2.1.1. X-ray Fluorescence (XRF) and X-ray Diffraction (XRD)
The XRF elemental analysis of cerium-incorporated samples is compiled in
Table 1. As can be seen, both materials have a similar cerium content, 5.89 and 5.24 wt.% for Ce-MCM-Cl and Ce-MCM-NO
3, respectively. In both cases, the Ce content is lower than the expected value, which led to Si/Ce molar ratios higher than the one used in the synthesis gel (Si/Ce = 25), i.e., Si/Ce = 31 for Ce-MCM-Cl and Si/Ce = 36 for Ce-MCM-NO
3. This result is not uncommon since the incorporation of heteroatom ions in silica matrices is strongly dependent on the synthetic procedure and precursors used [
39,
40,
41,
42].
Haller and Cesteros [
39] studied the effects of several synthesis parameters during the preparation of Al-MCM-41 materials with Si/Al = 24, including different aluminum and silicon sources. Some samples showed large deviations from the desired Si/Al ratio, reaching values between 27.48 and 37.61, an error of 15% and 57%, respectively. Also, not all heteroatom ions are incorporated into the framework of silica materials, even when the silicon-to-metal molar ratio obtained in the solid is the same as the one used in the synthesis gel [
43,
44]. For example, Matsumoto and co-workers [
43] studied Al-MCM-41 with different Si/Al ratios by solid state
27Al MAS-NMR and observed a signal at 53 ppm, assigned to tetrahedrally coordinated framework aluminum, and another at 0 ppm, associated with octahedrally coordinated non-framework aluminum.
The slightly higher Si/Ce ratio obtained for Ce-MCM-NO
3, when compared to Ce-MCM-Cl, suggests that cerium(III) precursors with different anions might influence the outcome in the silica-based molecular sieves’ synthesis. Indeed, XRD patterns of the calcined Ce-MCM-41 materials in the low angle region (
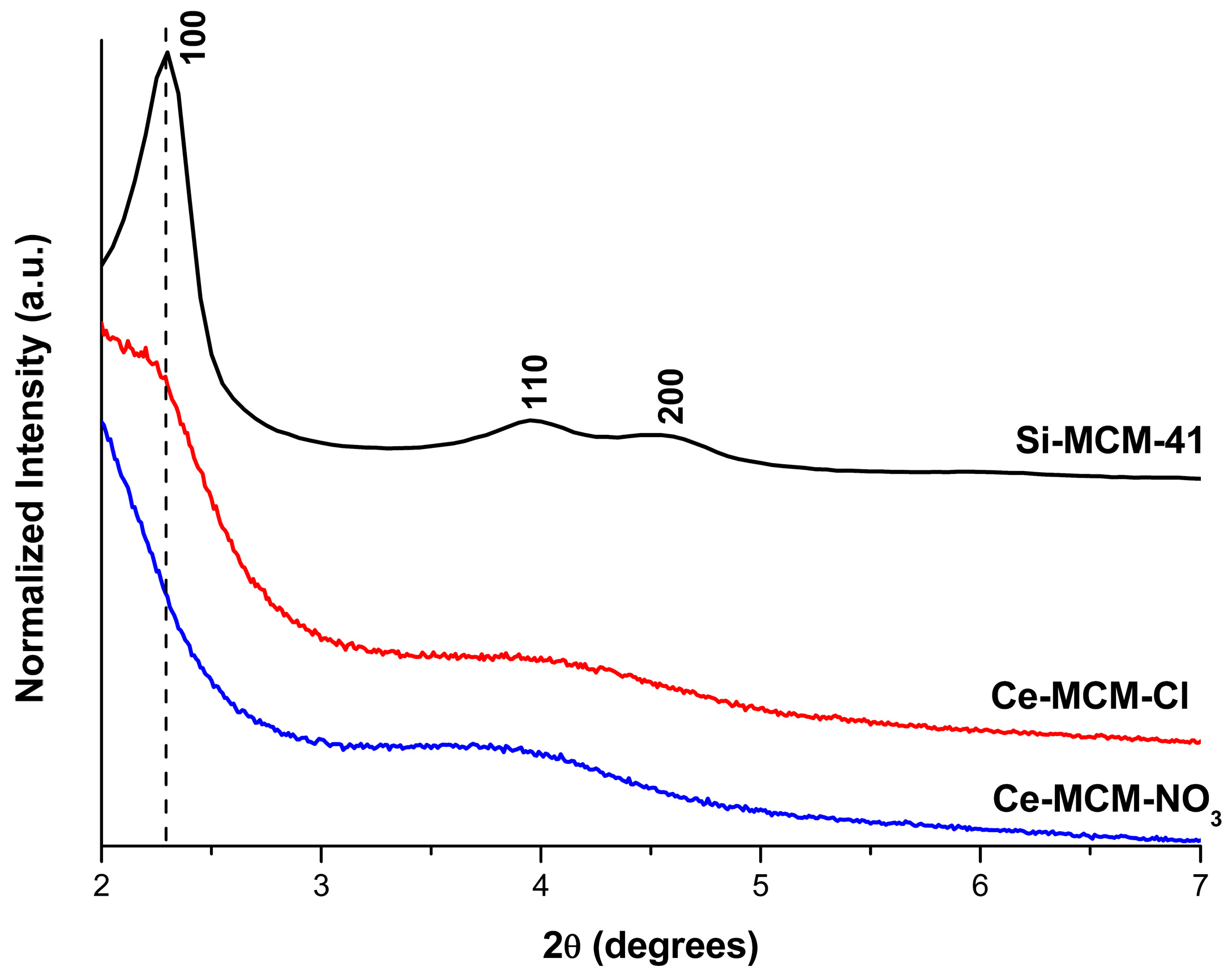
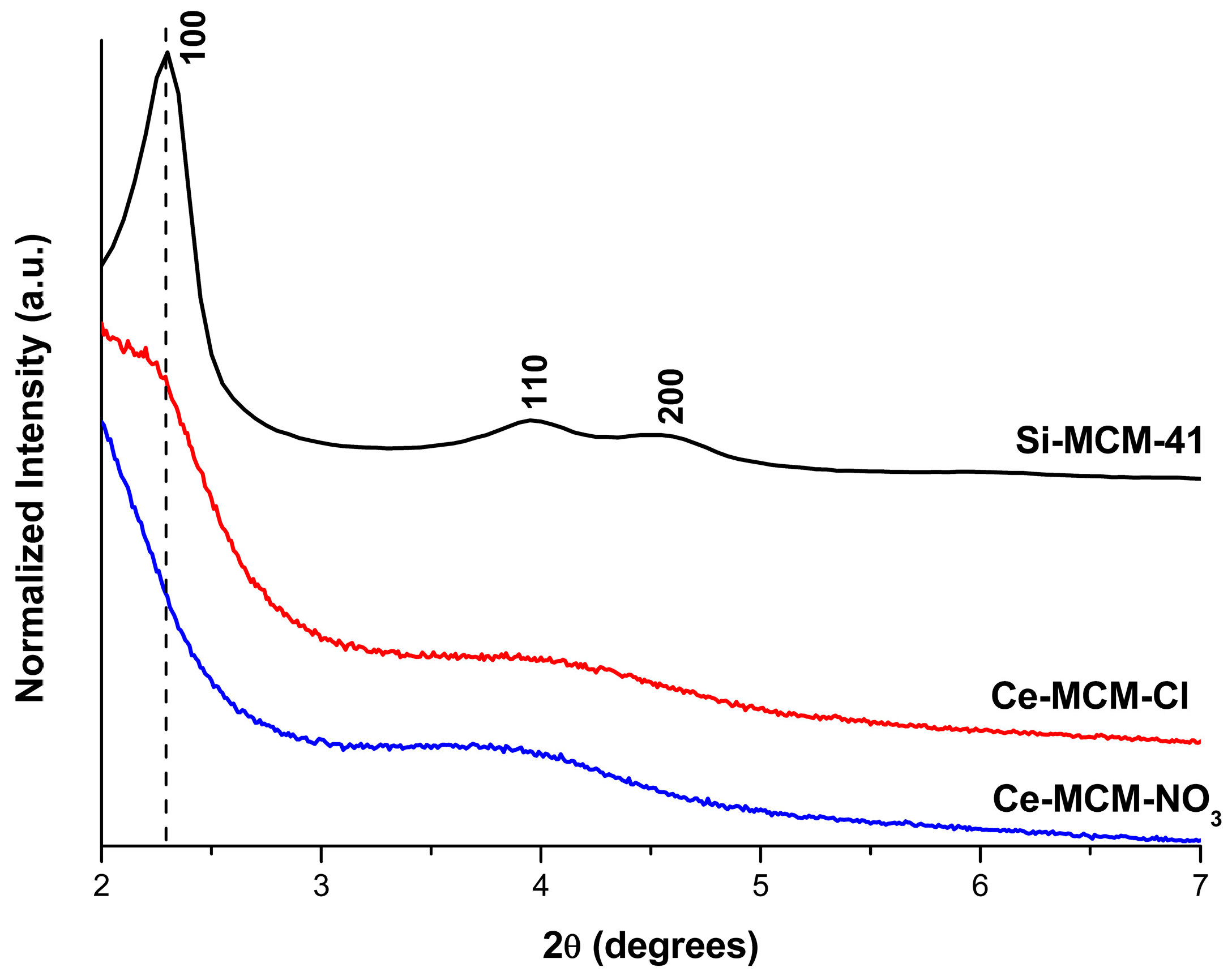
Figure 1) showed not only differences towards Si-MCM-41, but also among themselves. Si-MCM-41 displayed reflections at 2.30, 3.95, and 4.55°, corresponding to structural planes 100, 110, and 200 of the MCM-41 hexagonal mesophase (planar space group p6m) [
45]. For the prepared Ce-MCM-41, the 100 reflection was noticeably shifted towards lower 2θ values and the 110 and 200 reflections overlap with each other, forming a broad signal between 3 and 5°. The first indicates an expansion of the unit cell due to cerium incorporation in the MCM-41 framework, which is expected since the ionic radii of the Ce
4+ ions (87 pm for 6-coordination [
46]) is higher than Si
4+ (40 pm for 6-coordination [
46]). The second observation is characteristic of hexagonal disordered mesoporous materials [
47] and has been observed in the literature for Ce-MCM-41 [
48,
49,
50,
51]. This loss of mesophase ordering can be attributed to the partial substitution of Si
4+ for Ce
4+ ions due to changes in the Si-O-Si bond angles that lead to the emergence of local structural defects and bond strain [
37,
47]. However, metal precursor salts in the synthesis gel could cause changes in the assembly of CTA
+ molecules, leading to less organized materials when compared to Si-MCM-41. As Barkam and co-workers [
52] demonstrated, even the use of different anions for the same metal precursor can extensively affect the physicochemical properties of particles in solution [
52].
Since MCM-41 is a molecular sieve with a bidimensional structure, the calculation of the hexagonal unit cell parameter (
a) could be determined by using Equation (1) (see Materials and Methods). Unfortunately, the 100 reflection cannot be observed in the XRD pattern for the cerium-incorporated materials (
Figure 1), as a consequence of the experimental limitations (i.e., the signal of the beam source was only obtainable starting at 2θ = 2°). As mentioned before, the 110 and 200 reflections could be distinguished, but are overlapped. If the sole 200 reflection could be observed in the modified materials,
d100 could be calculated by a relationship between the interplanar spacing of the 200 and 100 reflections (Equation (2), see Materials and Methods). Therefore, deconvolution methods [
53] were applied in the overlapped signal to gain information towards the 2θ position of both the 110 and 200 reflections to find
d100 and, consequently, the unit cell,
a, for the modified materials (see
Figure 2).
The 110 and 200 reflections were found in 3.86 and 4.42° for Ce-MCM-Cl and in 3.56 and 4.10° for Ce-MCM-NO
3, respectively. The values obtained for
d100 and
a using the information of the 200 deconvoluted reflections are compiled in
Table 1. For Si-MCM-41, the
d100 value calculated from the 200 reflection (3.88 nm) was close to the one observed experimentally (3.84 nm), validating our data obtained from Equations (1) and (2). As can be observed in
Table 1, the values of
a calculated for the cerium materials confirmed the unit cell expansion mentioned before and this effect was more expressive towards Ce-MCM-NO
3.
Differences between both cerium-modified materials could also be observed at the high angle region (
Figure S1 in Supplementary Material). While the Ce-MCM-Cl pattern only exhibited the usual broad signal around 25° common to amorphous silica in the MCM-41 walls, Ce-MCM-NO
3 presented three reflections around 28, 47, and 57° that suggests the formation of CeO
2 (reflections 110, 220, and 311 according to JCPDS 34-0394, respectively [
54]). The surfacing of non-framework species is generally associated with the synthetic procedure [
37,
55] and the influence of a particular metal precursor is little discussed in the literature. However, the absence of peaks from segregated CeO
2 in the particles of the Ce-MCM-Cl pattern does not mean there are not any; they might be highly dispersed on the MCM-41 surface. Nonetheless, the XRD analysis demonstrated the clear presence of CeO
2 in one of the materials.
2.1.2. Spectroscopy Studies (FTIR, UV-Vis, Raman, and 29Si MAS-NMR)
To investigate the origin of non-framework CeO
2 species, as well as the consequences of using different cerium precursors, Raman spectroscopy was performed in both as-synthesized and calcined MCM-41 materials. The Raman spectra of as-synthesized materials can give information on how an assembly of surfactants is affected by other ionic species in the synthetic medium. Raman signals of organic molecules are dominant in this situation and the occluded CTA
+ cations can be in the same arrangement from the synthetic medium, counter-balancing the negative charges from deprotonated silanol groups. Also, the use of CTA
+ as a probe to access information towards microstructural characteristics from the framework of as-synthesized MCM-41 has been little explored in the literature [
51,
56,
57], encouraging the use of conformational-sensible spectroscopic techniques, such as Raman spectroscopy, in the unraveling of microstructural changes.
Figure 3 displays the spectra of the as-synthesized materials and, for means of comparison, the spectra of template molecules with two different molecular orderings were acquired: A 23 wt.% aqueous solution of cetyltrimethylammonium bromide (CTAB) and solid CTAB (
Figure S2 in Supplementary Material). The concentration of 23 wt.% was chosen because it is in the range of the concentration where surfactant molecules form an hexagonal liquid-crystal phase in solution [
58]. The comparison between the spectra of the as-synthesized materials and the two template standards clearly shows that CTA
+ cations are organized as cylindrical micelles inside the pores of as-synthesized materials, as it is expected based on the mechanism for MCM-41 crystallization [
59].
The assignment of each vibrational mode observable in the spectra of the as-synthesized materials and template standards is summarized in
Table S1 (see
Supplementary Material). For the solid CTAB, two of the most important Raman bands that contain information towards molecular configuration are those in 1061 and 1126 cm
−1, attributed to C-C symmetric and asymmetric stretching, respectively [
57,
60,
61]. These are characteristic of extended hydrocarbon chains with all-
trans ordered structures in which alternate carbon atoms move in the opposite direction. Other important vibrational modes at 2848/2880 and 2941/2958 cm
−1 were assigned to the C-H symmetric/asymmetric stretching of CH
2 and CH
3, respectively. These bands are the strongest in the Raman spectra and highly sensitive to molecular conformation [
57,
62]. In comparison with solid CTAB, the Raman spectra of CTAB solution and occluded CTA
+ in as-synthesized materials showed less and broader signals, as well as other relevant differences. First, a new signal appeared at 1076 cm
−1 for both as-synthesized materials and CTAB solution. This signal, which was not observed for solid CTAB, was assigned to the C-C stretching mode of hydrocarbon chains in the gauche configuration and is expected in more disordered systems [
57,
60]. Second, common signals found for solid CTAB were absent or became overlapped for CTA
+ molecules in as-synthesized materials or in solution, especially those at 1175 and 1464 cm
−1, associated to the dynamic motion of molecules in supramolecular assemblies [
57]. This result indicates that occluded CTA
+ molecules in as-synthesized materials act as it were in a liquid-like state [
57]. Finally, intensity and shift changes in conformational-sensitive peaks between 2850 and 3000 cm
−1 were also observed for CTA
+ occluded and in solution. The asymmetric methylene C-H mode around 2885 cm
−1 is less intense than in the solid CTAB and the signals assigned to methyl C-H modes were considerably displaced, both evidence of higher statistical chances for attaining the gauche configuration, i.e., a more disordered state [
57,
61,
62]. This was even more noticeable for the broad band around 2920 to 2928 cm
−1 for CTA
+ occluded and in solution. This band is a composite of two overlapping modes, one from the polymethylene chain (usually around 2920 cm
−l) and the other from the methyl group (at 2941 cm
−1 for solid CTAB), making it sensitive to conformational disorder of the long methylene chain, since the former contributes more than the latter [
61,
62].
As mentioned before, changes in the intensity of C-H and C-C stretching modes can reveal effects towards disordering since liquid-like states permit an increase in the number of molecules that can attain the gauche conformation. In the literature, it was demonstrated that the ratio between the intensities of certain long alkyl chain Raman bands can reflect their conformational state [
61,
62,
63]. Therefore, we employed such parameters to understand the influence of different cerium precursors in the assembly of the template molecules. Two of the most employed ratios are those between the C-C gauche stretching around 1080 cm
−1 and the C-C asymmetric stretching around 1120 cm
−1 [
57,
62]; and the C-H stretchings around 2920 and 2850 cm
−1 [
61,
63]. Higher values for these parameters are related to an increase of feasibility to attain the gauche conformation and, consequently, disordered the template assembly more in the synthetic medium that was retained in MCM-41 pores. Both ratios for the as-synthesized materials were calculated and are compiled in
Table 2.
The I
2920/I
2850 ratio demonstrates small changes in its value when compared to the I
1080/I
1120 ratio, which indicates that the latter is a more sensible parameter towards conformational disordering than the former [
62]. Nonetheless, the values presented for both ratios allowed the materials to be organized in increasing disorder of template assemblies: A-Si-MCM-41 < A-Ce-MCM-NO
3 < A-Ce-MCM-Cl. Thus, it was possible to perceive that: (i) The presence of cerium species altered the supramolecular arrangement of CTA
+ cations; (ii) the disordering was more prominent when utilizing cerium(III) chloride as the precursor; and (iii) the degree of disordering obtained for A-Ce-MCM-NO
3 was close to A-Si-MCM-41. To explain these observations, it is worth recalling the chemistry of cerium species in high pH aqueous mediums [
64,
65,
66] and the synthetic procedure used in this work to prepare Ce-MCM-41 materials (see Materials and Methods). Under basic conditions, the hydrolysis of Ce(III) generates [Ce(OH)
y]
(3−y) species, where
y goes from 0 to 3. During our synthesis, the dropwise addition of cerium(III) precursor turned the clear solution into a dark slurry before it became a light brown/purple solid. This could be addressed by assuming that [Ce(OH)]
2+, [Ce(OH)
2]
+, and [Ce(OH)
3] formation occurred progressively and continuously [
65]. In addition, the oxidation of Ce
3+ to Ce
4+ is not thermodynamically favored under the conditions used in this work (i.e., non-aerated solution and pH = 11.2) [
64] and [Ce(OH)
3] can be regarded as the main cerium compound in solution [
67].
Based on the above discussion and the well-known self-assembled systems used for synthesizing MCM-41 [
59], this work proposes the following mechanism for cerium incorporation: (i) CTA
+ cations are counterbalanced by basic silicate anions; (ii) the latter interacts through hydrogen bonds with [Ce(OH)
3] species; and (iii) the gradual condensation under basic conditions leads to silicate oligomers containing Si-O-Si and Ce-O-Si bonds. The higher disorder detected by Raman for occluded CTA
+ indicates that cerium incorporation into silica causes different electrostatic interactions between CTA
+ cations and the anionic surface. Since both cerium(III) precursors are highly soluble salts with similar stability constants [
68], the differences observed between Ce-MCM-Cl and Ce-MCM-NO
3 by the techniques used in this work must be related to the different anions. Berr and co-workers [
69] studied the effect of anions on alkyltrimethylammonium micelles and observed that micelle aggregation increases in the following order: OH
− << Cl
− < CH
3SO
4− < Br
− < NO
3−. For that reason, nitrate anions induce longer rods of CTA
+ cations in solution than chloride [
70]. In addition, the molar ratio used in the synthesis of Ce-MCM-41 materials presented a concentration of nitrate or chloride anions close to CTAB, exhibiting 1TEOS:0.04Ce:0.12A:0.152CTAB:2.8NH
4OH:141.2H
2O, where A = NO
3− or Cl
−. Since NO
3− ions have a higher ionic exchange constant on CTAB micelles than Cl
− [
71], they are expected to present a stronger binding to CTA
+ rods. For Ce-MCM-NO
3, nitrate ions bonded to CTA
+ could also be hydrogen bonded to [Ce(OH)
3]. This layer of concentrated cerium(III) species will aggregate during calcination to form non-framework CeO
2 species on the surface of the MCM-41 structure. The higher disordering level observed for Ce-MCM-Cl can be associated to a higher degree of framework Ce-O-Si due to the lower binding between chloride ions and CTA
+ molecules. Thus, the physicochemical differences among both cerium modified samples may arise fundamentally from an anion effect in the disturbance of CTA
+ molecule assemblies, especially the formation of different cerium species in the calcined materials.
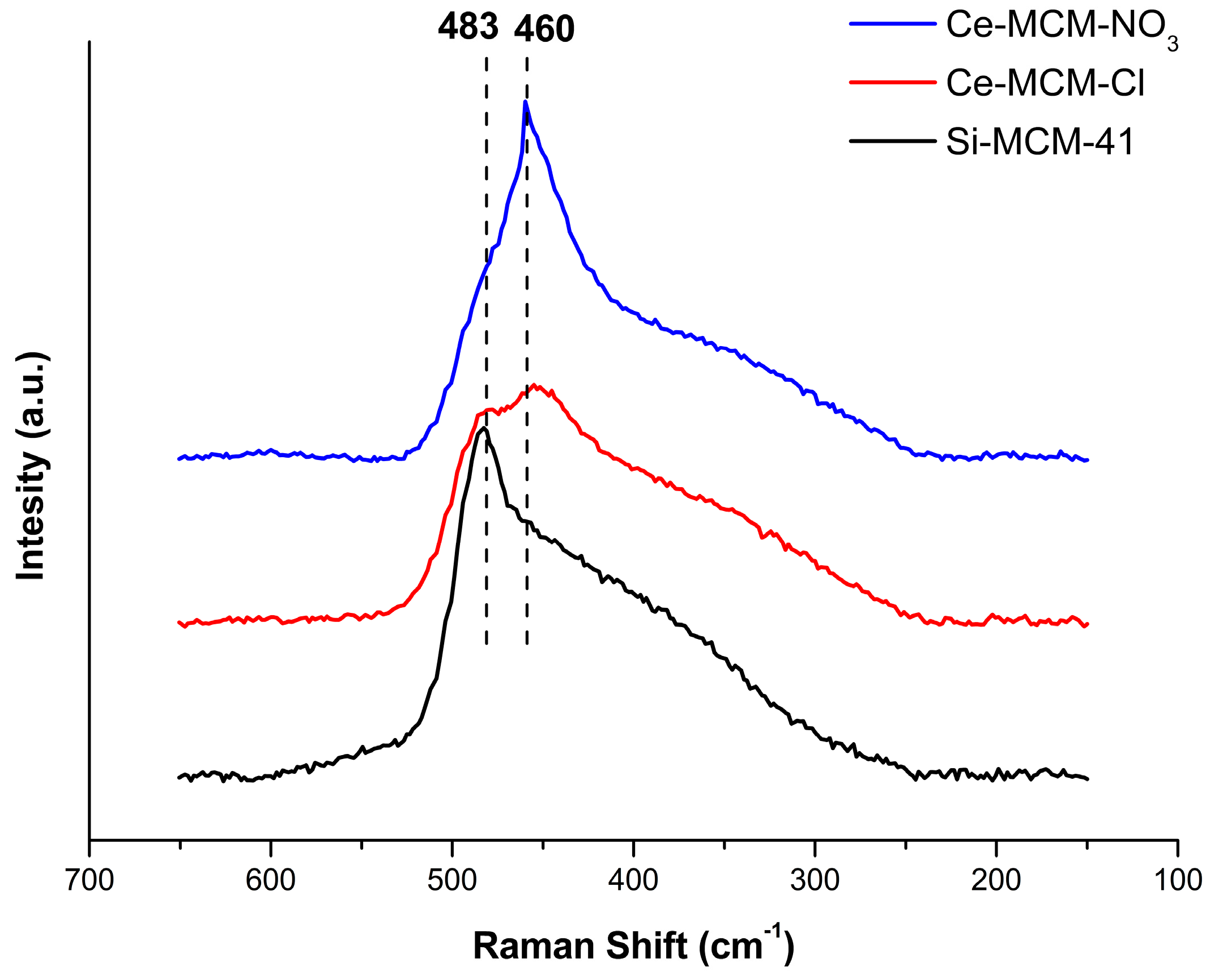
Further effects in the formation of cerium species can be perceived in the calcined materials.
Figure 4 displays the Raman spectra of the calcined materials between 650 and 150 cm
−1. In general, they all present signals common to the amorphous nature of the silica present in MCM-41. At the studied range, the Si-MCM-41 spectrum presents a signal at 483 cm
−1, typical of the stretching mode of Si-O bonds for fourfold cyclosiloxane groups [
72,
73,
74]. For Ce-MCM-Cl, a weak peak appears aside the cyclosiloxane group mode at 460 cm
−1 and this signal becomes dominant over the 483 cm
−1 mode in the spectrum of Ce-MCM-NO
3. This feature is commonly attributed in the literature to the symmetric stretching of Ce-O bonds of the fluorite crystal structure of CeO
2 [
75] and was assigned as the
F2g mode of CeO
2. The spectrum of crystalline CeO
2 was obtained for means of comparison (
Figure S3 in Supplementary Material) and the
F2g mode presents itself as a strong signal centered in 463 cm
−1. In the case of the modified materials, the presence of this extra signal in both samples indicates the existence of non-framework cerium and that Ce-MCM-NO
3 possess a higher content of these species due to a much stronger intensity for this vibrational mode, which is in agreement with the XRD data presented earlier. A similar observation in the
F2g mode appearance was made by Zhan and co-workers [
48] for Ce-MCM-48 materials, where the mode increased in intensity with the increase of cerium content. However, such a difference between two cerium-modified MCM-41 materials with similar Si/Ce ratios as it is presented in this work has not been observed in the literature so far.
Correlating with the information extracted for the as-synthesized materials, it is possible to ascertain the mechanism of crystallization proposed earlier. Thus, the disordering observed in the assembly of CTA
+ molecules of Ce-MCM-Cl was driven by isomorphic substitution. As for Ce-MCM-NO
3, part of the cerium added in the synthetic medium does not react with silicate oligomers and precipitates to form non-framework [Ce(OH)
3], which converts completely to CeO
2 after calcination. Such a hypothesis is scarcely pointed out to explain the difference in the reactivity of cerium precursors in the synthesis of oxides, but there is an extensive literature regarding the differences in the use of precursors that leads to singular microstructural properties of these materials [
52,
76,
77]. These works clearly suggest that the anions in cerium precursors have major roles in defining the properties of the final cerium-based materials more than just the mere electrostatic counterparts in the synthetic medium.
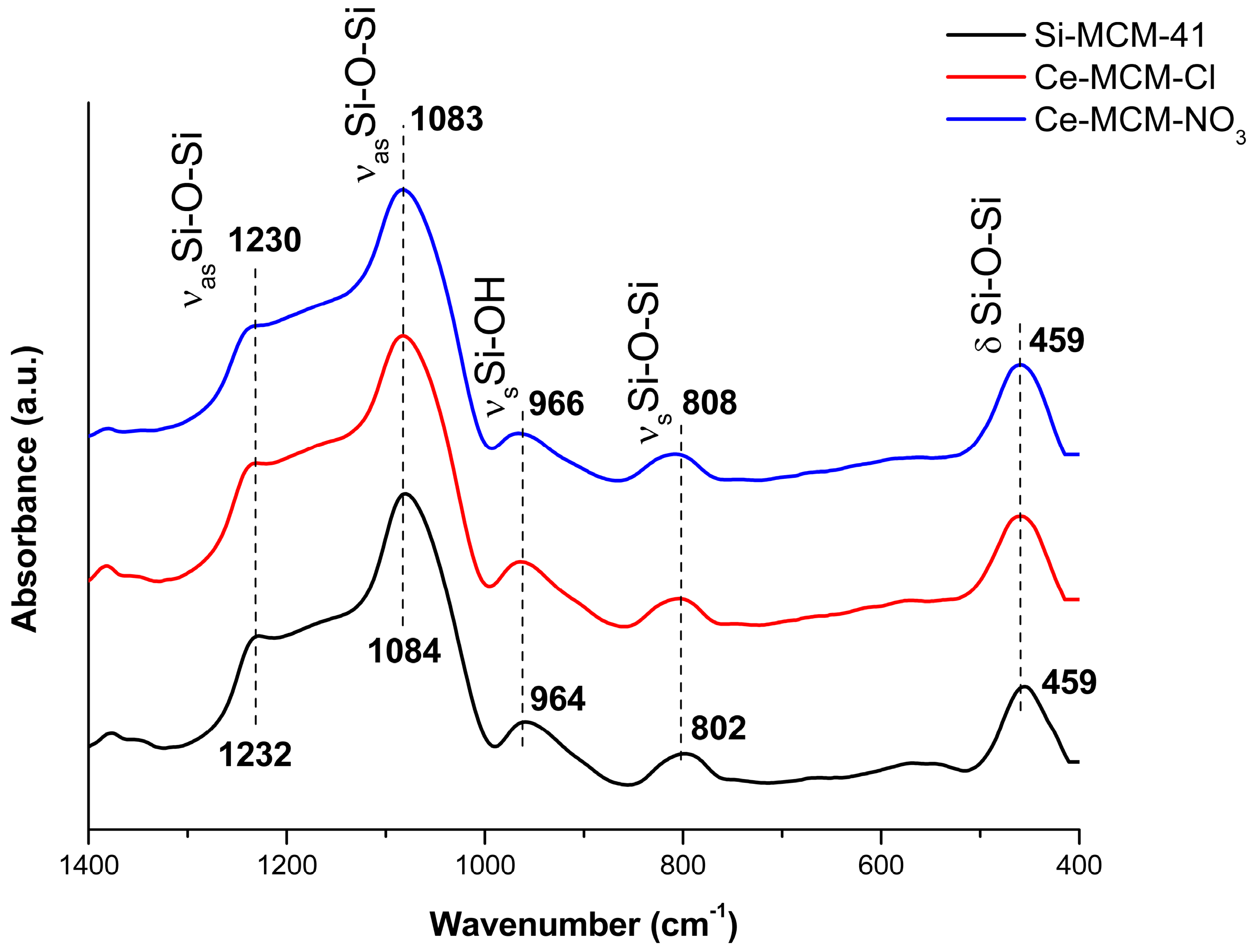
Figure 5 displays the Fourier transform infrared spectroscopy (FTIR) spectra for all calcined MCM-41 materials.
The vibrational modes between 1400 and 400 cm
−1 are typical of Si-O-Si bonds [
78]. In the case of Si-MCM-41: (i) Asymmetric stretching of Si-O-Si bonds at 1232 and 1084 cm
−1; (ii) symmetric stretching of Si-OH bonds at 964 cm
−1; (iii) symmetric stretching of Si-O-Si at 802 cm
−1; and (iv) the Si-O-Si bending mode at 459 cm
−1. Changes in the wavenumber of these modes are frequently associated to isomorphic substitution of heteroelements in the framework of silica-based materials, because the average length of T-O bonds (T corresponds to any heteroatom) in the walls of the MCM-41 is increased with the insertion of a larger atom than silicon [
37,
49,
51]. However, the shifts observed for the cerium-incorporated materials were too small to make any assumption of isomorphic substitution. In addition, no vibrational modes attributed to Ce-O bonds, between 1600 and 900 cm
−1 and below 700 cm
−1, were evidenced. These observations (i.e., small shifts and absence of Ce-O bonds) could be respectively attributed to the low concentration of framework Ce
4+ ions and non-framework CeO
2 when compared to the amount of silica in MCM-41.
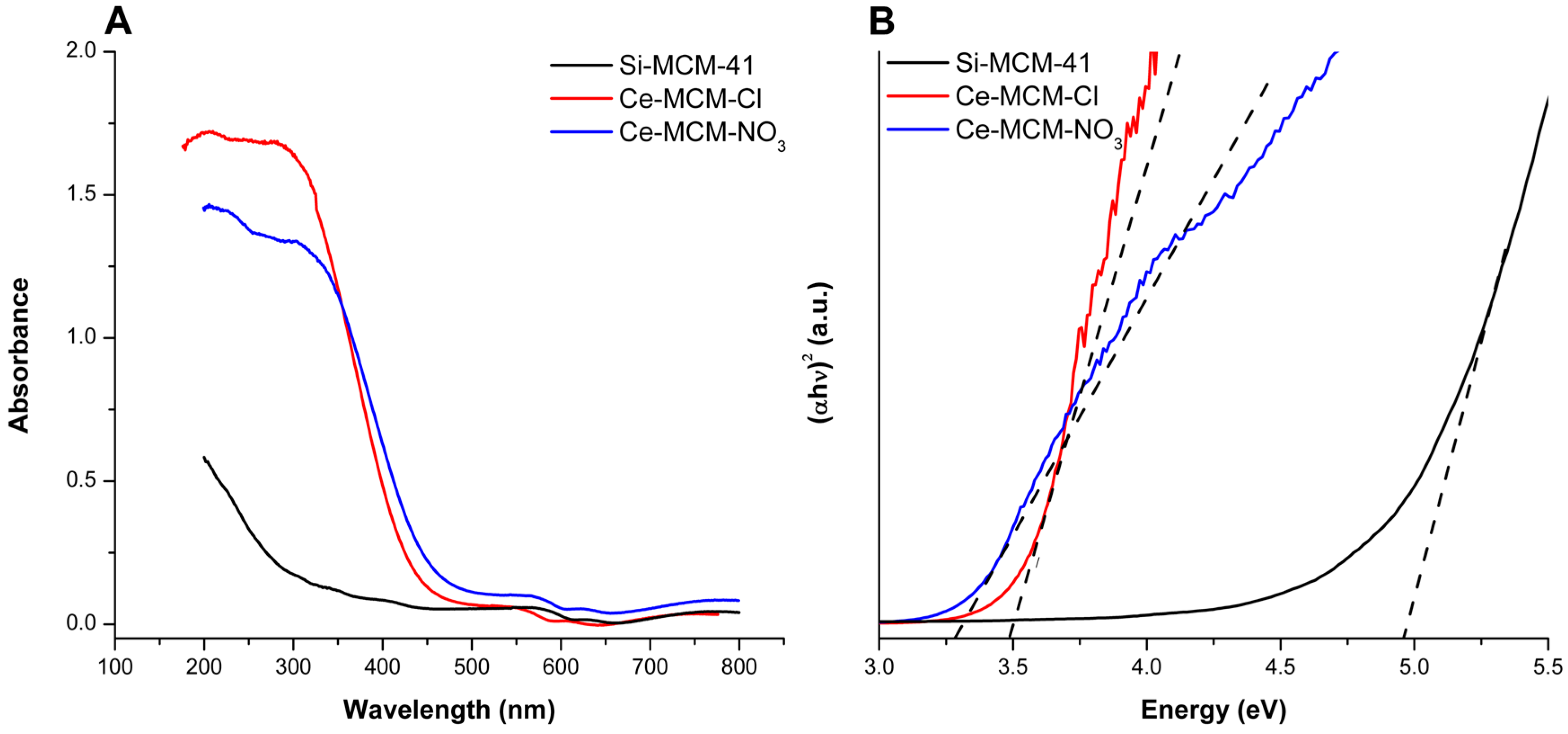
Diffuse reflectance UV-Vis (DRS UV-Vis) spectra for the calcined materials are displayed in
Figure 6A. When compared to Si-MCM-41, cerium-incorporated materials presented stronger absorption in the ultraviolet range and a red shift of the absorption edge due to O
2−→Ce
4+ charge transfer [
32]. These experimental observations are proof of cerium incorporation in the materials, as already described by Pal and co-workers [
32]. Moreover, the greater intensity of the Ce-MCM-Cl spectrum can be due to its slightly higher cerium content (see
Table 1).
When compared to a standard sample of CeO
2 (
Figure S4 in Supplementary Material), it is possible to gain information towards the coordination environment of the heteroatom, since the charge transfer transition is symmetry-sensible [
37,
51,
79]. The CeO
2 spectrum presented more intense bands at 380 and 250 nm, owing to Ce
4+ ions in octahedral/polymeric and tetrahedral coordination environments, respectively. The octahedral/polymeric environment is the main coordination type for cerium in the CeO
2 fluorite crystal structure. Its non-stoichiometric lattice [
80] gives the dioxide a high oxygen mobility and absorptions near or above 380 nm is strong evidence of non-framework species. The spectra of cerium-incorporated materials in
Figure 6A showed that UV-absorption finishes around 310 nm for Ce-MCM-Cl with a steep absorption edge until 450 nm, while for Ce-MCM-NO
3, it ends at 355 nm with a less steep edge until 500 nm. These results are evidence that the heteroatoms in Ce-MCM-Cl have more tetrahedral coordination sites than Ce-MCM-NO
3, which absorbs more radiation in the visible range of the spectrum due to non-framework species [
37,
51,
79].
The different positions found for the absorption edge in Ce-MCM-41 samples can be used to obtain important information towards the heteroatom by determining the optical band gap. The latter was calculated by Equation (3) (see Materials and Methods) using n = ½, due to direct allowed transitions, and Davis-Mott plots with suitable linear ranges were obtained (R
2 > 0.99). CeO
2 has an optical band gap of 3.02 eV, which is near to the values described in the literature for the bulk material [
81]. Ce-MCM-Cl and Ce-MCM-NO
3 showed band gap values of 3.50 and 3.33 eV, respectively, which were lower than Si-MCM-41 (4.97 eV). As studied by Casas-Orozco and co-workers [
73], higher band gap values of metal-incorporated MCM-41 than the metal oxide counterpart indicates high dispersion of the heteroatom in the siliceous framework. Also, the lower band gap value observed for Ce-MCM-NO
3 could be associated with the higher content of non-framework species identified by XRD and Raman, resulting in less dispersed CeO
2 particles.
29Si MAS-NMR spectra of the calcined materials are available in
Figure 7. As can be seen, the signals obtained for all samples were broad and overlapped in the range between −80 and −120 ppm. This interval is typical for the Q
n-groups with a general formula of Si(OSi)
n(OH)
4−n [
82]: (i) Geminal silanols or Q
2 groups (resonating around −90 ppm); (ii) terminal silanols or Q
3 groups (resonating around −100 ppm); and (iii) siloxane or Q
4 groups (resonating around −110 ppm). The overlapping effect is typical in systems based on amorphous silica due to the wide distribution of Si-O-Si bond angles and Si-O bond lengths [
82]. It was possible to perceive that, in the case of Ce-MCM-Cl, there was a slight diminishment in the signal of Q
3 groups, indicating a decrease in its content. This type of change was even more noticeable for Ce-MCM-NO
3, where all defect-related environments (Q
2 and Q
3) were significantly reduced when compared to Si-MCM-41. Such qualitative observations indicate that the cerium incorporation is modifying the condensation of Si-O-Si bonds in the structure of these materials. To access more information towards the latter, deconvolution methods were applied and are denoted in
Figure 7.
No additional peaks to those mentioned earlier were found after applying the deconvolution methods. Therefore, the traditional parameters associated to the condensation of Si-O-Si bonds were applied to further analyze the changes perceived for cerium modified materials. Among them, the percentage of each siliceous chemical environment was calculated based on the area of each peak and they were used to determine
Q4/(
Q2 +
Q3) ratios and the molar percentage of silanol groups (Si molar%) using Equation (4) (see Materials and Methods). Higher values for the mentioned ratio and lower ones for Si molar% indicates more condensed Si-O-Si bonds in the inorganic framework. These quantitative data were compiled in
Table 3.
From the values displayed in
Table 3, it was possible to infer that: (i) For Ce-MCM-Cl, even though the content of terminal silanols (Q
3) decreases, the siloxane group (Q
4) does not increase significantly when compared to geminal silanols (Q
2), suggesting that even if the condensation of Si-O-Si bonds slightly increases, the percentage of silanol groups (Si molar%) remains relatively constant if Q
2 increases; and (ii) for Ce-MCM-NO
3, the modification decreases drastically the content of silanol groups, indicating that the non-framework cerium species might be located in the mesopores’ wall or external surface, but Raman data suggests the former. Once again, there is little contribution from the literature to explain effectively the reason for differences in
29Si MAS NMR arising solely with varying the counterion in cerium precursor. Even though this is a scarce topic, it is well known that such effects in siliceous molecular sieves are dependent more in the synthetic procedure than in any other aspect and the fact that cerium incorporation can lead to a more or less condensed degree of chemical bonds has already been reported [
32,
51]. Thus, the MAS-NMR data is in good agreement with the observations made earlier due to the crystallization mechanism proposed: (i) The smaller aggregation effect of chloride anions on CTA
+ assemblies and the formation of framework Ce-O-Si bonds led to the emergence of silanol defects (Q
2); and (ii) the precipitation of Ce(OH)
3 and conversion to non-framework CeO
2 resulted in a coating effect in the MCM-41 surface, leading to the decrease of Q
2.
2.1.3. Morphological (TEM and HRTEM), Textural (Surface Area and Pore Volume), and Thermal Analyses (TG/DTG/DSC)
The morphology of the calcined MCM-41 materials was accessed through low-resolution transmission electron microscopy (TEM). Representative images are selected and displayed in
Figure 8.
For Si-MCM-41, perfectly spherical nanoparticles were obtained (mean size of 61.8 ± 15.9 nm). There is little aggregation between the particles, possibly a synthetic consequence of the rapid formation of ethanol as a side product during TEOS hydrolysis [
83]. On the other hand, the modified materials present smaller and spherical nanoparticles, with a mean size of around 34.2 ± 14.5 nm and 38.7 ± 9.1 nm for Ce-MCM-Cl and Ce-MCM-NO
3, respectively. Also, the nanoparticles have a more aggregated aspect when compared with Si-MCM-41, exhibiting more irregular shapes. These observations are possibly an evidence of a faster nucleation process, a consequence of the presence of cerium precursors in the synthetic medium. This supports the loss of long-range ordering observed previously by XRD, as well as the disordering effect of the supramolecular assembly of CTA
+ cations evidenced by Raman spectroscopy.
To further study the modified materials, high-resolution transmission electron microscopy (HRTEM) was used to access the formation of CeO
2 species on Ce-MCM-41 materials and representative images are displayed in
Figure 9. As one can see, some of the irregular entities observed in TEM for both materials showed dark nanorods on their surface that were not observable on Si-MCM-41. High magnifications of these nanorods (
Figure S5 in Supplementary Material) displayed lattices fringes around 0.33 nm, consistent with the interplanar distance characteristic to 110 XRD reflection of CeO
2, thus confirming their identity. For Ce-MCM-Cl, these nanorods displayed lengths between 22.0 and 70.0 nm and widths between 1.75 and 2.50 nm, while for Ce-MCM-NO
3, these entities were bigger and thicker, with lengths between 100 and 250 nm and widths between 5.0 and 6.7 nm. The differences observed in their measures were consistent with the crystallization mechanism proposed for non-framework cerium species, where longer and thicker rods were expected for Ce-MCM-NO
3 due to the aggregation effect of micelles attributed to the nitrate anion. Also, the formation of these nanorods was consistent with the mechanism proposed for non-framework cerium species. The formation of ceria nanorods is widely studied in the literature [
84,
85] and includes surfactant-assisted synthetic procedures using CTAB. However, these methodologies are normally performed under hydrothermal conditions with high temperatures (80–160 °C), longer time periods (8–96 h), and low Ce/CTAB molar ratios (0.33–0.66) [
84,
85]. The results observed in this work suggest that nanorods can also form under milder conditions (30–35 °C and 4 h) and high Ce/CTAB molar ratios (3.8).
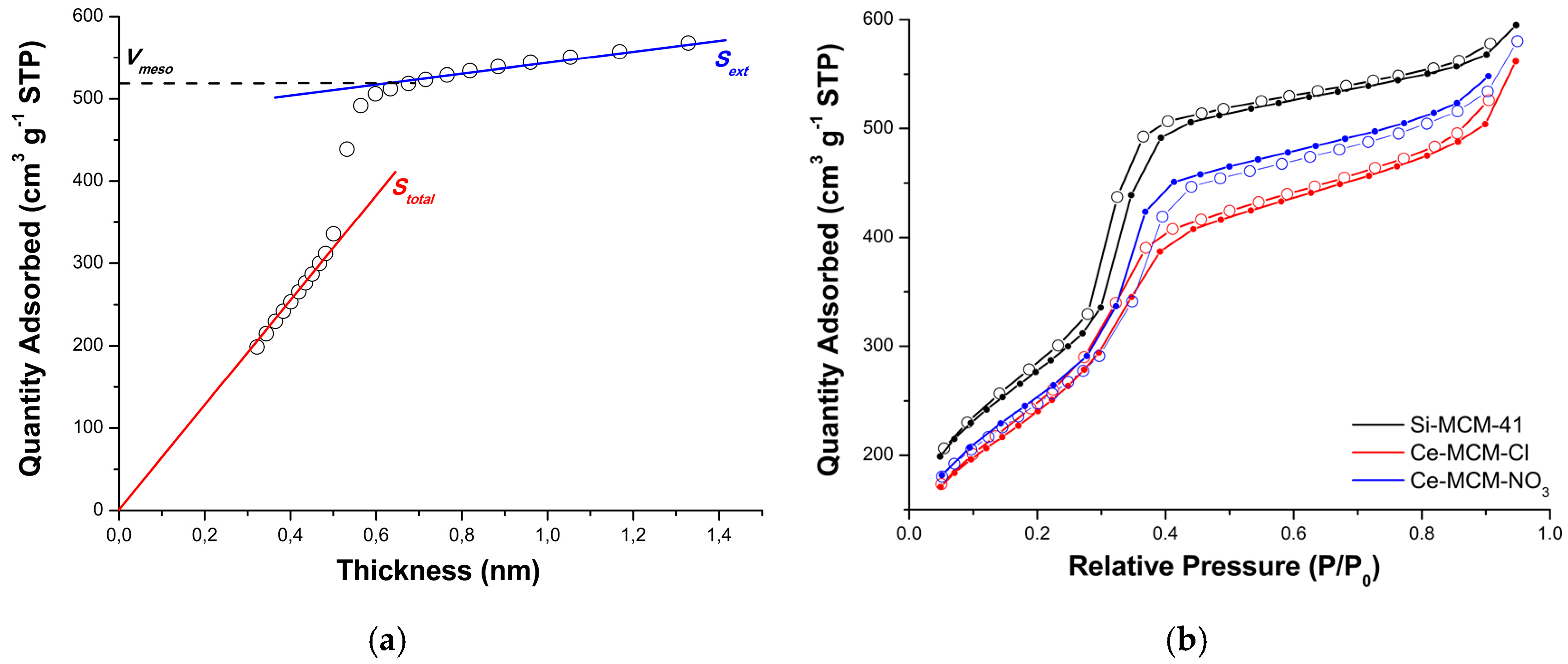
The textural characterization of the calcined MCM-41 materials was performed by N
2 physisorption using t-plot curves obtained from analytical functions for each portion of the
P/
P0 domain (see Equations (5)–(8) in Materials and Methods) [
86]. The initial part of the curve was used to calculate the total specific surface area (
Stotal) of the materials using the slope of the linear fit going through the origin (red line in
Figure 10a). On the other hand, the final part of the curve was used to calculate the external specific surface area (
Sext), indicated in
Figure 10a by the blue line.
Figure 10b exhibits the N
2 adsorption/desorption isotherms obtained for Si-MCM-41, Ce-MCM-Cl, and Ce-MCM-NO
3, respectively. According to the IUPAC classification [
87], all curves were type IV isotherms, presenting a characteristic hysteresis loop associated with capillary condensation in mesopores. This hysteresis was type H1 according to the IUPAC classification [
87] and could be associated with a narrow and uniform pore size distribution. This result implies that the MCM-41 pore system in cerium-incorporated materials was preserved and that the long-range disorder was responsible for the loss of the mesophase observed by XRD. This is not uncommon and even in highly disordered materials, such as titanosilicate ETS-10, short-range ordering could not be affected by long-range disordering [
88].
The initial part of the isotherm, before capillary condensation, is associated to monolayer-multilayer adsorption on both mesopores and external surface, and consequently, was used to calculate
Stotal, as described above [
86]. Similarly, in the final part of the isotherm, after capillary condensation, adsorption occurs only on the external surface and, for that reason, was used to determine
Sext. The capillary condensation is identified by an increase of adsorbed N
2 at the relative pressure range of P/P
0 = 0.30 to 0.45, due to the filling of mesopores.
The data obtained by N
2 physisorption experiments (
Table 4) showed a reduction of
Stotal, mesoporous surface area (
Smeso), and mesopore volume (
Vmeso) for the cerium-incorporated samples, which could be attributed to the loss of long-range order. The sharp increase observed in all isotherms at the relative pressure range of P/P
0 = 0.9 to 1.0 is related to the presence of interparticle porosity [
32] and its effect can be observed in a higher value of the total pore volume (
Vtotal) than
Vmeso. For that reason, the average pore diameter (
PD) was calculated using the penultimate point from the adsorption isotherm (see Materials and Methods) to avoid contributions from interparticle porosities, since the materials showed different particle sizes. The values obtained for
PD were almost identical, which is in agreement with the narrow and uniform pore size distribution expected for the type H1 hysteresis loop observed for all materials and discussed above. However, the sample of Ce-MCM-NO
3 showed a small increase that could be attributed to its different mechanism of pore formation, possibly due to the coating effect of non-framework CeO
2. Additionally,
PD values were used to determine the wall thickness (
WT) of MCM-41 materials, evidencing that cerium incorporation affects this parameter due to framework cerium and non-framework CeO
2. In the case of Ce-MCM-Cl, the increase in
WT was consistent with the incorporation of Ce in the inorganic framework, causing an expansion of the unit cell (
a) and, ultimately, of the pore walls. However, in the case of Ce-MCM-NO
3, not only isomorphic substitution could cause such an increase, but also the existence of non-framework CeO
2 and the higher value of
WT corroborates the possible coating effect mentioned previously, which can make the pore walls appear thicker. These results are in accordance with the higher value of
a found for Ce-MCM-NO
3 (4.97 nm) when compared to Ce-MCM-Cl (4.61 nm), explaining the cell expansion observed by XRD. Finally, the higher
Sext value obtained for cerium-incorporated materials are linked to their smaller particle size when compared to Si-MCM-41, as observed by TEM.
Thermal analyses were performed for the as-synthesized and calcined MCM-41 materials to further analyze the consequences of texture changes towards microstructural aspects. The thermogravimetry (TG), differential thermogravimetry (DTG), and differential scanning calorimetry (DSC) curves for the as-synthesized materials are displayed in
Figure 11,
Figure 12 and
Figure 13. The values of mass loss, temperature range, and DTG maximum were collected in
Table S2 (see
Supplementary Material).
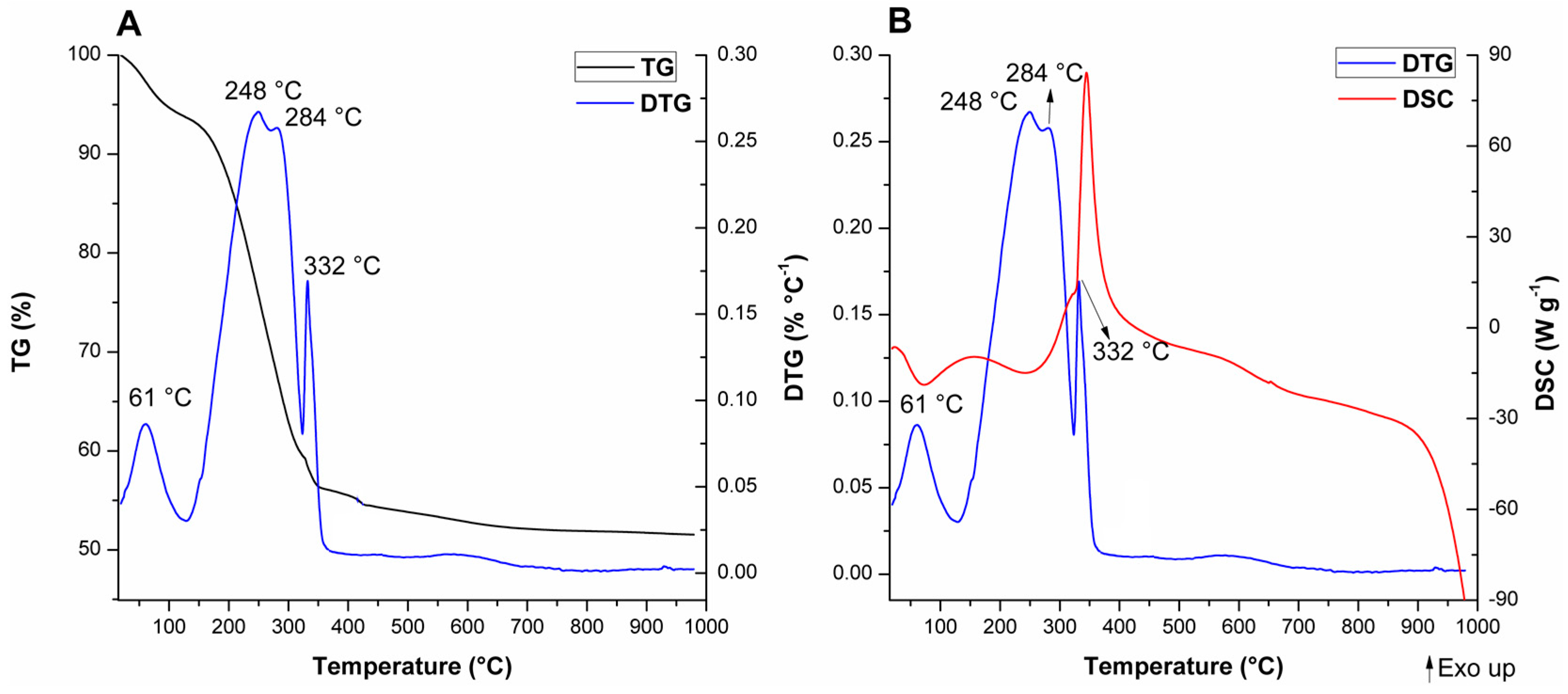
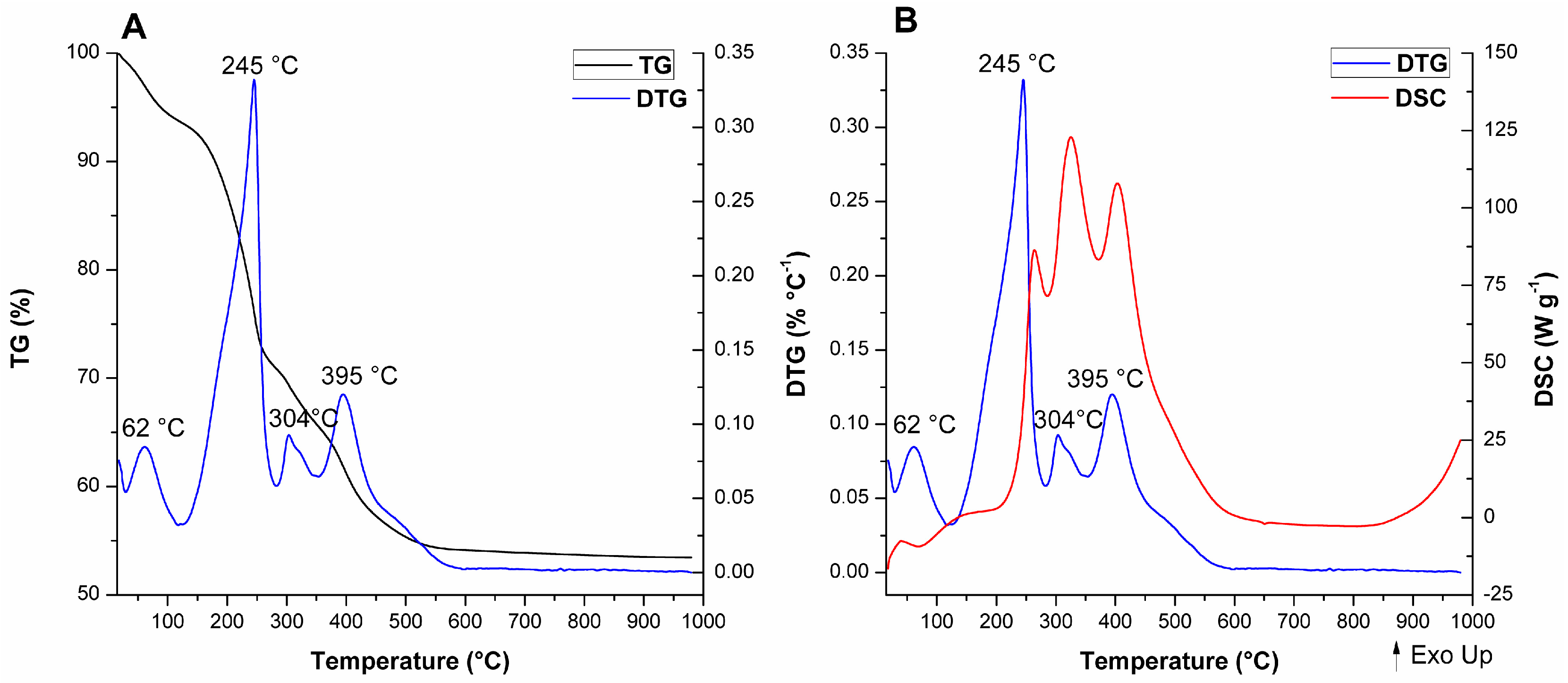
For A-Si-MCM-41, five thermal events were observed and are consistent with previously reported events for similar as-synthesized MCM-41 materials [
89]: (i) Evaporation of physically adsorbed water (endothermic, from room temperature to 129 °C); (ii) initial decomposition of occluded CTA
+ molecules through Hofmann elimination (endothermic, from 129 to 270 °C), generating triethylamine and alkenes; (iii) further fragmentation of carbon chains that resisted Hofmann elimination (exothermic; from 270 to 322 °C); (iv) oxidation of reminiscent organic residues from previous thermal events, like coke (exothermic; from 322 to 482 °C); and (v) condensation of silanol groups (with almost undetectable DTG and DSC maximums, from 482 to 1000 °C).
The comparison between the modified as-synthesized materials and A-Si-MCM-41 showed differences in the DTG and DSC curves, even though the TG curves display similar profiles and CTA
+/SiO
2 ratios. The different thermal behavior for both cerium incorporated materials, especially after the Hofmann elimination, revealed that: (i) In the temperature range associated to CTA
+ decomposition, one extra endothermic event occurs at 304 °C for A-Ce-MCM-Cl and 309 °C for A-Ce-MCM-NO
3; (ii) these extra events were overlapped with the first oxidation process, now shifted in the range of 315 to 360 °C; (iii) increase in the temperature of the DTG maximum for the final oxidation process, occurring around 70 °C higher than for A-Si-MCM-41; and (iv) increase in mass loss for the final oxidation processes, 5.59% for A-Si-MCM-41 and above 8.50% for both A-Ce-MCM-41 samples. Overall, this evidence demonstrates clearly that template molecules in the modified materials are thermally more resistant than in the as-synthesized Si-MCM-41. The extra thermal event might be due to an extra Hofmann elimination of molecules that survived the first decomposition process, delaying both exothermic events. Such phenomena can be either related to an increase of the interaction stability between CTA
+ and the inorganic framework or to diffusion limitations of products from thermal processes, since there was a decrease in
Vmeso after cerium incorporation. Similar observations have been made by Khalil [
55] and de Souza and co-workers [
90], especially as further evidence of isomorphic substitution of heteroelements in the inorganic framework.
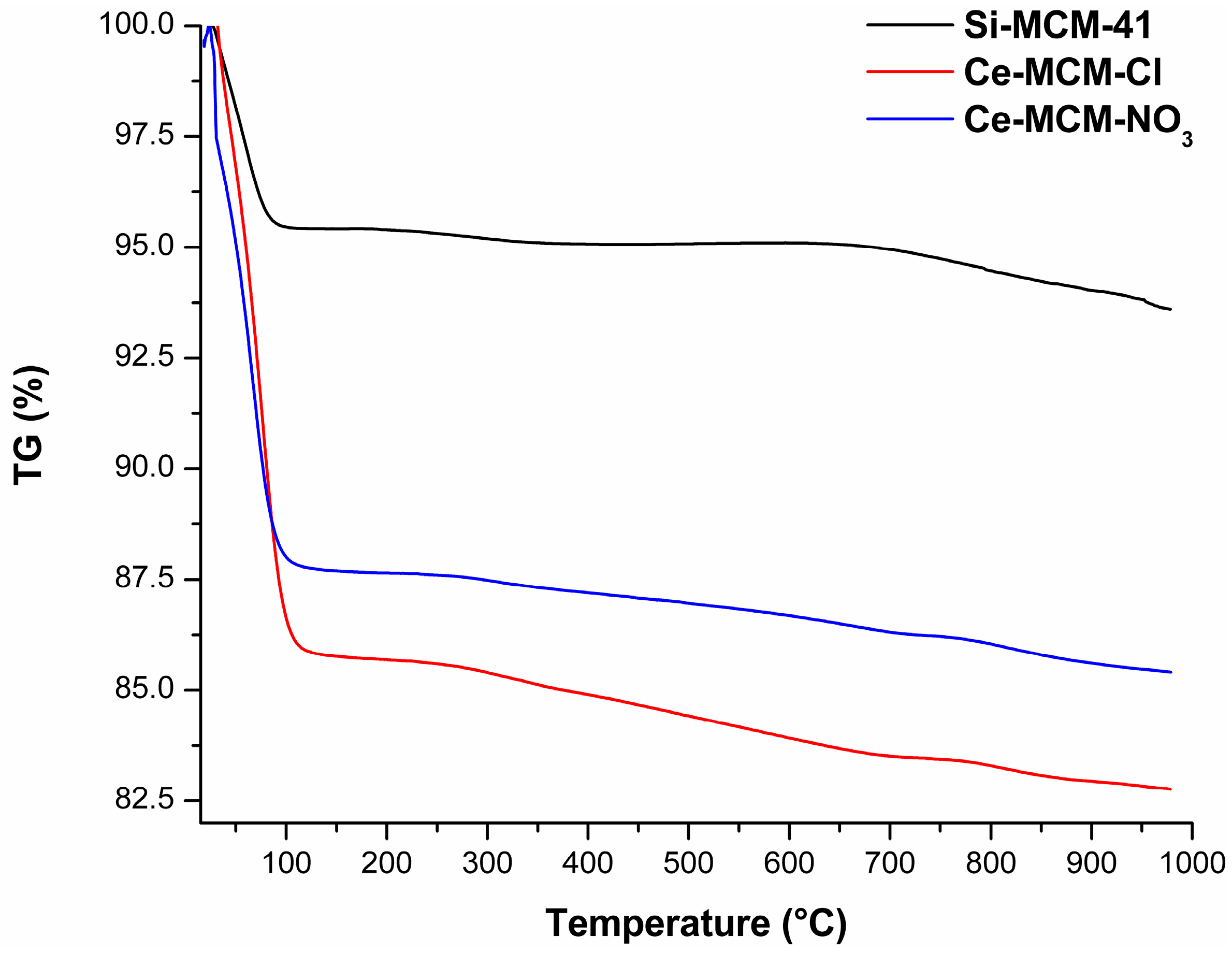
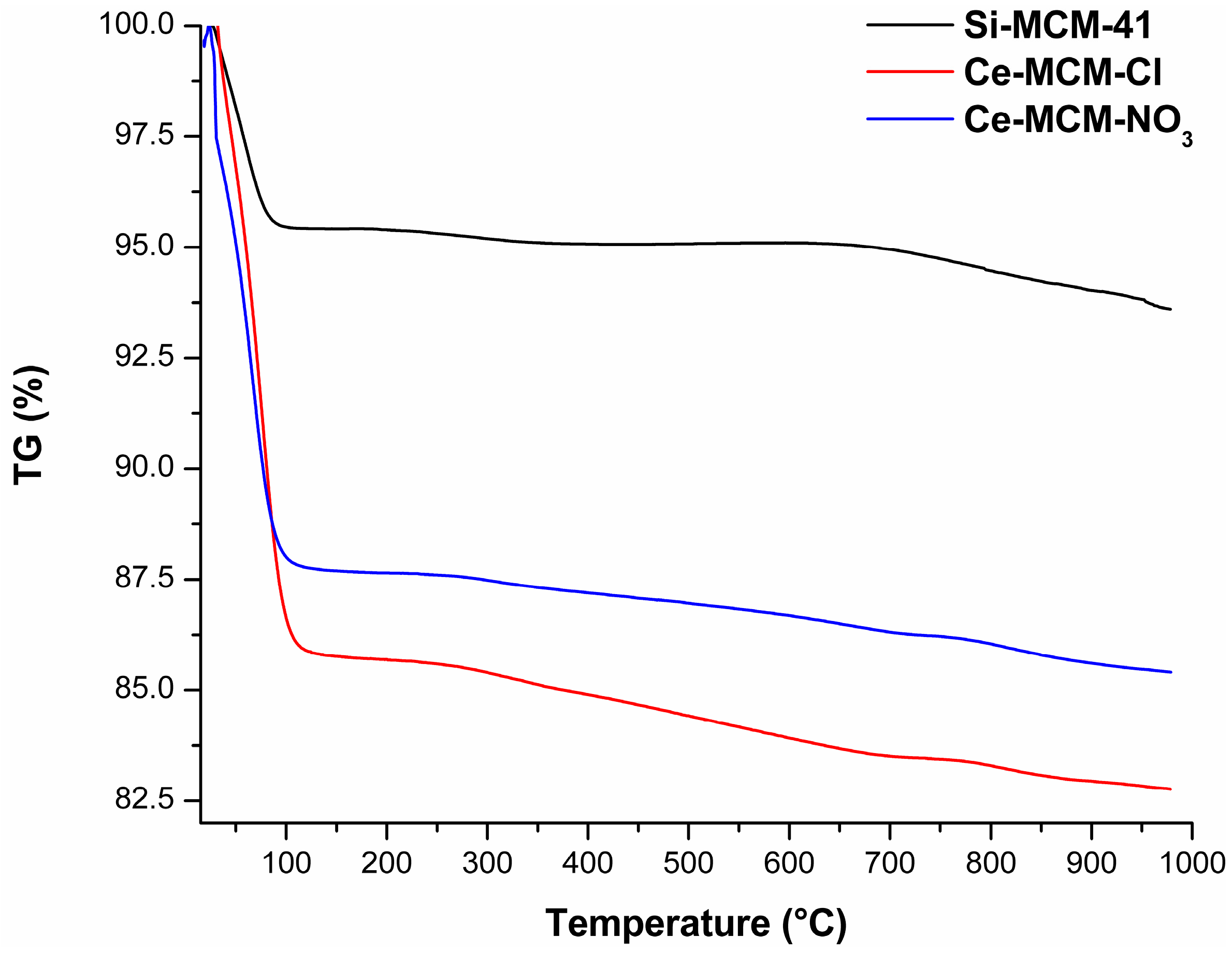
The TG curves of the calcined MCM-41 materials (
Figure 14) and the mass loss associated to all processes (
Table S3) are displayed in the
Supplementary Material. These curves provided significant information to differentiate the materials among themselves, where a difference in total mass loss from room temperature to 1000 °C was observed in all solids. The two thermal events were associated to the evaporation of adsorbed water (endothermic event from room temperature to temperatures around 115 to 150 °C) and the condensation of silanol groups (water evaporation until 1000 °C). The major mass loss was associated to the initial evaporation of water: 4.58%, 14.23%, and 12.29% for Si-MCM-41, Ce-MCM-Cl, and Ce-MCM-NO
3, respectively. Also, this event ends in higher temperatures for the cerium-modified samples than for Si-MCM-41 (usually above 130 °C). Altogether, these observations indicate that cerium incorporation increases the surface polarity of the materials, somewhat expected since cerium is less electronegative than silicon. Also, the mass loss from silanol condensation was different for the three materials: 1.70%, 3.50%, and 2.67% for Si-MCM-41, Ce-MCM-Cl, and Ce-MCM-NO
3, respectively. Thus, it is noticeable that the isomorphic substitution leads to less thermally stable materials, highlighting the fact that Ce-MCM-Cl is less stable than Ce-MCM-NO
3, evidence of its higher degree of silicon substitution in the framework. This is expected once the insertion of heteroelements causes bond angle tension and loss of the crystalline mesophase as indicated by XRD data, which is in accordance with the disordering effect proposed for the crystallization of the materials and corroborated previously with other characterization techniques.

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}