Abstract
The software tool CaRMeN (Catalytic Reaction Mechanism Network) was exemplarily used to analyze several surface reaction mechanisms for the combustion of H2, CO, and CH4 over Rh. This tool provides a way to archive and combine experimental and modeling information as well as computer simulations from a wide variety of sources. The tool facilitates rapid analysis of experiments, chemical models, and computer codes for reactor simulations, helping to support the development of chemical kinetic models and the analysis of experimental data. In a comparative study, experimental data from different reactor configurations (channel, annular, and stagnation flow reactors) were modeled and numerically simulated using four different catalytic reaction mechanisms from the literature. It is shown that the software greatly enhanced productivity.
1. Introduction
Computer-aided design using chemical kinetics software has become essential in reaction engineering, as it provides valuable guidance in scale-up. In particular, the prediction of the reactor performance based on kinetics is a crucial issue. Using chemical kinetics, reactive flows can be simulated numerically on a technical scale, helping to reduce elaborate and expensive experiments. Furthermore, kinetics can also lead to a profound understanding of the underlying elementary processes.
In chemical engineering, a macroscopic kinetic approach was used for many years. In the macroscopic regime, the rate of catalytic reaction is modeled by fitting empirical equations such as power laws to experimental data to describe its dependence on concentration and pressure and to determine rate constants that depend exponentially on temperature. The downside of this approach is its very limited extrapolation to conditions that were not covered by the fit to experimental data. A more robust approach is to use microkinetics, where the processes are described by a sequence of elementary reaction steps of the catalytic cycle as they occur on a microscopic scale. These steps include adsorption, surface diffusion, reactions between adsorbed species, and desorption. A major advantage of the microkinetic approach versus the macrokinetic approach are its improved prediction capabilities beyond the experimental data that were used in its development.
However, the development of microkinetic models is a difficult and very time-consuming task. A hierarchical approach is commonly followed to develop reaction kinetics. Starting from a single fuel, the complexity of the reaction scheme is augmented by increasing the number of reactive components. For example, H2 oxidation, CO oxidation, preferential oxidation of CO in H2 and O2 mixtures, water–gas shift (WGS), and reverse water–gas shift (RWGS) reactions as well as total and partial oxidation reactions of CH4 are added consecutively. To optimize the reaction kinetics, reactions are examined for varying fuel/oxygen ratios over a wide range of temperatures and compared to experimental data. Catalytic ignition studies are also conducted to understand the adsorption and desorption kinetics of the reactive species.
Hence, a major part of the development process towards a validated, reliable mechanism is comprised of iteratively comparing simulation results with experimental data. In each refinement step, changes are made to the reaction mechanism until the simulation results match measurements from experiments. Due to its repetitive nature, this approach is time-consuming and error prone. CaRMeN (Catalytic Reaction Mechanism Network) is a recently developed software tool [1] that addresses these problems by automating experiment vs. model comparisons in a graphical user interface. This is achieved by providing a platform to archive and evaluate structured experimental data, kinetic models, and simulation software. These data can be conveniently compared with the results of any simulation code under the matching experimental conditions.
There are several related projects described in the literature, such as PrIMe (Process Informatics Model), RESPECTH (short for reaction kinetics, spectroscopy, thermochemistry), or CloudFlame [2,3,4]. However, these projects have a very specific focus on combustion research that does not include the use of catalysts.
The high diversity of data in the field of catalysis makes such automation software far from trivial. Many reactor types and measurement techniques have been developed in the last years that are each capable of elucidating certain facets of the catalytic system. Ideally, these techniques are used in tandem when developing a detailed kinetic scheme. Accordingly, the simulation software used to model the reactions in these reactors is equally diverse. Furthermore, the verification of the surface kinetic mechanisms can itself be a challenging task, given the complications of performing experiments under a purely kinetically controlled regime. This requirement can be difficult to fulfill, especially for very fast processes, such as oxidation reactions. Therefore, special attention for evaluation datasets has to be paid to choosing accurate, reproducible experiments, which were carried out under appropriate operating conditions, so as to minimize the influence of transport phenomena. In addition, there are various commonly used metrics to assess the performance of a catalyst that can be extracted from a single experiment. These metrics can therefore be seen as different “views” of the data. An example is a “light-off” experiment, in which the temperature is varied and species concentrations are determined at the end of the reactor. This data can either be viewed directly, or the data can be transformed to conversions as a function of temperature, from which, in turn, to values can be derived.
In this contribution, we illustrate how the existing hierarchical approach to develop reaction mechanisms is significantly improved using the recently released software tool CaRMeN [1]. This refined methodology improves the quality and applicability of the model considerably because the model can be validated against a larger experimental database than before. Furthermore, reaction mechanisms can be developed and evaluated more quickly.
2. Combustion over Rh-Based Catalysts
Systems of catalytic oxidation CO in hydrogen-rich mixtures and fuel-lean methane oxidation have key applications for automotive and factory exhaust gas after treatment. Even though the catalytic combustion systems have been studied intensively, both experimentally and theoretically, accurate and reliable kinetic models for these systems are not readily available. In this article, this system is used as an example to show case various features of the CaRMeN software. The software is, however, neither limited to rhodium based catalysts nor to catalytic combustion. For example, catalysts based on Pt, Pd or Ni, which also play a very important role in industry [5,6,7,8,9,10], have also been studied with the software. Furthermore, CaRMeN can also be used to develop gas-phase mechanisms.
2.1. Overview of Mechanisms in the Literature
A selection of microkinetic models for CO–H2–O2 mixtures and methane combustion over Rh-based catalysts, developed by several groups, are summarized in Table 1. The following four detailed surface reaction mechanisms were used in this work as examples to demonstrate the capabilities of the software tool and are therefore described in more detail below:

Table 1.
Selection of surface reaction mechanisms for CO and H2 oxidation, preferential oxidation of CO, and CH4 combustion over Rh-based catalysts (R = number of reactions).
• Maier–Deutschmann (2001) [11]
The detailed surface reaction mechanism for methane oxidation, produced by Deutschmann and co-workers, assumes dissociative adsorption of oxygen and two different methane activation paths. The first pyrolytic path involves the stepwise abstraction of hydrogen from CHx* (x = 0–4) species on the free Rh sites down to surface carbon C*. The second path considers oxygen-assisted methane activation through pre-adsorbed O*. The reaction mechanism consists of six gas-phase species including the reactants and products (H2, CO, H2O, and CO2), 11 surface intermediates and a total of 38 elementary-step reactions. The mechanism was then further improved by Schwiedernoch et al. [12] including coverage-dependent heats of chemisorption for CO and oxygen and validated against own light-off experiments and transient measurements by Williams et al. [13].
• Karakaya–Deutschmann (2016) [14]
The model was developed on the basis of former kinetic scheme of Deutschmann et al. [11] using the same dual methane activation route, including additional CO–H2 coupling reactions via carboxyl COOH* related pathways, which are important in the water gas shift (WGS) reaction. The 44-step mechanism contains elementary reaction for H2 oxidation, CO oxidation, preferential oxidation, and WGS. Both methane models [11,14] were developed and extensively validated for fuel-rich partial oxidation and reforming of CH4 with water and CO2. One of the objectives of this work was to test the mechanisms against the experimental datasets at fuel-lean combustion conditions.
• Deshmukh–Vlachos (2007) [15]
The model presents a reduced mechanism for fuel-lean methane/air catalytic combustion on a Rh catalyst. It was developed from a detailed microkinetic model of Mhadeshwar and Vlachos [16]. The original 104-elementary-step mechanism [16] was reduced (to 15 reversible reactions) using reaction path, sensitivity, and partial equilibrium analysis to deduce the most abundant reaction intermediate and the rate-determining step. The mechanism was evaluated against the methane catalytic combustion experiments in microreactor on Rh/Al2O3 catalyst [15].
• Rankovic–Da Costa (2011) [17]
The mechanism contains elementary reaction for H2 oxidation, CO oxidation, CO–H2 coupling, and NOx chemistry on Rh. It was derived from the previous modeling works of Deutschmann and co-workers. Most of the kinetic data were taken from Schwiedernoch et al. [12] and Boll [18] with the exception of two parameters: the pre-exponential factor of oxygen adsorption was changed from 1 × 10−2 to 6 × 10−3 and the coverage-dependence of the CO desorption step was increased from 15 to 55 so that the model reproduces the CO conversion curves obtained in experimental measurements in packed bed flow reactor with Rh/Al2O3 [19] and Rh/SiO2 [20].
2.2. Overview of Experimental Setups
Data from different experimental setups were used in the present work: A stagnation flow reactor, an annular duct reactor, and an optically accessible single channel-flow reactor.
Stagnation flow reactors offer a simple configuration for the investigation of reactions on catalytic surfaces. The setup allows microprobe sampling of the gas-phase boundary layer adjacent to the catalyst surface. A reactive gas mixture (e.g., O2 and H2) enters the reactor and impinges upon the heated catalyst surface (e.g., Rh/Al2O3 coated disk) [27]. The catalyst surface is approximately 5 cm in diameter and the separation distance between the porous-frit gas inlet and the catalyst surface is approximately cm. This configuration also enables efficient modeling of the surface chemistry, coupled with convective and diffusive transport within the boundary layer. In the past, manifold measurements in stagnation–flow reactor over rhodium were used in model verification for H2 oxidation [21,27], CO oxidation [28,29], WGS [30], methane partial oxidation [14] and reforming [14,31].
Appel et al. [23] introduced the methodology of in situ spatially-resolved Raman measurements of species concentrations in gas phase over the catalyst boundary layer as a direct way to assess the catalytic reactivity at realistic operating conditions. Here, experiments were performed in a rectangular, optically accessible reactor, which comprises two horizontal, non-porous ceramic plates coated with Rh/Al2O3 and vertical quartz windows. This technique represents a powerful method to gain detailed insight into the reactor during operation in a non-intrusive manner. Sui et al. [24,25] studied the hetero-/homogeneous combustion of CH4/O2/N2 mixtures over rhodium in this set up.The experiments included in situ spatially-resolved Raman measurements of gas phase species concentrations for evaluating the catalytic processes, and planar laser induced fluorescence (LIF) of the OH radical for assessing homogeneous combustion.
2.3. Overview of Simulation Tools
CaRMeN can be configured to run any simulation software. The simulations are run on a central server and the results are cached to avoid multiple runs of the same input parameters. In this work, two codes based on DETCHEM were used for the numerical simulations. These are briefly described in the following.
DETCHEM Stagnation calculates a catalytically active stagnation point flow reactor. Within a boundary layer above the surface, the general fluid flow problem can be reduced to a one-dimensional model. Thus, temperature, axial velocity and gas composition only depend on the axial position, i.e., the distance from the surface. Concentrations at the gas-surface interface and coverages are independent of the position on the plate [28,32,33].
DETCHEM Channel simulates the steady state chemically reacting gas flow through a cylindrical channel. If radial velocity gradients inside a tube cannot be neglected, it is necessary to resolve another dimension of the flow field. In typical channel flow configurations, axial transport is dominated by convection instead of diffusion. Then, the general Navier–Stokes equations can be parabolized, resulting in a partial differential equation system for conservation of mass, species, momentum and energy, which can be solved efficiently by a method-of-lines integration [34]. If cylindrical symmetry is exploited, channel flows and annular duct flows can be described. Furthermore, the case of parallel plates in a micro reactor channel can be covered by the same model, when the radius of the inner duct is chosen much larger than the distance of the two walls.
3. Illustrative Examples
In this section, the capabilities of CaRMeN are illustrated using combustion data for the CH4, CO, and H2 over rhodium catalysts. All results presented in this section were generated using the CaRMeN software. The numerical simulations were carried out using four different mechanisms available in the literature and are described in the previous section. The experiments were conducted in an annular reactor (Tavazzi et al. [35]), in a rectangular shaped and optically accessible channel reactor (Sui et al. [25]), and two different stagnation flow reactors (Karakaya et al. [14] and Pery et al. [29]).
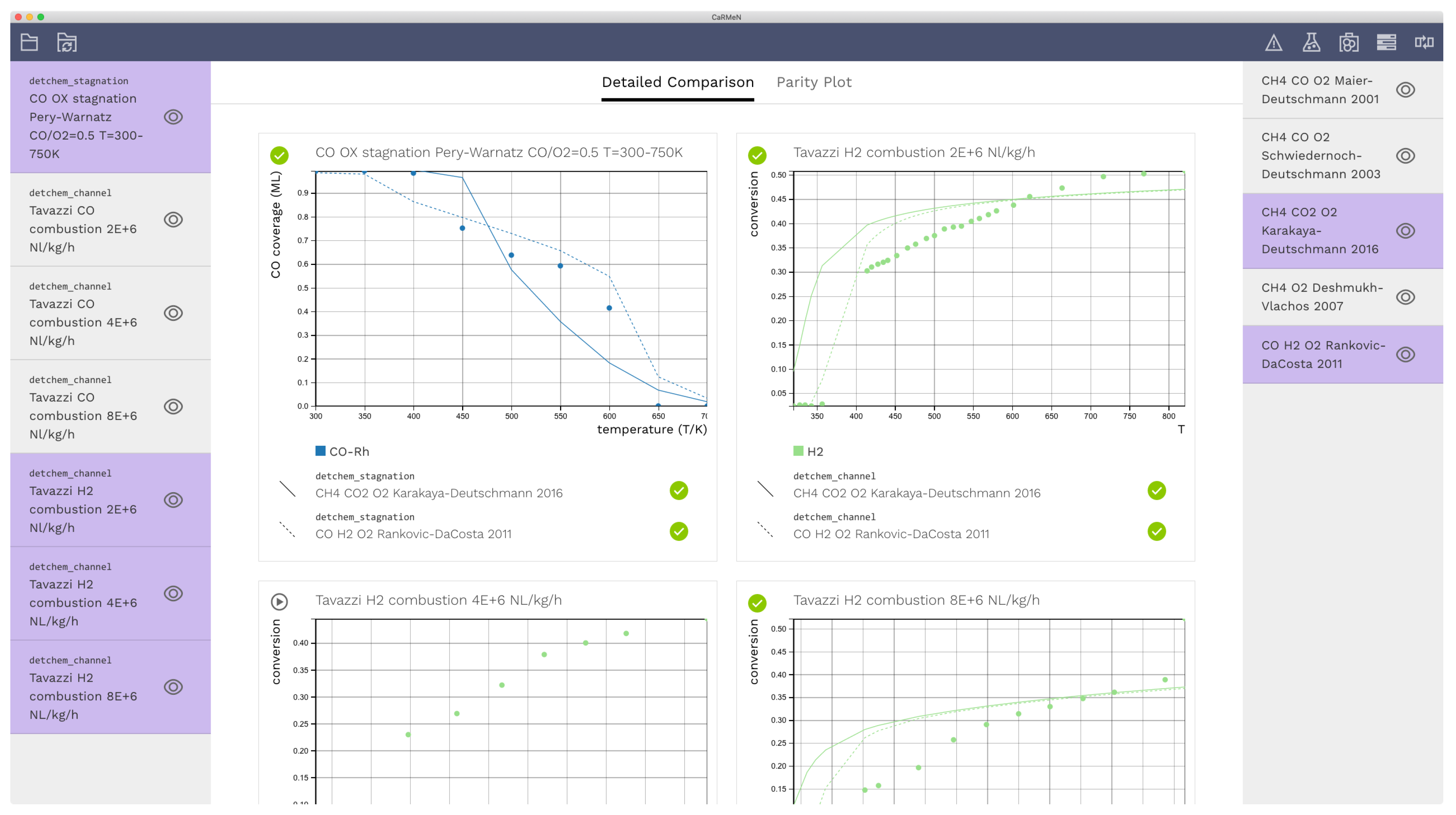
A screen shot of the graphical user interface is shown in Figure 1. Experimental data are listed on the left sidebar, while the right side bar contains mechanisms. The selections on the left control which experiments to display. These items can be combined with models on the right by making selections. Four exemplary cases was selected, and each case was combined with the Karakaya–Deutschmann [14] and the Rankovic–Da Costa [17] mechanisms. In this example, only mechanisms are shown on the right-hand side. However, any other model, e.g., various diffusion models can be added to the sidebar to evaluate diffusion models.
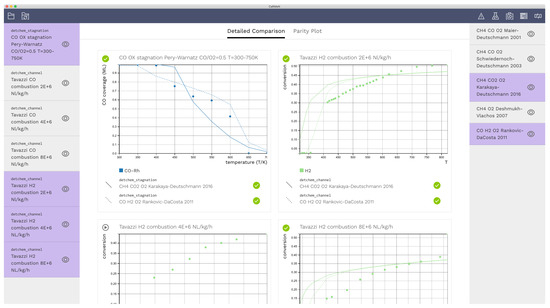
Figure 1.
Screen shot of the detailed comparison view. Experimental data are shown on the left sidebar, and reaction mechanisms on the right. The selections on the left control which data to display. These items can then be combined with the mechanisms on the right. The dots in the graphs represent experimental data points while lines correspond to simulation data.
3.1. Detailed Comparison—CO Combustion
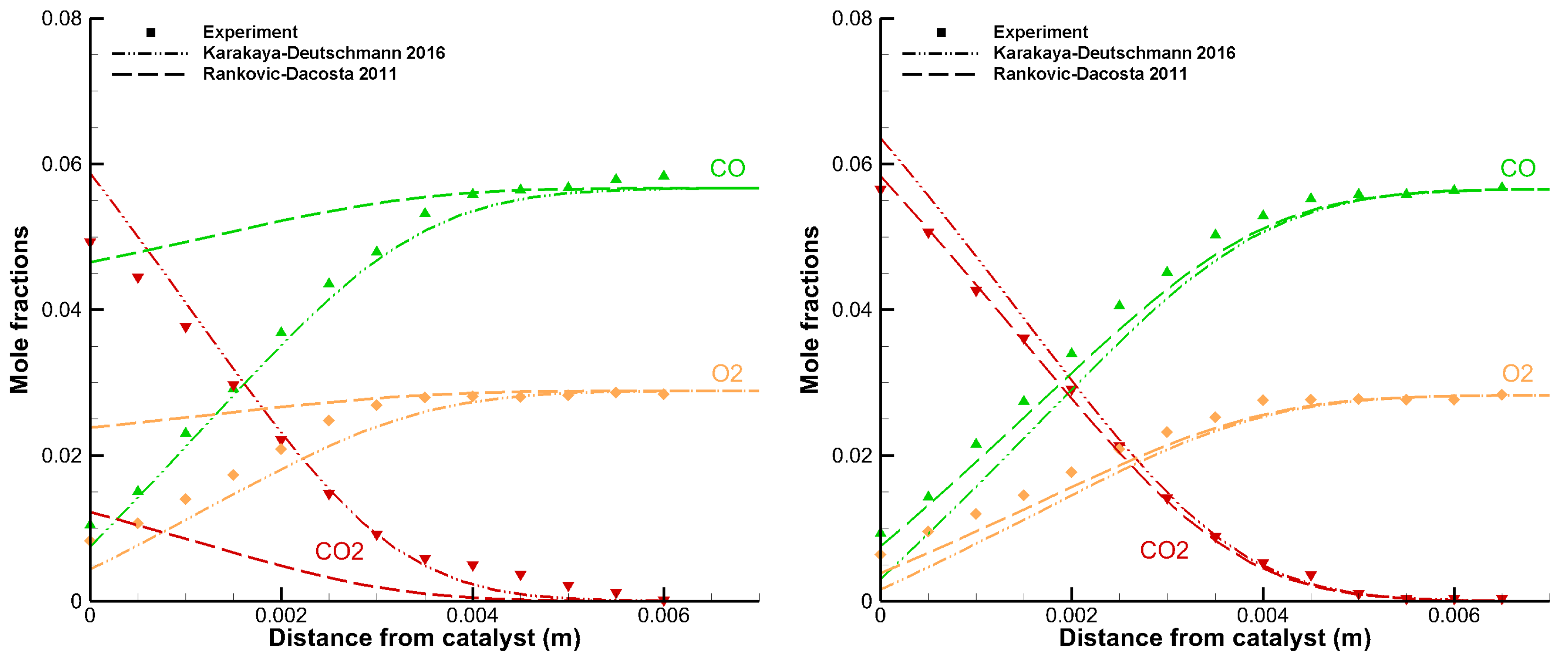
A detailed comparison between the simulation output and the corresponding experimental data is very indicative to evaluate the quality of the model. The following example in Figure 2 shows two spatially resolved concentration profiles from a stagnation flow reactor. Both experiments were carried out under the same conditions, except for the surface temperatures.
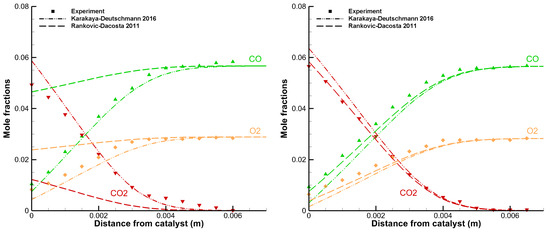
Figure 2.
Data from a stagnation flow experiment showing mole fractions of CO (green triangles pointing up), O2 (yellow diamonds) and CO2 (red triangles pointing down) as functions of distance from the catalytic surface ((left) ; and (right) ).
The inlet gas feed had a temperature of 313 K and contained a CO/O2 ratio of 2 diluted in argon, corresponding to a stoichiometric mixture for total oxidation of CO. The surface temperatures were 673 K (Figure 2, left) and 873 K (Figure 2, right). The lines in the figures represent model predictions by the Karakaya–Deutschmann (dash-dotted) and the Rankovic–DaCosta (dashed) mechanisms that were obtained using DETCHEM Stagnation. Good agreement between model and experiment is obtained with the Karakaya–Deutschmann mechanism, while the other fails to predict the experimental data in the case of 673 K. This is, however, not surprising, as the Rankovic–DaCosta mechanism was developed for use in high temperature combustion applications. This mechanism matches the experimental points with a surface temperature of 873 K and otherwise same conditions perfectly as shown in the right figure.
From a practical perspective, these comparisons are fairly straightforward, because the experimental data can be mapped directly to the simulation data without post processing. Hence, only a single simulation must be carried out for each mechanism and each experiment. Figure 2 therefore shows the results of four simulations. The situation is slightly more complex in the next example, where each data point is the result of one simulation. Here, CaRMeN provides a significant benefit to the work flow.
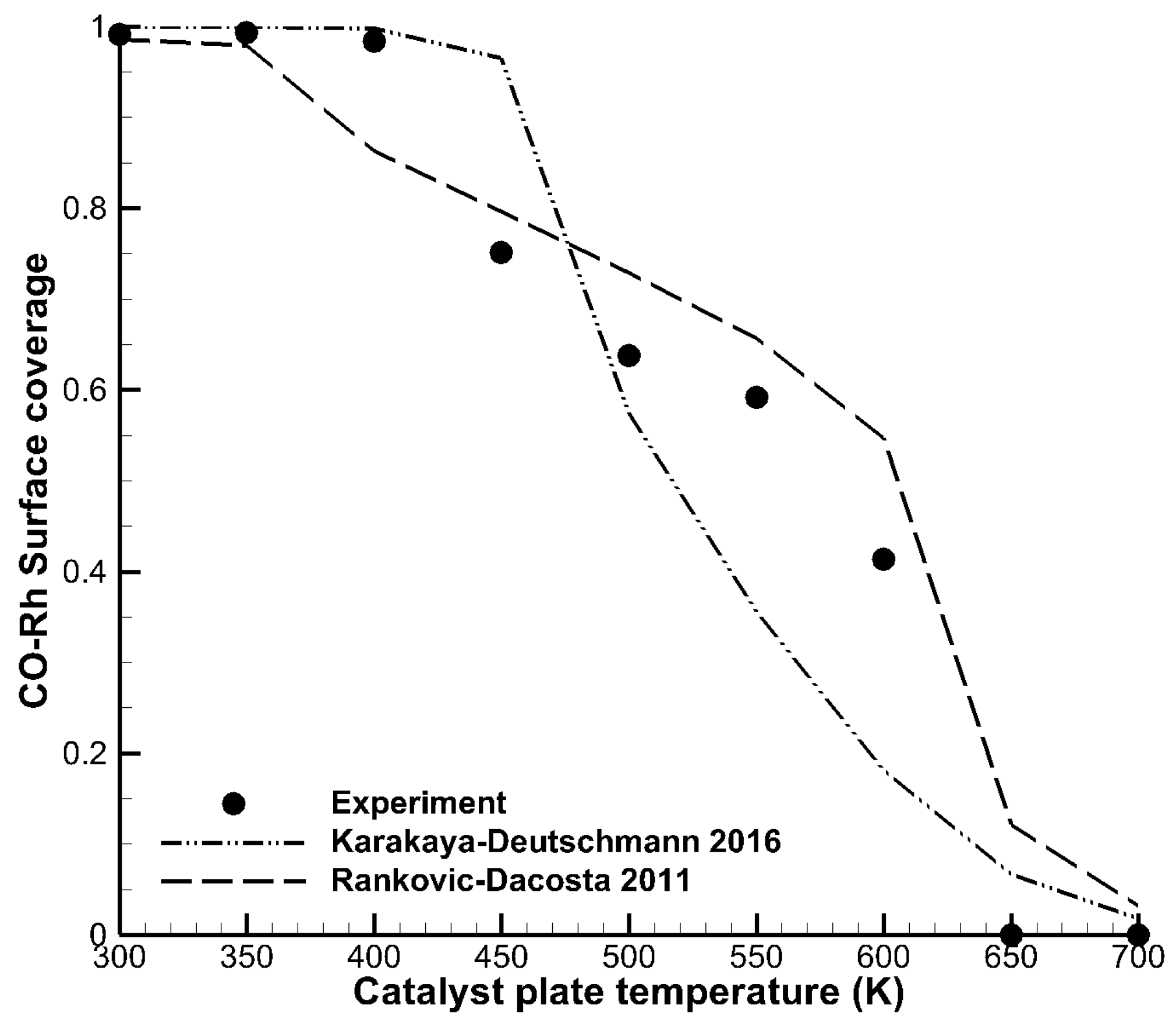
Figure 3 shows nine measured data points of surface coverages normalized to unity as a function of surface temperature. Surface coverages of adsorbates can reveal important information for the development of elementary step kinetics on catalytic surfaces. Pery et al. [29] carried out CO surface coverage experiments in a stagnation flow reactor. In these experiments, the surface temperature of the catalytic plate was varied in the range of 300 K to 700 K. At intervals of 50 K, the surface coverage of CO on the rhodium surface was characterized by sum-frequency generation in-situ spectroscopy. The lines in the figure denote the same mechanisms as in Figure 2, and again using DETCHEM Stagnation. Both mechanisms showed reasonable agreement with the experiment, although the overall shape, i.e., the step-like appearance, was reproduced better by the Rankovic–DaCosta mechanism.
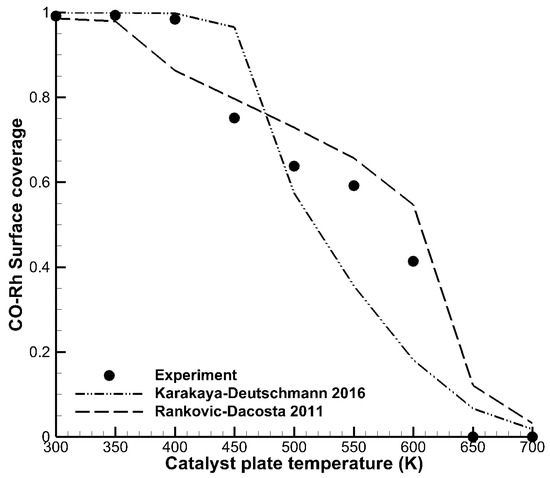
Figure 3.
Surface coverages of CO on rhodium as a function of catalyst plate temperature. The experiment (dots) was carried out by Pery et al. [29].
The catalyst plate temperature enters the simulation as an input parameter, hence a separate simulation must be carried out for each of the nine data points. A post processing step then extracts the CO-coverage (y value) from each simulation and maps it to the appropriate surface temperature (x value). Because two mechanisms are shown in the figure, a total of 18 simulations were required for the corresponding model data. CaRMeN can execute each simulation in parallel because each simulation is independent of the others.
3.2. Light-Off Curves—H2 Combustion
Light-off curves are a very common experiment type used to assess the performance of a catalyst. In these experiments, the temperature is varied and species concentrations are determined at the end of the reactor (“end-of-pipe” measurement), effectively resulting in species concentrations as functions of temperature. It is also common to plot the conversions instead of the concentrations directly. Similar to the last example in Figure 3, the data themselves (the temperature) enter the simulation as an input parameter. Consequently, one simulation was again required for each measured point.
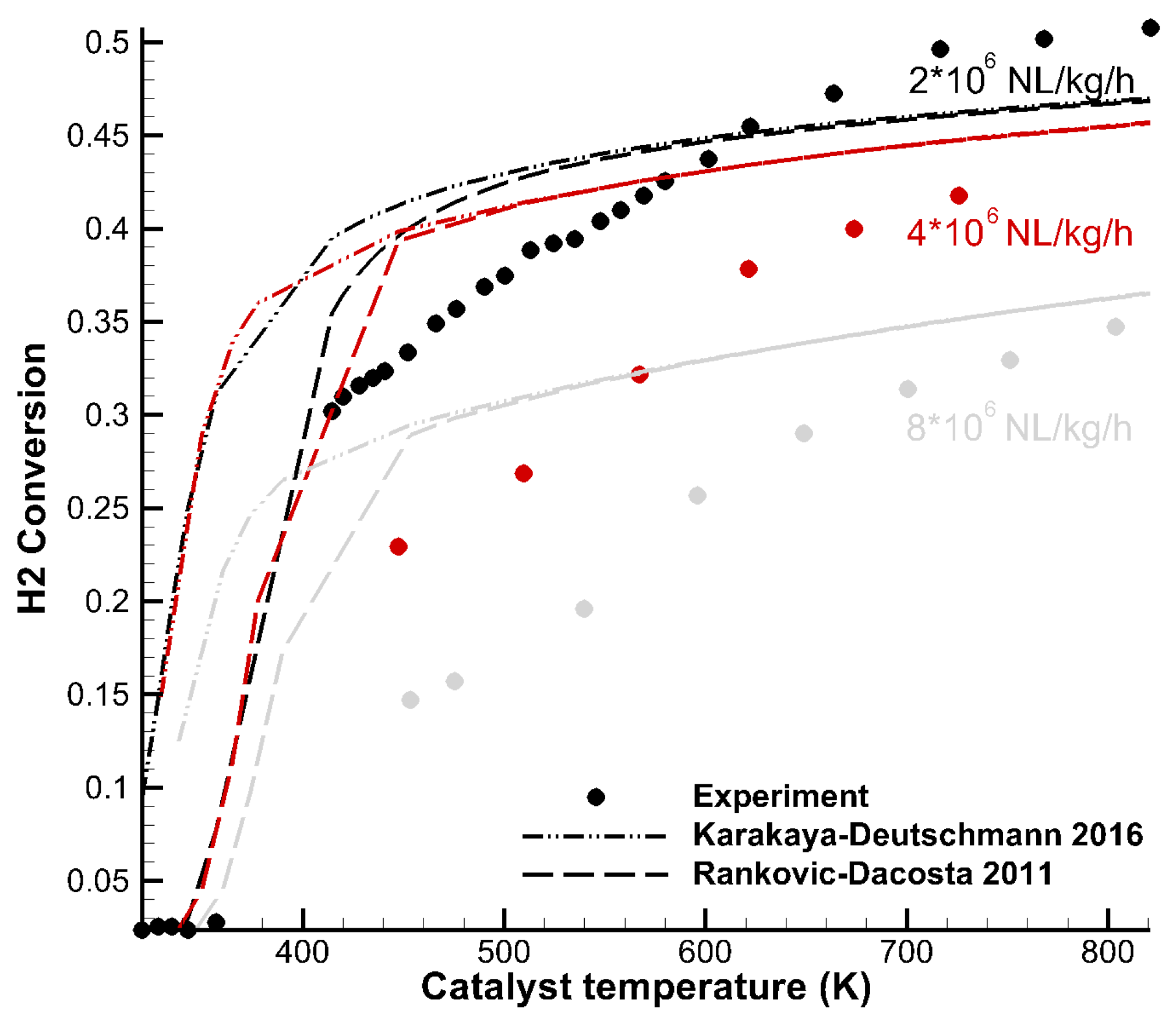
Tavazzi et al. [35] carried out H2 combustion experiments under rich conditions in an annular duct reactor. The temperature of the enclosing oven was varied, and the conversion of H2 at the end of the reactor was determined. Figure 4 shows a total of about 45 experimental data points (dots) at three different space velocities compared to model predictions of the Karakaya–Deutschmann (dash-dotted) and Rankovic–DaCosta (dashed) mechanisms. Conversion tended towards a maximum of 50% due to lack of oxygen in the rich conditions. The Rankovic–DaCosta mechanism reproduced the light-off temperature reasonably well, whereas the Karakaya–Deutschmann mechanism overpredicted H2 conversion. At approximately 500 K, both models gave the same results for all flow configurations. Furthermore, both mechanisms were less accurate at higher space velocities. This behavior may be due to diffusion effects within the washcoat, which were not captured accurately by the used diffusion model of the simulation code (DETCHEM Channel). However, it may also permit the conclusion that it is important to take into account various flow velocities and accurate diffusion models during mechanism development.
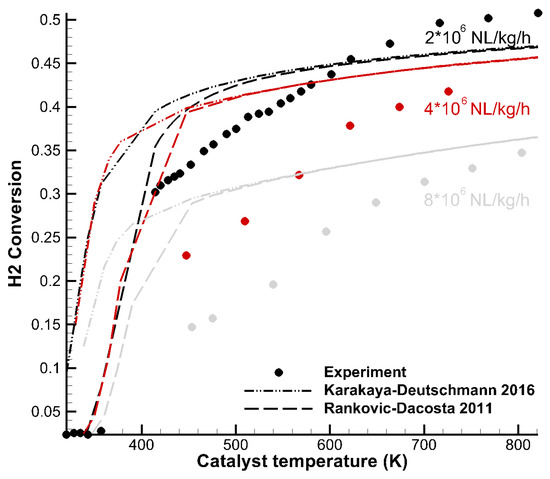
Figure 4.
Hydrogen conversion over temperature at different GHSV reported by Tavazzi et al. [35]. The lines correspond to numerical data using Karakaya–Deutschmann (2016) (dash-dotted) and Rankovic–DaCosta (2011) (dashed).
To reproduce the lines in Figure 4, approximately 90 simulations were required. In the case of the used simulation code DETCHEM Channel, an additional post processing step was required to calculate the H2 conversions from the format that was output by the code to be able to map the value to the experimental data. At this point, it is clear that tools to automate such comparisons are essential for a productive work flow. CaRMeN generates the input files, manages the intermediate results (90 individual simulations), carries out the post processing and handles the visualization. Adding another mechanism or testing the influence of a changed rate parameter then becomes trivial.
3.3. Parity Plot—CH4 Combustion
The previously shown detailed comparisons are useful to gain a comprehensive understanding of how the model reproduces experimental data. However, as more experimental data are used, it becomes difficult to evaluate all the individual plots. In these cases, parity plots can be more useful, as they allow quickly judging the quality of a model against large sets of experimental data.
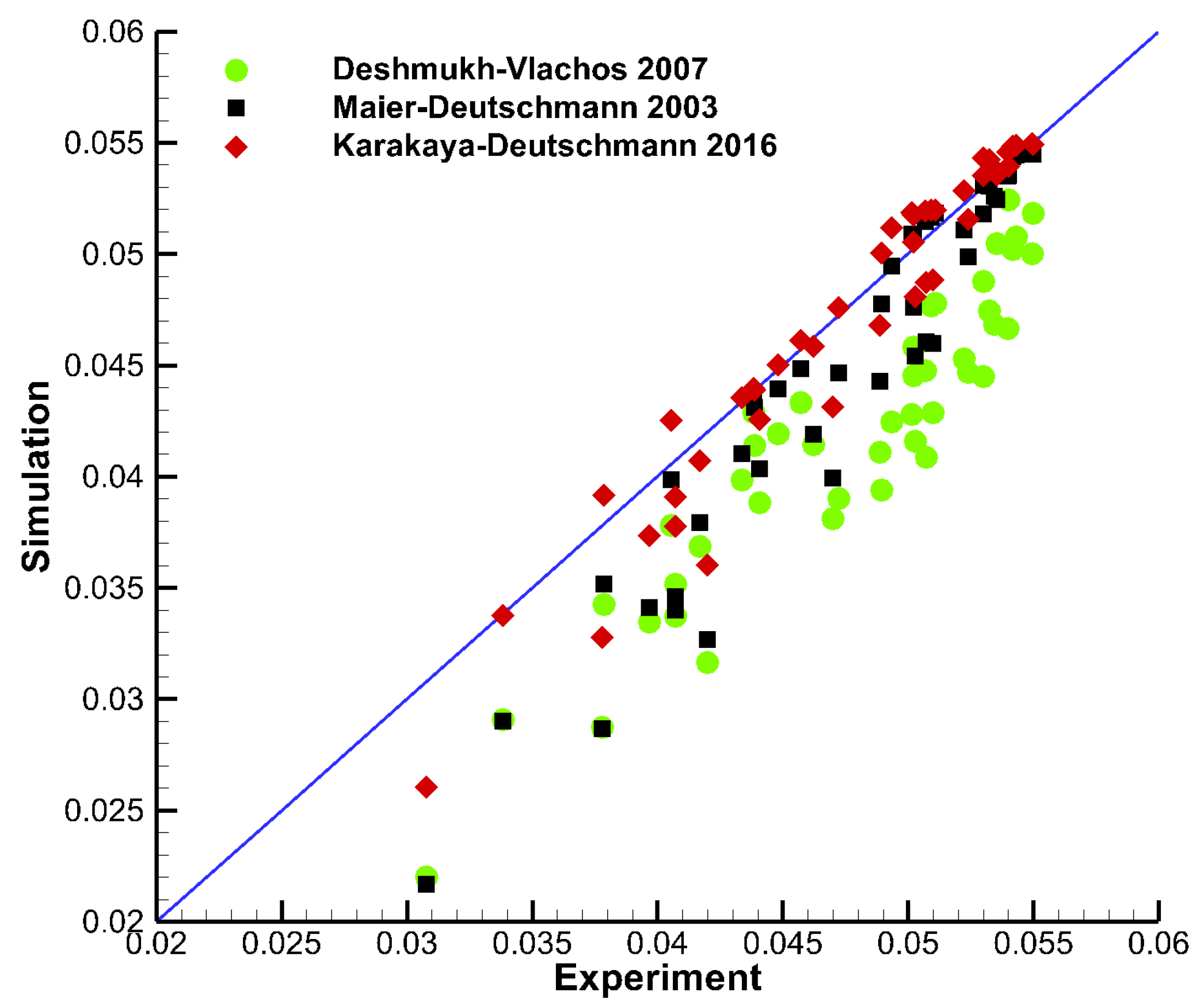
Figure 5 shows a parity plot with points from the experiments by Sui et al. [25] plotted against model data using three different mechanisms: Deshmukh–Vlachos (2007), Maier–Deutschmann (2003) and Karakaya–Deutschmann (2016). Here, x is the experimental value, and y is the corresponding value from the simulation. The experiments were conducted in a wide pressure range (2 bar to 12 bar), under total oxidation conditions with C/O2 ratios from 0.15 to 0.2, and dilution in N2. The 2D experimental data points extracted were transversely averaged and simulated using DETCHEM Channel considering the average temperature profile of the two coated plates. Overall, Karakaya–Deutschmann (2016) (red diamonds) showed best agreement with the experimental data, while the other two mechanisms generally overpredicted the CH4 conversion. However, the experimental data used for this comparison span a large pressure range up to 12 bar, which are conditions none of the mechanisms were tested under during their development. Furthermore, the simulation code used for the comparison (DETCHEM Channel) does not account for axial diffusion. Therefore, some of the discrepancies between model and experiment can likely be attributed to the simplified flow model, and not to the chemical model.
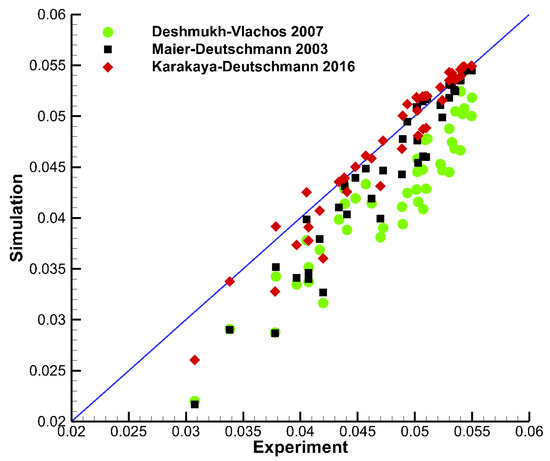
Figure 5.
Parity plot showing points from the experiments by Sui et al. [25] plotted against model data using mechanisms Deshmukh–Vlachos (2007) (green circles), Maier–Deutschmann (2003) (black squares), and Karakaya–Deutschmann (2016) (red diamonds). The experiments were conducted in a wide pressure range (2 bar to 12 bar), with C/O ratios from 0.15 to 0.2, and dilution in N2.
4. Conclusions
Elementary-step based reaction mechanisms are a powerful tool in predictive simulations of catalytic reactors. In the work presented, the methodology that was developed and implemented in the computer code CaRMeN [1] was exemplarily applied to analyze and evaluate catalytic reaction mechanisms developed for the combustion of CH4, H2 and CO over rhodium-coated surfaces. Appropriate sets of experimental data, reaction mechanisms and reactor simulators (flow solvers) for this specific chemical system were able to be compared in an automated fashion. CaRMeN generates the input files for the simulation code, manages the intermediate results (90 individual simulations), carries out the post processing and handles the visualization.
Without surprise, the mechanisms worked well to reproduce the experimental data, which were originally used in their development process. Mostly they were also able to predict experiments by other groups and reactor configurations conducted at similar operating conditions; sometimes, however, they failed completely, which implies that the microkinetic model was not truly intrinsic, i.e. physical or reactor-specific features were also represented in the kinetics. The more recent models, in particular those that obey thermodynamic consistency, usually performed much better and were often applicable to a wider range of conditions.
The tool CaRMeN can also be used to easily compare different physical models, for example the flow field, diffusion, heat transfer and physical parameters, such as catalyst loadings and structure, as well as inlet and boundary conditions. Furthermore, special features of a certain catalyst structure/support and also systematic errors in certain experimental set-ups can be identified more easily by comparing huge datasets from different sources and reactor types. The biggest hurdle for archiving much more data, however, is the incompleteness of literature information for reproduction of the experiments and models described. CaRMeN can be downloaded for free from www.detchem.com.
Author Contributions
Conceptualization: H.G. and O.D.; methodology: H.G. and O.D.; literature survey: L.M. and S.A.; original draft preparation: H.G., L.M., S.A., and S.T.; review and editing: H.G., L.M., and O.D.; supervision: O.D.; funding acquisition: O.D.
Funding
This work was supported by Steinbeis GmbH & Co. KG.
Acknowledgments
The authors would like to thank Robert J. Kee, Huayang Zhu, Canan Karakaya, and Greg Jackson, Colorado School of Mines, for fruitful discussions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gossler, H.; Maier, L.; Angeli, S.; Tischer, S.; Deutschmann, O. CaRMeN: A Tool for Analysing and Deriving Kinetics in the Real World. Phys. Chem. Chem. Phys. 2018, 20, 10857–10876. [Google Scholar] [CrossRef] [PubMed]
- PrIMe. Available online: http://primekinetics.org (accessed on 9 December 2018).
- RESPECTH. Available online: http://respecth.chem.elte.hu/respecth/ (accessed on 9 December 2018).
- Goteng, G.; Speight, M.; Nettyam, N.; Farooq, A.; Franklach, M.; Sarathy, S. A Hybrid Cloud System for Combustion Kinetics Simulation. In Proceedings of the 23rd International Symposium on Gas Kinetics and Related Phenomena, Szeged, Hungary, 20–24 July 2014. [Google Scholar]
- Lunsford, J.H. Catalytic Conversion of Methane to More Useful Chemicals and Fuels: A Challenge for the 21st Century. Catal. Today 2000, 63, 165–174. [Google Scholar] [CrossRef]
- Abbasi, R.; Wu, L.; Wanke, S.E.; Hayes, R.E. Kinetics of Methane Combustion over Pt and Pt–Pd Catalysts. Chem. Eng. Res. Des. 2012, 90, 1930–1942. [Google Scholar] [CrossRef]
- Chen, D.; Lødeng, R.; Svendsen, H.; Holmen, A. Hierarchical Multiscale Modeling of Methane Steam Reforming Reactions. Ind. Eng. Chem. Res. 2011, 50, 2600–2612. [Google Scholar] [CrossRef]
- Osman, A.I.; Abu-Dahrieh, J.K.; Laffir, F.; Curtin, T.; Thompson, J.M.; Rooney, D.W. A Bimetallic Catalyst on a Dual Component Support for Low Temperature Total Methane Oxidation. Appl. Catal. B Environ. 2016, 187, 408–418. [Google Scholar] [CrossRef]
- Osman, A.I.; Meudal, J.; Laffir, F.; Thompson, J.; Rooney, D. Enhanced Catalytic Activity of Ni on η-Al2O3 and ZSM-5 on Addition of Ceria Zirconia for the Partial Oxidation of Methane. Appl. Catal. B Environ. 2017, 212, 68–79. [Google Scholar] [CrossRef]
- Maier, L.; Schädel, B.; Delgado, K.H.; Tischer, S.; Deutschmann, O. Steam Reforming of Methane Over Nickel: Development of a Multi-Step Surface Reaction Mechanism. Top. Catal. 2011, 54, 845. [Google Scholar] [CrossRef]
- Deutschmann, O.; Schwiedemoch, R.; Maier, L.I.; Chatterjee, D. Natural Gas Conversion in Monolithic Catalysts: Interaction of Chemical Reactions and Transport Phenomena. In Studies in Surface Science and Catalysis; Iglesia, E., Spivey, J.J., Fleisch, T.H., Eds.; Elsevier: Amsterdam, The Netherlands, 2001; Volume 136, Natural Gas Conversion VI; pp. 251–258. [Google Scholar]
- Schwiedernoch, R.; Tischer, S.; Correa, C.; Deutschmann, O. Experimental and Numerical Study on the Transient Behavior of Partial Oxidation of Methane in a Catalytic Monolith. Chem. Eng. Sci. 2003, 58, 633–642. [Google Scholar] [CrossRef]
- Williams, K.A.; Leclerc, C.A.; Schmidt, L.D. Rapid Lightoff of Syngas Production from Methane: A Transient Product Analysis. AIChE J. 2005, 51, 247–260. [Google Scholar] [CrossRef]
- Karakaya, C.; Maier, L.; Deutschmann, O. Surface Reaction Kinetics of the Oxidation and Reforming of CH4 over Rh/Al2O3 Catalysts. Int. J. Chem. Kinet. 2016, 48, 144–160. [Google Scholar] [CrossRef]
- Deshmukh, S.R.; Vlachos, D.G. A Reduced Mechanism for Methane and One-Step Rate Expressions for Fuel-Lean Catalytic Combustion of Small Alkanes on Noble Metals. Combust. Flame 2007, 149, 366–383. [Google Scholar] [CrossRef]
- Mhadeshwar, A.B.; Vlachos, D.G. Hierarchical Multiscale Mechanism Development for Methane Partial Oxidation and Reforming and for Thermal Decomposition of Oxygenates on Rh. J. Phys. Chem. B 2005, 109, 16819–16835. [Google Scholar] [CrossRef] [PubMed]
- Rankovic, N.; Nicolle, A.; Berthout, D.; Da Costa, P. Kinetic Modeling Study of the Oxidation of Carbon Monoxide–Hydrogen Mixtures over Pt/Al2O3 and Rh/Al2O3 Catalysts. J. Phys. Chem. C 2011, 115, 20225–20236. [Google Scholar] [CrossRef]
- Boll, W. Korrelation Zwischen Umsatzverhalten Und Katalytischer Oberfläche von Dieseloxidationskatalysatoren Unter Variation von Beladung Und Alterungszustand. Ph.D. Thesis, Karlsruhe Institute of Technology, Karlsruhe, Germany, 2011. [Google Scholar]
- Cai, Y.; Stenger, H.G., Jr.; Lyman, C.E. Catalytic CO Oxidation over Pt–Rh/γ-Al2O3Catalysts. J. Catal. 1996, 161, 123–131. [Google Scholar] [CrossRef]
- Ito, S.I.; Fujimori, T.; Nagashima, K.; Yuzaki, K.; Kunimori, K. Strong Rhodium–Niobia Interaction in Rh/Nb2O5, Nb2O5–Rh/SiO2 and RhNbO4/SiO2 Catalysts: Application to Selective CO Oxidation and CO Hydrogenation. Catal. Today 2000, 57, 247–254. [Google Scholar] [CrossRef]
- Zum Mallen, M.P.; Williams, W.R.; Schmidt, L.D. Steps in Hydrogen Oxidation on Rhodium: Hydroxyl Desorption at High Temperatures. J. Phys. Chem. 1993, 97, 625–632. [Google Scholar] [CrossRef]
- Hickman, D.A.; Schmidt, L.D. Steps in CH4 Oxidation on Pt and Rh Surfaces: High-Temperature Reactor Simulations. AIChE J. 1993, 39, 1164–1177. [Google Scholar] [CrossRef]
- Appel, C.; Mantzaras, J.; Schaeren, R.; Bombach, R.; Inauen, A.; Tylli, N.; Wolf, M.; Griffin, T.; Winkler, D.; Carroni, R. Partial Catalytic Oxidation of Methane to Synthesis Gas over Rhodium: In Situ Raman Experiments and Detailed Simulations. Proc. Combust. Inst. 2005, 30, 2509–2517. [Google Scholar] [CrossRef]
- Sui, R.; Mantzaras, J.; Bombach, R. A Comparative Experimental and Numerical Investigation of the Heterogeneous and Homogeneous Combustion Characteristics of Fuel-Rich Methane Mixtures over Rhodium and Platinum. Proc. Combust. Inst. 2017, 36, 4313–4320. [Google Scholar] [CrossRef]
- Sui, R.; Mantzaras, J.; Bombach, R.; Denisov, A. Hetero-/Homogeneous Combustion of Fuel-Lean Methane/Oxygen/Nitrogen Mixtures over Rhodium at Pressures up to 12 bar. Proc. Combust. Inst. 2017, 36, 4321–4328. [Google Scholar] [CrossRef]
- Maestri, M.; Beretta, A.; Faravelli, T.; Groppi, G.; Tronconi, E.; Vlachos, D.G. Two-Dimensional Detailed Modeling of Fuel-Rich H2 Combustion over Rh/Al2O3 Catalyst. Chem. Eng. Sci. 2008, 63, 2657–2669. [Google Scholar] [CrossRef]
- Karakaya, C.; Deutschmann, O. Kinetics of Hydrogen Oxidation on Rh/Al2O3 Catalysts Studied in a Stagnation-Flow Reactor. Chem. Eng. Sci. 2013, 89, 171–184. [Google Scholar] [CrossRef]
- Karadeniz, H.; Karakaya, C.; Tischer, S.; Deutschmann, O. Numerical Modeling of Stagnation-Flows on Porous Catalytic Surfaces: CO Oxidation on Rh/Al2O3. Chem. Eng. Sci. 2013, 104, 899–907. [Google Scholar] [CrossRef]
- Pery, T.; Schweitzer, M.G.; Volpp, H.R.; Wolfrum, J.; Ciossu, L.; Deutschmann, O.; Warnatz, J. Sum-Frequency Generation in Situ Study of CO Adsorption and Catalytic CO Oxidation on Rhodium at Elevated Pressures. Proc. Combust. Inst. 2002, 29, 973–980. [Google Scholar] [CrossRef]
- Karakaya, C.; Otterstätter, R.; Maier, L.; Deutschmann, O. Kinetics of the Water-Gas Shift Reaction over Rh/Al2O3 Catalysts. Appl. Catal. A Gen. 2014, 470, 31–44. [Google Scholar] [CrossRef]
- McGuire, N.E.; Sullivan, N.P.; Deutschmann, O.; Zhu, H.; Kee, R.J. Dry Reforming of Methane in a Stagnation-Flow Reactor Using Rh Supported on Strontium-Substituted Hexaaluminate. Appl. Catal. A Gen. 2011, 394, 257–265. [Google Scholar] [CrossRef]
- Behrendt, F.; Deutschmann, O.; Maas, U.; Warnatz, J. Simulation and Sensitivity Analysis of the Heterogeneous Oxidation of Methane on a Platinum Foil. J. Vac. Sci. Technol. A 1995, 13, 1373–1377. [Google Scholar] [CrossRef]
- Kee, R.J.; Coltrin, M.E.; Glarborg, P. Chemically Reacting Flow: Theory and Practice; Wiley Interscience: Hoboken, NJ, USA, 2003. [Google Scholar]
- Tischer, S.; Correa, C.; Deutschmann, O. Transient Three-Dimensional Simulations of a Catalytic Combustion Monolith Using Detailed Models for Heterogeneous and Homogeneous Reactions and Transport Phenomena. Catal. Today 2001, 69, 57–62. [Google Scholar] [CrossRef]
- Tavazzi, I.; Beretta, A.; Groppi, G.; Forzatti, P. Development of a Molecular Kinetic Scheme for Methane Partial Oxidation over a Rh/α-Al2O3 Catalyst. J. Catal. 2006, 241, 1–13. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).