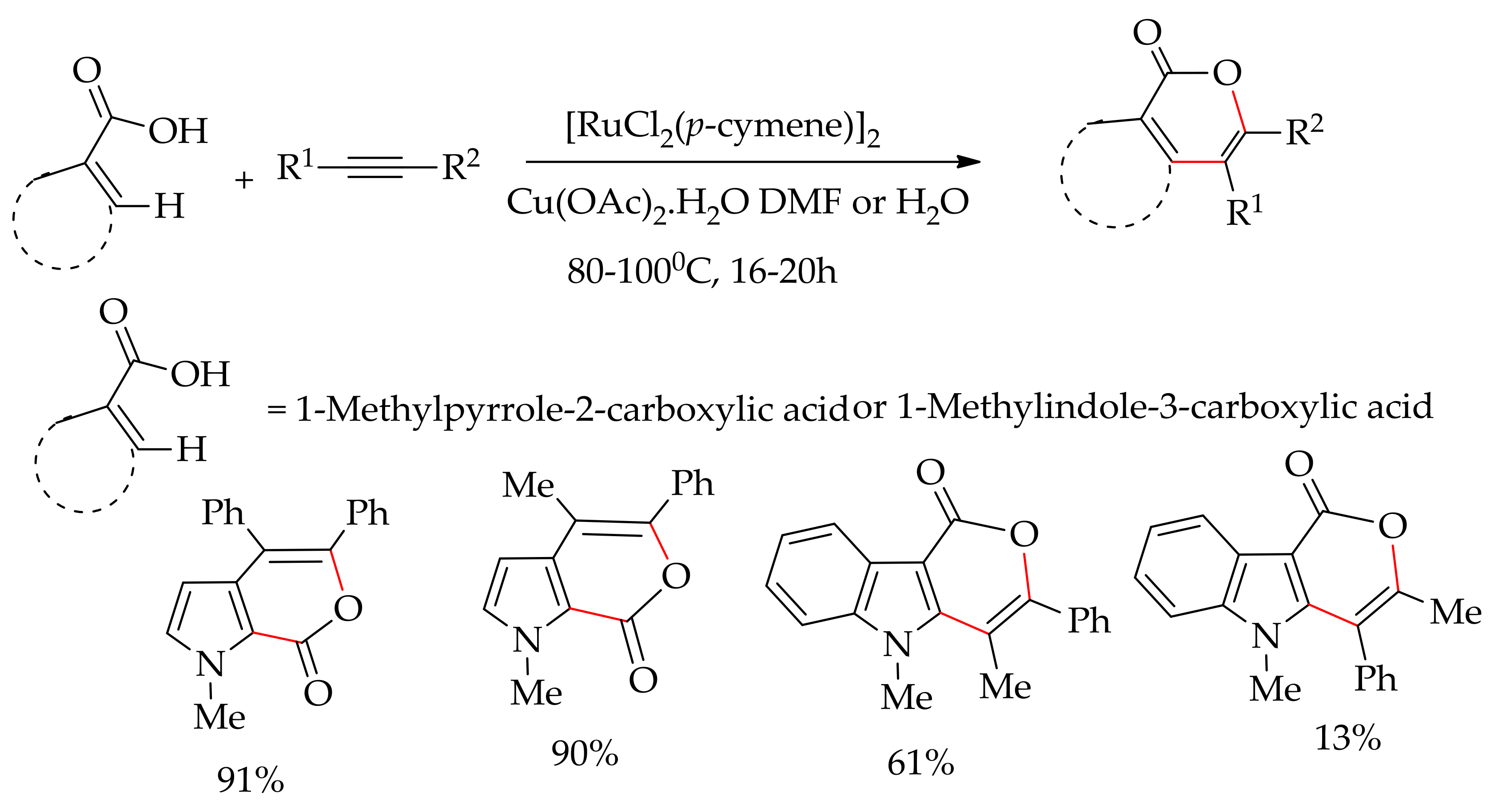
Scheme 86.
Annulation fo N-heteroaromatic acids and alkynes with N^O chelate ruthenium(II) catalyst.
Scheme 86.
Annulation fo N-heteroaromatic acids and alkynes with N^O chelate ruthenium(II) catalyst.

Scheme 87.
Ruthenium-catalysed aerobic annulation of aromatic nitriles with alkynes.
Scheme 88.
Ruthenium-catalysed hydroxy assisted annulation of naphthols or 4-hydroxycoumarins with alkynes.
Scheme 88.
Ruthenium-catalysed hydroxy assisted annulation of naphthols or 4-hydroxycoumarins with alkynes.
Scheme 89.
Ruthenium(II)-catalysed cyclization of ketimines with alkynes promoted by NHC ligands.
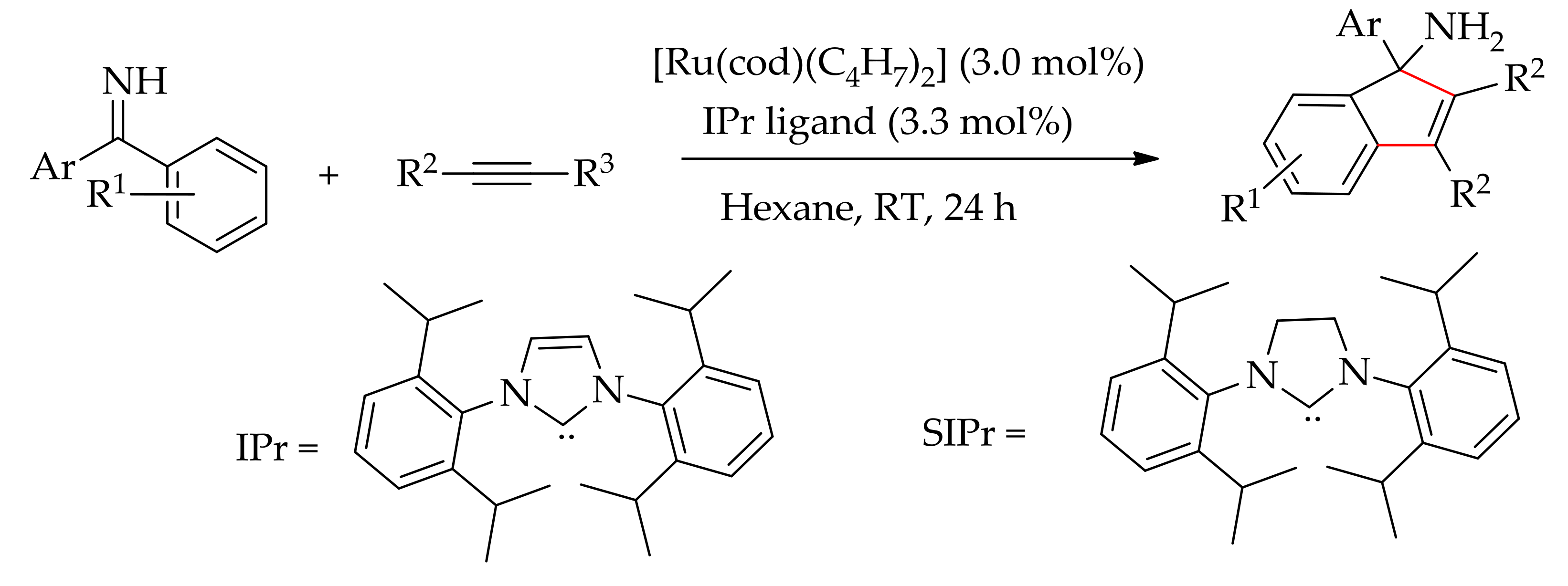
Scheme 90.
Ruthenium(II)-catalysed oxidative annulation of arylbenzimidazoles with alkynes.
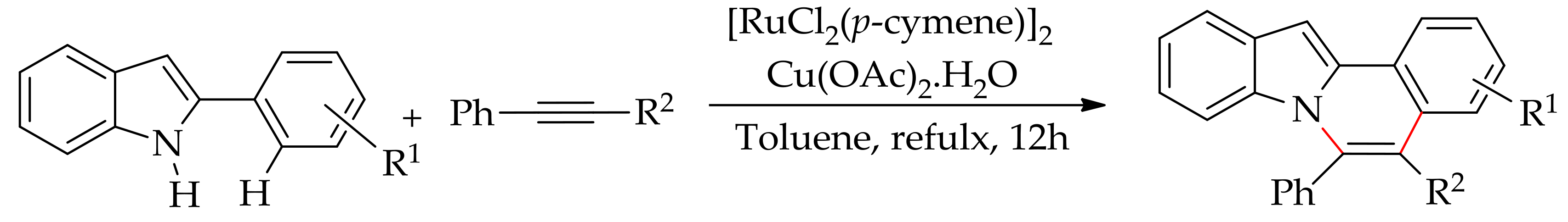
Scheme 91.
Ruthenium catalysed oxidative annulation of primary amines and alkynes.

Scheme 92.
Ruthenium(II)-catalysed annulation of 6-aminopurines with alkynes.

Scheme 93.
Ruthenium-catalysed annulation of alkyne and amine containing a removable benzothiazole group.
Scheme 93.
Ruthenium-catalysed annulation of alkyne and amine containing a removable benzothiazole group.

Scheme 94.
Ruthenium-catalysed decarbonylative annulation of 3-hydroxy-2-phenylchromones with alkynes.
Scheme 94.
Ruthenium-catalysed decarbonylative annulation of 3-hydroxy-2-phenylchromones with alkynes.
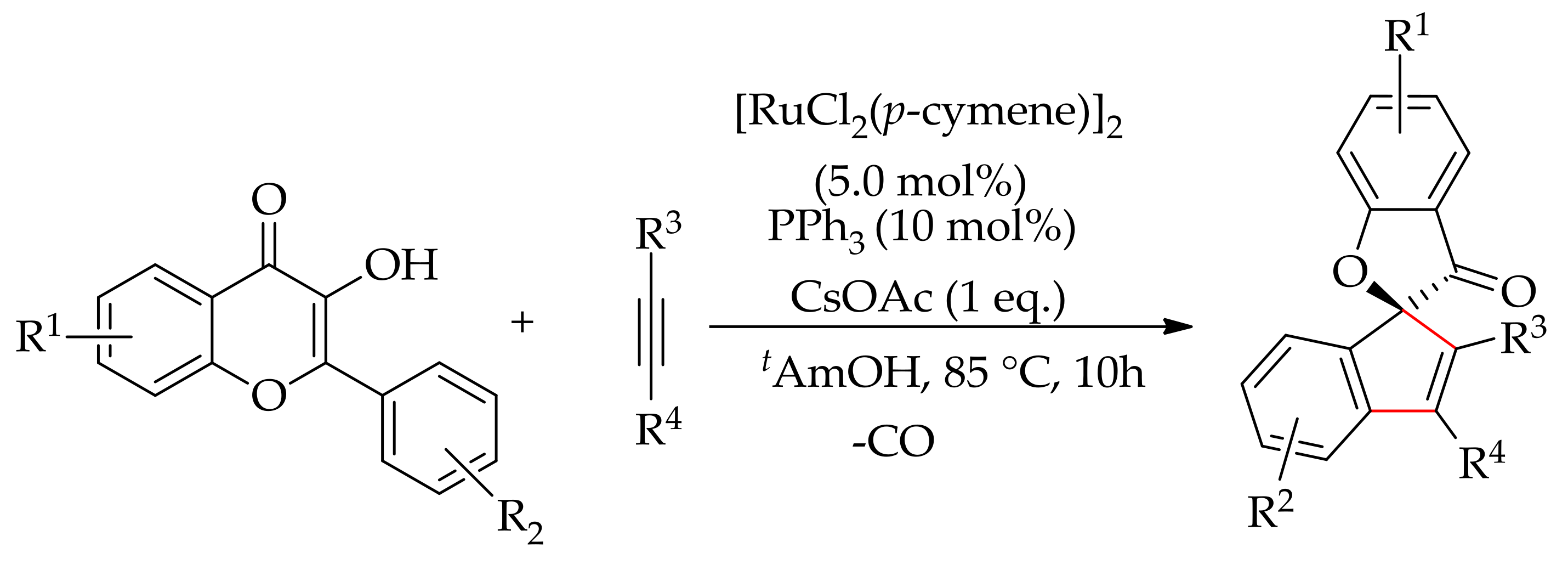
Scheme 95.
Ruthenium(II) catalysed regiosepecific C-H/O-H annulations of directing arenes and alkynes via assistance of weak coordination.
Scheme 95.
Ruthenium(II) catalysed regiosepecific C-H/O-H annulations of directing arenes and alkynes via assistance of weak coordination.

Scheme 96.
Ruthenium-catalysed electrochemical dehydrogenative annulation of aniline derivatives and alkynes.
Scheme 96.
Ruthenium-catalysed electrochemical dehydrogenative annulation of aniline derivatives and alkynes.
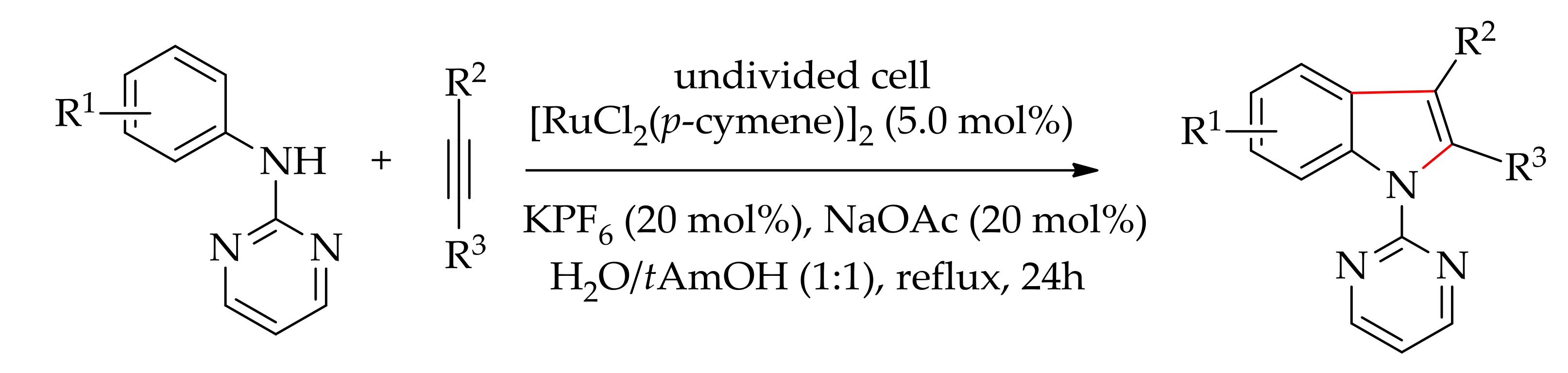
Scheme 97.
Ruthenium-catalysed annulation of indole with propargyl alcohol.
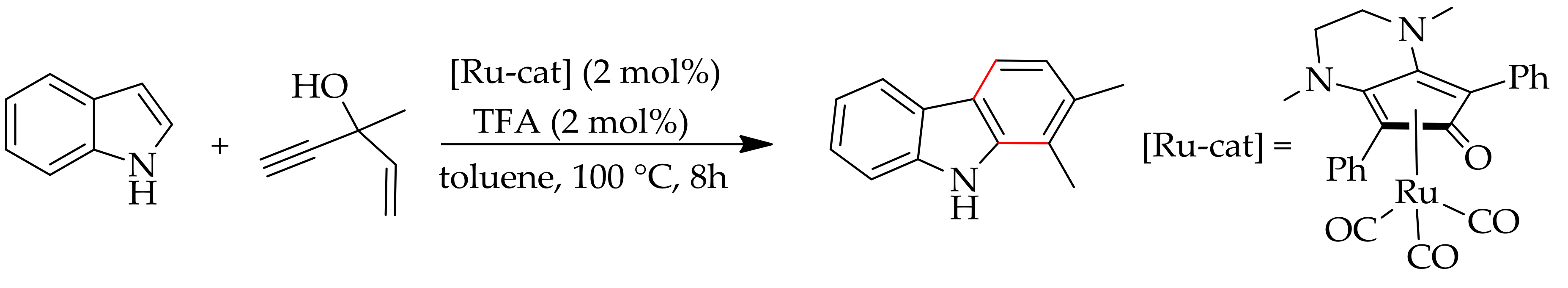

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Solvent and oxidant effects on mono-diarylated product formation in the arylation of 2-phenylpyridine with aryl silanes.
Table 1.
Solvent and oxidant effects on mono-diarylated product formation in the arylation of 2-phenylpyridine with aryl silanes.
| Additive | Oxidant | Solvent | Mono/Di | Yield |
|---|---|---|---|---|
| CuF2 | − | DEC | 6:94 | >95 |
| CuF2 | − | Toluene | 69:31 | >95 |
| CuF2 | − | THF | 75:25 | >95 |
| CuF2 | − | NMP | − | <2 |
| CuF2 | AgF | DCE | − | <2 |
| CuF2 | Ag2O | DCE | 95:5 | 40 |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Singh, K.S. Recent Advances in C–H Bond Functionalization with Ruthenium-Based Catalysts. Catalysts 2019, 9, 173. https://doi.org/10.3390/catal9020173
AMA Style
Singh KS. Recent Advances in C–H Bond Functionalization with Ruthenium-Based Catalysts. Catalysts. 2019; 9(2):173. https://doi.org/10.3390/catal9020173
Chicago/Turabian StyleSingh, Keisham S. 2019. "Recent Advances in C–H Bond Functionalization with Ruthenium-Based Catalysts" Catalysts 9, no. 2: 173. https://doi.org/10.3390/catal9020173
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.