Catalytic Behaviour of Flame-Made CuO-CeO2 Nanocatalysts in Efficient CO Oxidation

 , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Catalysts’ Preparation
2.2. Catalysts’ Characterization
2.3. Catalytic Activity Measurement
3. Results and Discussion
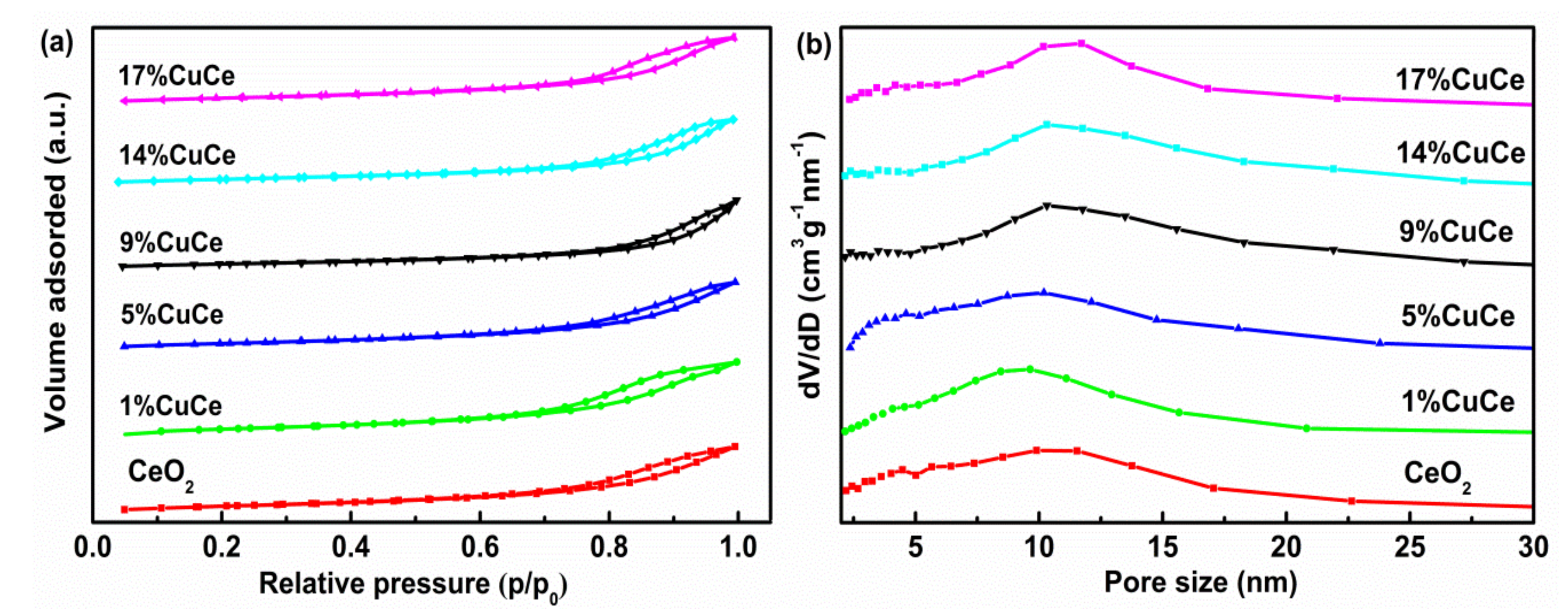
3.1. Microstructure of the Prepared Catalysts
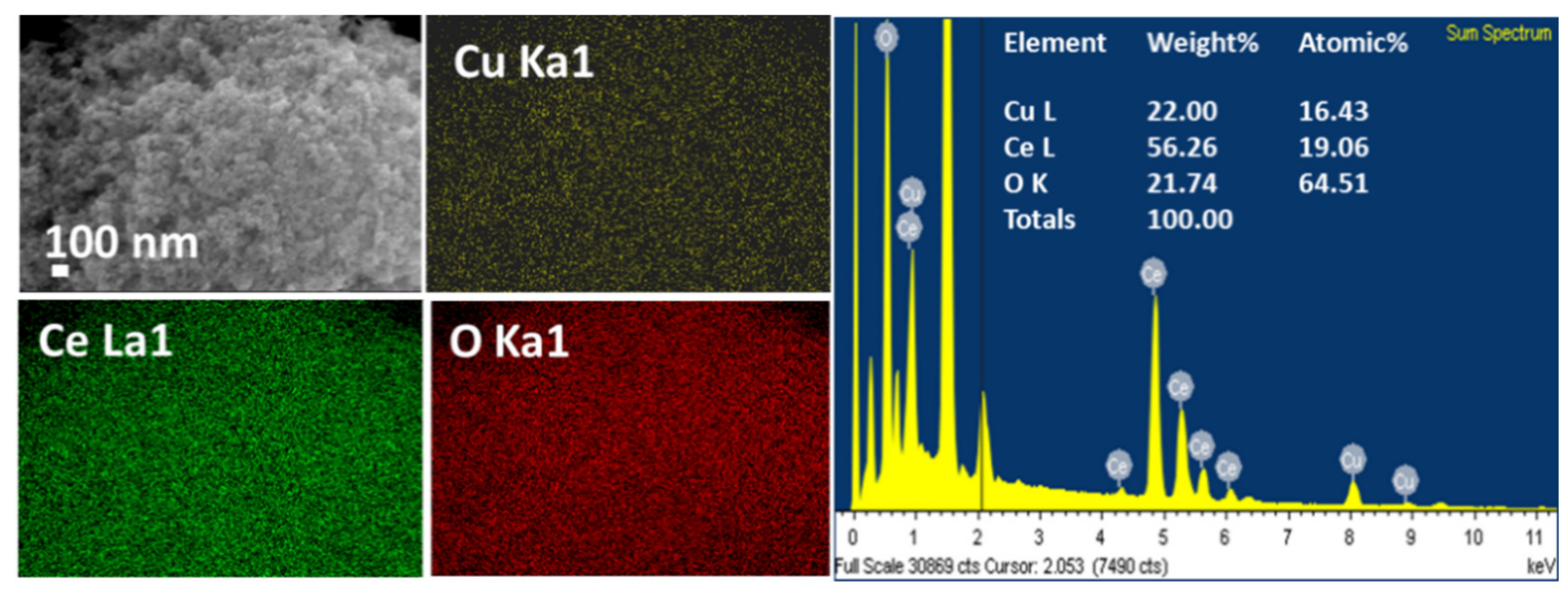
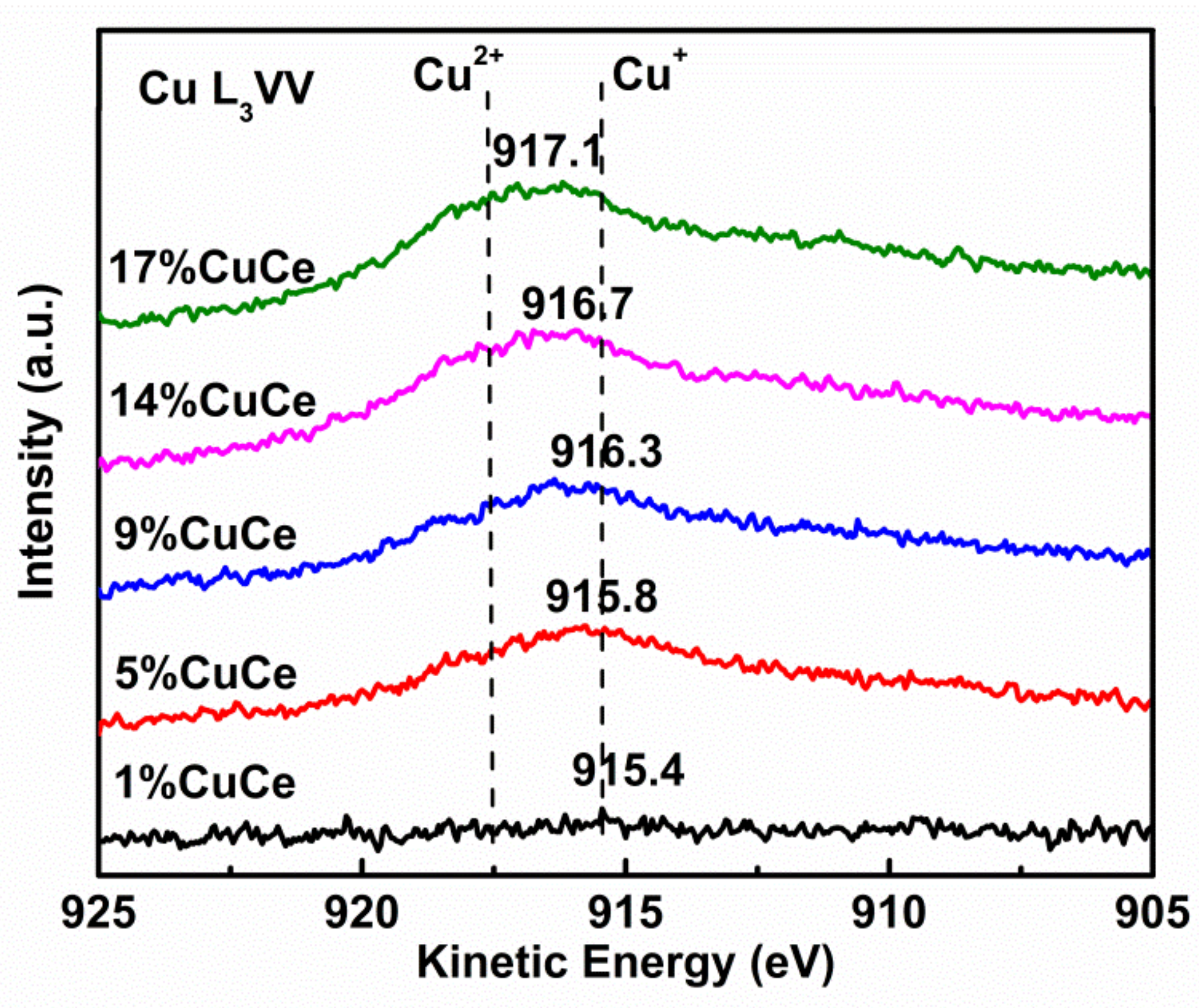
3.2. The Surface Composition of Catalysts
3.3. Redox Properties of the Prepared Catalysts
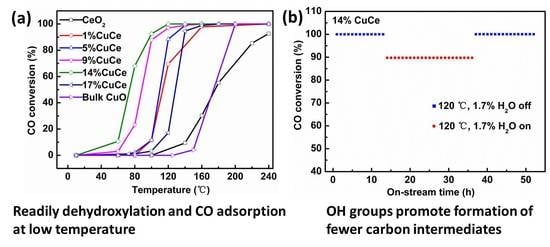
3.4. The CO Catalytic Oxidation Performance
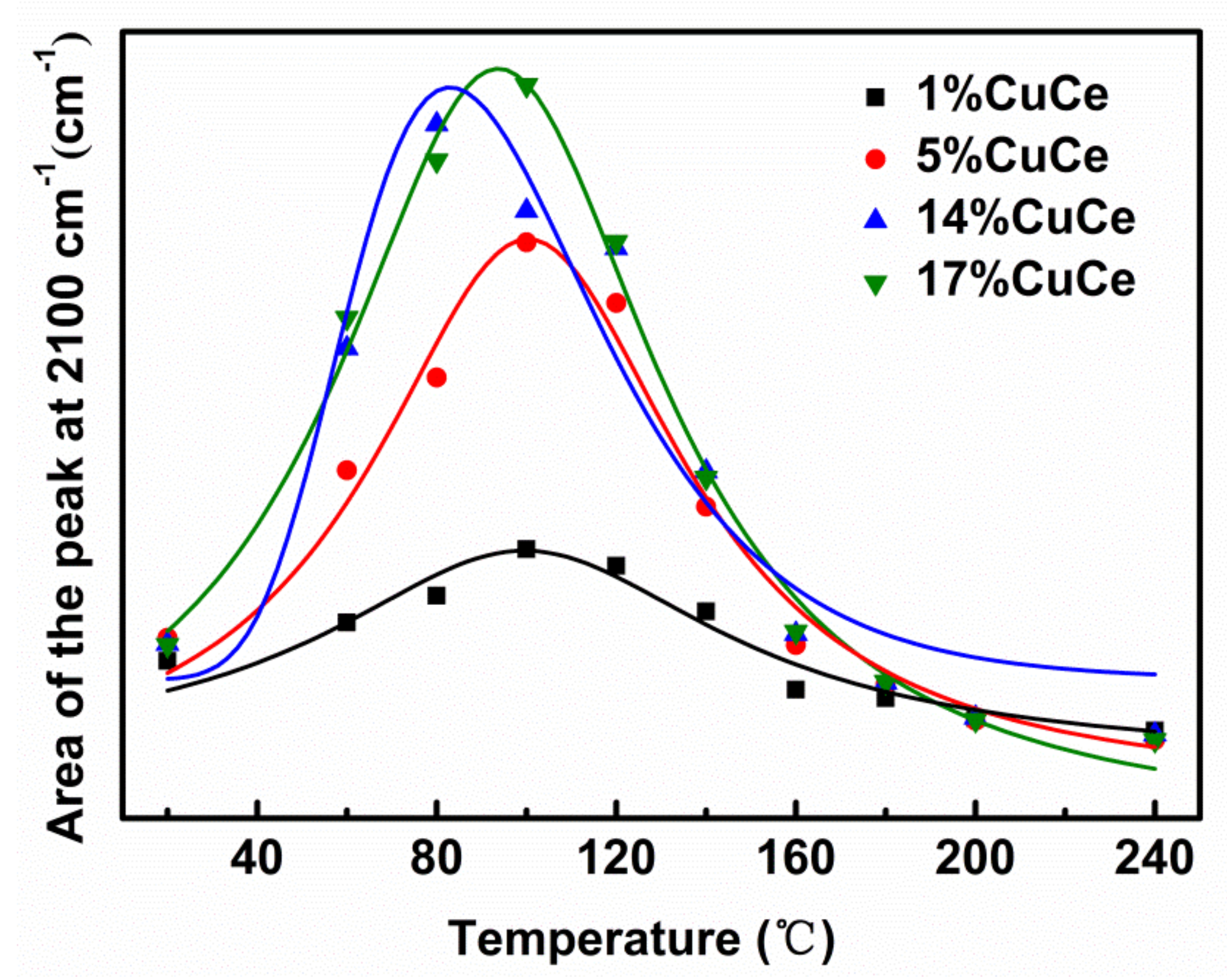
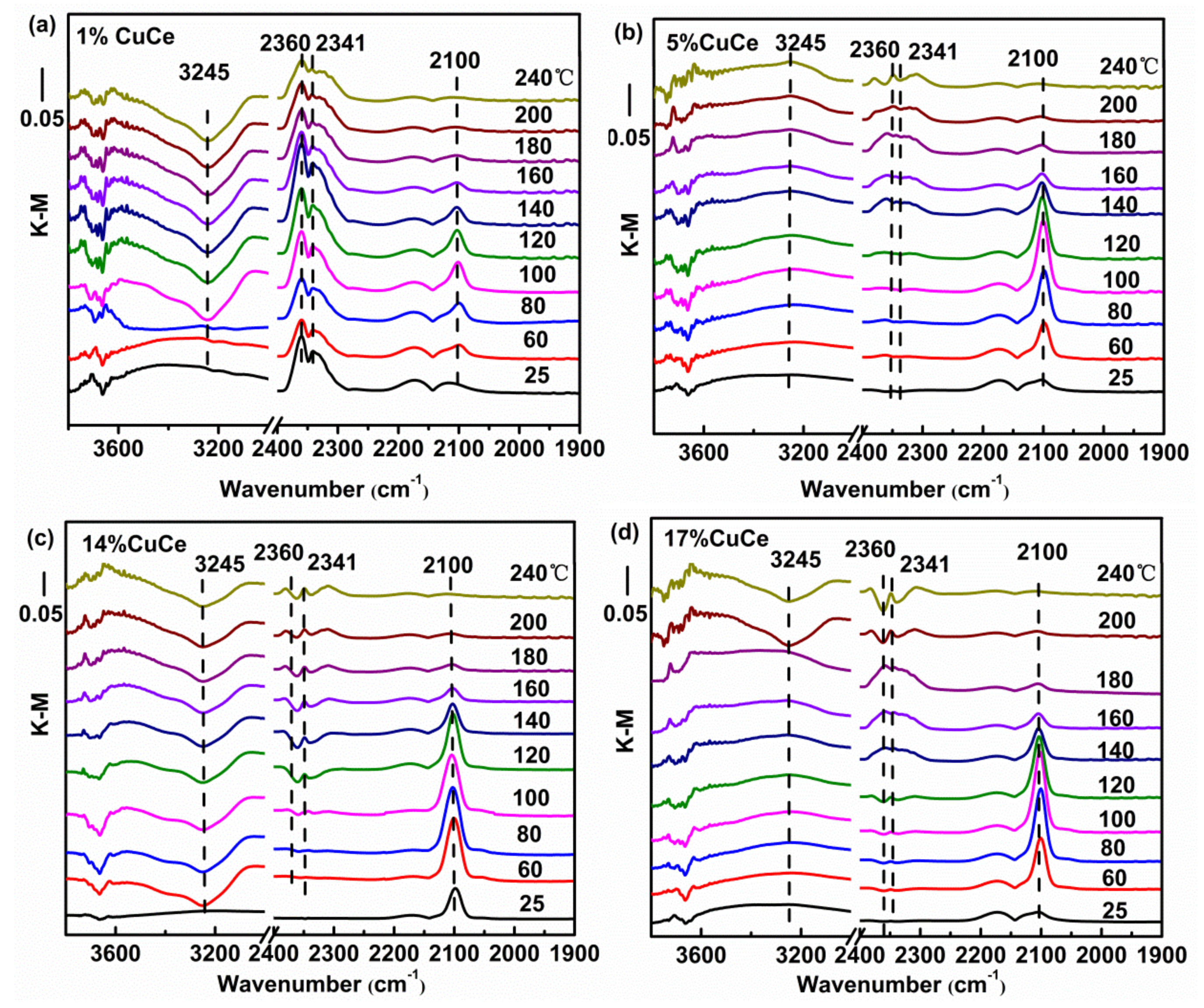
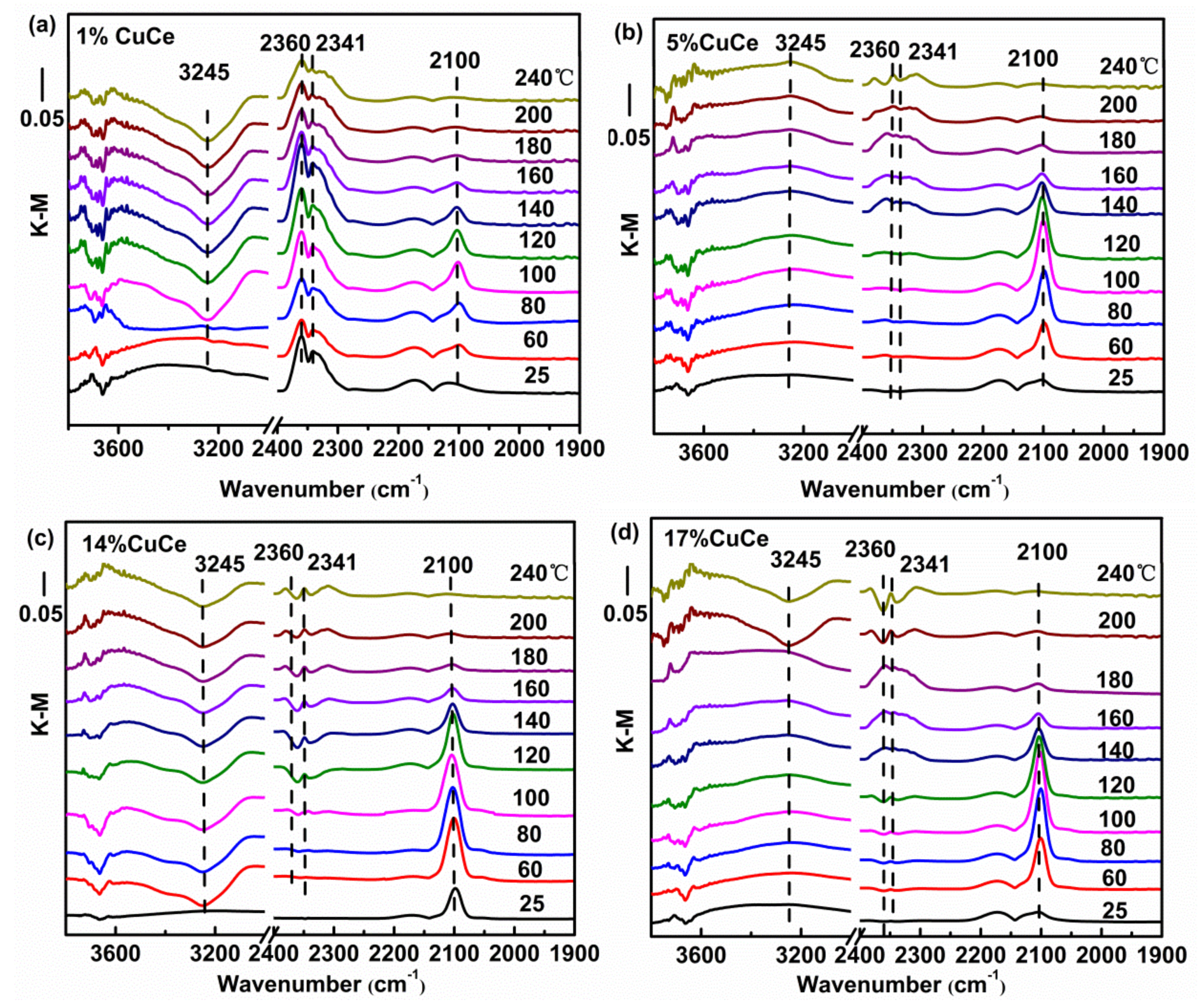
3.5. CO and O2 Co-Adsorption In Situ Diffuse Reflectance Infrared Fourier Transform Spectroscopy (DRIFTS) Analysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A

References
- Pitkäaho, S.; Matejova, L.; Jiratova, K.; Ojala, S.; Keiski, R.L. Oxidation of perchloroethylene-activity and selectivity of Pt, Pd, Rh, and V2O5 catalysts supported on Al2O3, Al2O3-TiO2 and Al2O3-CeO2. Part 2. Appl. Catal. B 2012, 126, 215–224. [Google Scholar] [CrossRef]
- Varghese, S.; Cutrufello, M.G.; Rombi, E.; Cannas, C.; Monaci, R.; Ferino, I. CO oxidation and preferential oxidation of CO in the presence of hydrogen over SBA-15-templated CuO-Co3O4 catalysts. Appl. Catal. A 2012, 443–444, 161–170. [Google Scholar] [CrossRef]
- Qiao, D.; Lu, G.; Liu, X.; Guo, Y.; Wang, Y.; Guo, Y. Preparation of Ce1−XFeXO2 solid solution and its catalytic performance for oxidation of CH4 and CO. J. Mater. Sci. 2011, 46, 3500–3506. [Google Scholar] [CrossRef]
- Sedmak, G.; Hočevar, S.; Levec, J. Kinetics of selective CO oxidation in excess of H2 over the nanostructured Cu0.1Ce0.9O2-y catalyst. J. Catal. 2003, 213, 135–150. [Google Scholar] [CrossRef]
- Avgouropoulos, G.; Ioannides, T.; Papadopoulou, C.; Batista, J.; Hocevar, S.; Matralis, H.K. A comparative study of Pt/γ-Al2O3, Au/α-Fe2O3 and CuO- CeO2 catalysts for the selective oxidation of carbon monoxide in excess hydrogen. Catal. Today 2002, 75, 157–167. [Google Scholar] [CrossRef]
- Sudarsanam, P.; Hillary, B.; Mallesham, B.; Rao, B.G.; Amin, M.H.; Nafady, A.; Alsalme, A.M.; Reddy, B.M.; Bhargava, S.K. Designing CuOx nanoparticle-decorated CeO2 nanocubes for catalytic soot oxidation: Role of the nanointerface in the catalytic performance of heterostructured nanomaterials. Langmuir 2016, 32, 2208–2215. [Google Scholar] [CrossRef]
- Mukherjee, D.; Rao, B.G.; Reddy, B.M. CO and soot oxidation activity of doped ceria: Influence of dopants. Appl. Catal. B 2016, 197, 105–115. [Google Scholar] [CrossRef]
- Davó-Quiñonero, A.; Navlani-García, M.; Lozano-Castelló, D.; Bueno-López, A.; Anderson, J.A. Role of hydroxyl groups in the preferential oxidation of CO over copper oxide–cerium oxide catalysts. ACS Catal. 2016, 6, 1723–1731. [Google Scholar] [CrossRef]
- Di Benedetto, A.; Landi, G.; Lisi, L. Improved CO-PROX performance of CuO/CeO2 catalysts by using nanometric ceria as support. Catalysts 2018, 8, 209. [Google Scholar] [CrossRef]
- Aneggi, E.; Boaro, M.; Leitenburg, C.D.; Dolcetti, G.; Trovarelli, A. Insights into the redox properties of ceria-based oxides and their implications in catalysis. J. Alloys Compd. 2006, 408–412, 1096–1102. [Google Scholar] [CrossRef]
- Bae, C.M.; Ko, J.B.; Kim, D.H. Selective catalytic oxidation of carbon monoxide with carbon dioxide, water vapor and excess hydrogen on CuO-CeO2 mixed oxide catalysts. Catal. Commun. 2005, 6, 507–511. [Google Scholar] [CrossRef]
- Wang, J.; Pu, H.; Wan, G.; Chen, K.; Lu, J.; Lei, Y.; Zhong, L.; He, S.; Han, C.; Luo, Y. Promoted the reduction of Cu2+ to enhance CuO-CeO2 catalysts for CO preferential oxidation in H2-rich streams: Effects of preparation methods and copper precursors. Int. J. Hydrogen Energy 2017, 42, 21955–21968. [Google Scholar] [CrossRef]
- Shang, H.; Zhang, X.; Xu, J.; Han, Y. Effects of preparation methods on the activity of CuO/CeO2 Catalysts for CO oxidation. Front. Chem. Sci. Eng. 2017, 11, 603–612. [Google Scholar] [CrossRef]
- Cecilia, J.; Arango-Díaz, A.; Marrero-Jerez, J.; Núñez, P.; Moretti, E.; Storaro, L.; Rodríguez-Castellón, E. Catalytic behaviour of CuO-CeO2 systems prepared by different synthetic methodologies in the CO-PROX reaction under CO2-H2O feed stream. Catalysts 2017, 7, 160. [Google Scholar] [CrossRef]
- Liu, G.; Li, J.; Yang, K.; Tang, W.; Liu, H.; Yang, J.; Yue, R.; Chen, Y. Effects of cerium incorporation on the catalytic oxidation of benzene over flame-made perovskite La1−XCexMnO3 catalysts. Particuology 2015, 19, 60–68. [Google Scholar] [CrossRef]
- Liu, G.; Yue, R.; Jia, Y.; Ni, Y.; Yang, J.; Liu, H.; Wang, Z.; Wu, X.; Chen, Y. Catalytic oxidation of benzene over Ce–Mn oxides synthesized by flame spray pyrolysis. Particuology 2013, 11, 454–459. [Google Scholar] [CrossRef]
- Zhao, F.; Li, S.; Wu, X.; Yue, R.; Li, W.; Chen, Y. Synergetic effect over flame-made manganese doped CuO–CeO2 Nanocatalyst for enhanced CO oxidation performance. RSC Adv. 2019, 9, 2343–2352. [Google Scholar] [CrossRef]
- Wang, Z.; Qu, Z.; Quan, X.; Li, Z.; Wang, H.; Fan, R. Selective catalytic oxidation of ammonia to nitrogen over CuO-CeO2 mixed oxides prepared by surfactant-templated method. Appl. Catal. B 2013, 134–135, 153–166. [Google Scholar] [CrossRef]
- Solsona, B.; Sanchis, R.; Dejoz, A.; García, T.; Ruiz-Rodríguez, L.; López Nieto, J.; Cecilia, J.; Rodríguez-Castellón, E. Total oxidation of propane using CeO2 and CuO-CeO2 catalysts prepared using templates of different nature. Catalysts 2017, 7, 96. [Google Scholar] [CrossRef]
- Strobel, R.; Pratsinis, S.E. Flame aerosol synthesis of smart nanostructured materials. J. Mater. Chem. 2007, 17, 4743–4756. [Google Scholar] [CrossRef]
- Feng, X.; Sayle, D.C.; Wang, Z.L.; Paras, M.S.; Santora, B.; Sutorik, A.C.; Sayle, T.X.T.; Yang, Y.; Ding, Y.; Wang, X.; et al. Converting ceria polyhedral nanoparticles into single-crystal nanospheres. Science 2006, 312, 1504. [Google Scholar] [CrossRef]
- Liu, T.; Yao, Y.; Wei, L.; Shi, Z.; Han, L.; Yuan, H.; Li, B.; Dong, L.; Wang, F.; Sun, C. Preparation and evaluation of copper–manganese oxide as a high-efficiency catalyst for CO oxidation and NO reduction by CO. J. Phys. Chem. C 2017, 121, 12757–12770. [Google Scholar] [CrossRef]
- Balducci, G.; Kaspar, J.; Fornasiero, P.; Graziani, M.; Islam, M.S. Surface and reduction energetics of the CeO2-ZrO2 catalysts. J. Phys. Chem. C 1998, 102, 557–561. [Google Scholar] [CrossRef]
- Fornasiero, P.; Balducci, G.; Di Monte, R.; Kašpar, J.; Sergo, V.; Gubitosa, G.; Ferrero, A.; Graziani, M. Modification of the redox behaviour of CeO2 induced by structural doping with ZrO2. J. Catal. 1996, 164, 173–183. [Google Scholar] [CrossRef]
- Dosa, M.; Piumetti, M.; Bensaid, S.; Andana, T.; Novara, C.; Giorgis, F.; Fino, D.; Russo, N. Novel Mn-Cu-containing CeO2 nanopolyhedra for the oxidation of CO and diesel soot: Effect of dopants on the nanostructure and catalytic activity. Catal. Lett. 2017, 148, 298–311. [Google Scholar] [CrossRef]
- Du, L.; Wang, W.; Yan, H.; Wang, X.; Jin, Z.; Song, Q.; Si, R.; Jia, C. Copper-ceria sheets catalysts: Effect of copper species on catalytic activity in CO oxidation reaction. J. Rare Earths 2017, 35, 1186–1196. [Google Scholar] [CrossRef]
- Piumetti, M.; Bensaid, S.; Fino, D.; Russo, N. Nanostructured ceria-zirconia catalysts for CO oxidation: Study on surface properties and reactivity. Appl. Catal. B 2016, 197, 35–46. [Google Scholar] [CrossRef]
- Binet, C.; Badri, A.; Boutonnet-Kizling, M.; Lavalley, J.C. FTIR study of carbon monoxide adsorption on ceria: CO carbonite dianion adsorbed species. J. Chem. Soc. Faraday Trans. 1994, 90, 1023–1028. [Google Scholar] [CrossRef]
- Avgouropoulos, G.; Ioannides, T. Selective CO oxidation over CuO-CeO2 catalysts prepared via the urea-nitrate combustion method. Appl. Catal. A 2003, 244, 155–167. [Google Scholar] [CrossRef]
- Hočevar, S.; Krašovec, U.O.; Orel, B.; Aricó, A.S.; Kim, H. CWO of phenol on two differently prepared CuO-CeO2 Catalysts. Appl. Catal. B 2000, 28, 113–125. [Google Scholar] [CrossRef]
- Liu, W.; Flytzani-stephanopoulos, M. Total oxidation of carbon-monoxide and methane over transition metal fluorite oxide composite catalysts: II. catalyst characterization and reaction-kinetics. J. Catal. 1995, 153, 317–332. [Google Scholar] [CrossRef]
- Xie, Y.; Yin, Y.; Zeng, S.; Gao, M.; Su, H. Coexistence of Cu+ and Cu2+ in star-shaped CeO2/CuXO catalyst for preferential CO oxidation. Catal. Commun. 2017, 99, 110–114. [Google Scholar] [CrossRef]
- Gómez, L.E.; Miró, E.E.; Boix, A.V. Spectroscopic characterization of Mn-Co-Ce mixed oxides, active catalysts for COPROX reaction. Int. J. Hydrogen Energy 2013, 38, 5645–5654. [Google Scholar] [CrossRef]
- Qi, L.; Yu, Q.; Dai, Y.; Tang, C.; Liu, L.; Zhang, H.; Gao, F.; Dong, L.; Chen, Y. Influence of cerium precursors on the structure and reducibility of mesoporous CuO-CeO2 catalysts for CO oxidation. Appl. Catal. B 2012, 119–120, 308–320. [Google Scholar] [CrossRef]
- Li, S.; Wang, H.; Li, W.; Wu, X.; Tang, W.; Chen, Y. Effect of Cu substitution on promoted benzene oxidation over porous CuCo-based catalysts derived from layered double hydroxide with resistance of water vapor. Appl. Catal. B 2015, 166–167, 260–269. [Google Scholar] [CrossRef]
- Zhang, R.; Teoh, W.Y.; Amal, R.; Chen, B.; Kaliaguine, S. Catalytic reduction of NO by CO over Cu/CexZr1−XO2 prepared by flame synthesis. J. Catal. 2010, 272, 210–219. [Google Scholar] [CrossRef]
- Dow, W.P.; Huang, T.J. Effects of oxygen vacancy of yttria-stabilized zirconia support on carbon monoxide oxidation over copper catalyst. J. Catal. 1994, 147, 322–332. [Google Scholar] [CrossRef]
- Piumetti, M.; Bensaid, S.; Russo, N.; Fino, D. Nanostructured ceria-based catalysts for soot combustion: Investigations on the surface sensitivity. Appl. Catal. B 2015, 165, 742–751. [Google Scholar] [CrossRef]
- Zhu, P.; Li, J.; Zuo, S.; Zhou, R. Preferential oxidation properties of CO in excess hydrogen over CuO-CeO2 catalyst prepared by hydrothermal Method. Appl. Surf. Sci. 2008, 255, 2903–2909. [Google Scholar] [CrossRef]
- Delimaris, D.; Ioannides, T. VOC oxidation over CuO–CeO2 catalysts prepared by a combustion Method. Appl. Catal. B 2009, 89, 295–302. [Google Scholar] [CrossRef]
- Jia, A.P.; Deng, Y.; Hu, G.S.; Luo, M.F.; Lu, J.Q. Kinetic and activity study of CO oxidation over CuO-MnOx-CeO2 catalysts. React. Kinet. Mech. Catal. 2015, 117, 503–520. [Google Scholar] [CrossRef]
- Piumetti, M.; Bensaid, S.; Andana, T.; Russo, N.; Pirone, R.; Fino, D. Cerium-copper oxides prepared by solution combustion synthesis for total oxidation reactions: from powder catalysts to structured reactors. Appl. Catal. B 2017, 205, 455–468. [Google Scholar] [CrossRef]
- Sun, S.; Mao, D.; Yu, J.; Yang, Z.; Lu, G.; Ma, Z. Low-temperature CO oxidation on CuO/CeO2 catalysts: The significant effect of copper precursor and calcination temperature. Catal. Sci. Technol. 2015, 5, 3166–3181. [Google Scholar] [CrossRef]
- Chen, G.; Xu, Q.; Yang, Y.; Li, C.; Huang, T.; Sun, G.; Zhang, S.; Ma, D.; Li, X. Facile and mild strategy to construct mesoporous CeO2-CuO nanorods with enhanced catalytic activity toward CO oxidation. ACS Appl. Mater. Interfaces 2015, 7, 23538–23544. [Google Scholar] [CrossRef]
- Xiao, W.; Yang, S.; Zhang, P.; Li, P.; Wu, P.; Li, M.; Chen, N.; Jie, K.; Huang, C.; Zhang, N.; et al. Facile synthesis of highly porous metal oxides by mechanochemical nanocasting. Chem. Mater. 2018, 30, 2924–2929. [Google Scholar] [CrossRef]
- Spezzati, G.; Benavidez, A.D.; DeLaRiva, A.T.; Su, Y.; Hofmann, J.P.; Asahina, S.; Olivier, E.J.; Neethling, J.H.; Miller, J.T.; Datye, A.K.; Hensen, E.J.M. CO oxidation by Pd supported on CeO2(100) and CeO2(111) facets. Appl. Catal. B Evniron. 2019, 243, 36–46. [Google Scholar] [CrossRef]
- Wang, H.; Mao, D.; Qi, J.; Zhang, Q.; Ma, X.; Song, S.; Gu, L.; Yu, R.; Wang, D. Hollow multishelled structure of heterogeneous Co3O4-CeO2−X nanocomposite for CO catalytic oxidation. Adv. Funct. Mater. 2019, 1806588. [Google Scholar] [CrossRef]
- Gong, S.; Chen, J.; Wu, X.; Han, N.; Chen, Y. In-situ synthesis of Cu2O/reduced graphene oxide composite as effective catalyst for ozone decomposition. Catal. Commun. 2018, 106, 25–29. [Google Scholar] [CrossRef]
- Sanchez, M.G.; Gazquez, J.L. Oxygen vacancy model in strong metal-support interaction. J. Catal. 1987, 104, 120–135. [Google Scholar] [CrossRef]
- Wan, H.; Wang, Z.; Zhu, J.; Li, X.; Liu, B.; Gao, F.; Dong, L.; Chen, Y. Influence of CO pretreatment on the activities of CuO/γ-Al2O3 catalysts in CO+O2 reaction. Appl. Catal. B 2008, 79, 254–261. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhu, Yi.; Asakura, H.; Zhang, B.; Zhang, J.; Zhou, M.; Han, Y.; Tanaka, T.; Wang, A.; Zhang, T.; et al. Thermally stable single atom Pt/M-Al2O3 for selective hydrogenation and CO oxidation. Nat. Commun. 2017, 8, 16100. [Google Scholar] [CrossRef] [PubMed]
- Gamarra, D.; Martínez-Arias, A. Preferential oxidation of CO in rich H2 over CuO/CeO2: Operando-drifts analysis of deactivating effect of CO2 and H2O. J. Catal. 2009, 263, 189–195. [Google Scholar] [CrossRef]
- Binet, C.; Daturi, M.; Lavalley, J.C. Ir study of polycrystalline ceria properties in oxidised and reduced states. Catal. Today 1999, 50, 207–225. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Crystallite Size a (nm) | Cell Parameter (nm) | Surface Area (m2 × g−1) | Pore Volume (cm3 × g−1) | Pore Size (nm) |
|---|---|---|---|---|---|
| CeO2 | 25.6 | 0.5421 | 54.2 | 0.18 | 9.46 |
| 1% CuCe | 26.5 | 0.5420 | 48.5 | 0.16 | 9.10 |
| 5% CuCe | 30.1 | 0.5420 | 43.9 | 0.17 | 10.30 |
| 9% CuCe | 31.6 | 0.5419 | 42.6 | 0.20 | 13.60 |
| 14% CuCe | 31.4 | 0.5417 | 39.4 | 0.15 | 11.50 |
| 17% CuCe | 32.8 | 0.5422 | 38.9 | 0.16 | 10.46 |
| Samples | T50 (°C) | T90 (°C) | Ea (kJ × mol−1) | Cu/Ce (at. %) | Cu2+/Cu (%) | Ce3+/Ce (%) | |
|---|---|---|---|---|---|---|---|
| Nominal | XPS | ||||||
| CeO2 | 176 | 233 | 63.2 | - | - | - | - |
| 1% CuCe | 114 | 149 | 124.4 | 0.02 | 0.32 | 0 | 17.4 |
| 5% CuCe | 110 | 123 | 99.4 | 0.11 | 0.56 | 63.9 | 33.1 |
| 9% CuCe | 88 | 104 | 40.8 | 0.19 | 0.69 | 64.3 | 33.5 |
| 14% CuCe | 74 | 98 | 28.9 | 0.30 | 1.04 | 66.5 | 35.4 |
| 17% CuCe | 129 | 139 | 87.0 | 0.37 | 1.08 | 67.2 | 27.1 |
| Bulk CuO | 174 | 195 | 121.8 | - | - | - | - |
| Sample | Preparation | CO Conversion (%) | Reaction Condition | Ref. |
|---|---|---|---|---|
| 14 wt % CuO-CeO2 | FSP | 100 (120 °C) | 1% CO/0.6% O2/N2, 60,000 mL × g−1 × h−1 | This work |
| 1MnOx-5CuO-CeO2 | FSP | 100 (160 °C) | 1% CO/0.6% O2/N2, 60,000 mL × g−1 × h−1 | [17] |
| Cu0.40Ce0.60 | SCS a | 100 (130 °C) | 1000 ppm CO/10%O2/N2, 30,000 mL × g−1 × h−1 | [42] |
| 10 wt % CuO-CeO2 | IMP b | 99 (117 °C) | 4% CO/10% O2/N2, 9000 mL × g−1 × h−1 | [43] |
| 14.1 mol % CuO-CeO2NR | ST c | 100 (150 °C) | 1% CO/10% O2/N2, 60,000 mL × g−1 × h−1 | [44] |
| 5 wt % CuO-CeO2 | MN d | 100 (200 °C) | 1% CO/Air, 30,000 mL × g−1 × h−1 | [45] |
| 1 wt % Pd/CeO2-rod | IMP | 100 (150 °C) | 2% CO/2% O2/He, 60,000 mL × g−1 × h−1 | [46] |
| 4Co3O4-CeO2−x | STA e | 100 (160 °C) | 1% CO/1.6% O2/He, 30,000 mL × g−1 × h−1 | [47] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, F.; Li, S.; Wu, X.; Yue, R.; Li, W.; Zha, X.; Deng, Y.; Chen, Y. Catalytic Behaviour of Flame-Made CuO-CeO2 Nanocatalysts in Efficient CO Oxidation. Catalysts 2019, 9, 256. https://doi.org/10.3390/catal9030256
Zhao F, Li S, Wu X, Yue R, Li W, Zha X, Deng Y, Chen Y. Catalytic Behaviour of Flame-Made CuO-CeO2 Nanocatalysts in Efficient CO Oxidation. Catalysts. 2019; 9(3):256. https://doi.org/10.3390/catal9030256
Chicago/Turabian StyleZhao, Feng, Shuangde Li, Xiaofeng Wu, Renliang Yue, Weiman Li, Xicuo Zha, Yuzhou Deng, and Yunfa Chen. 2019. "Catalytic Behaviour of Flame-Made CuO-CeO2 Nanocatalysts in Efficient CO Oxidation" Catalysts 9, no. 3: 256. https://doi.org/10.3390/catal9030256
APA StyleZhao, F., Li, S., Wu, X., Yue, R., Li, W., Zha, X., Deng, Y., & Chen, Y. (2019). Catalytic Behaviour of Flame-Made CuO-CeO2 Nanocatalysts in Efficient CO Oxidation. Catalysts, 9(3), 256. https://doi.org/10.3390/catal9030256