Iridium-Catalyzed Transfer Hydrogenation of Ketones and Aldehydes Using Glucose as a Sustainable Hydrogen Donor



Abstract
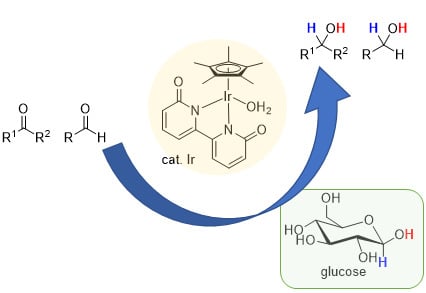
:
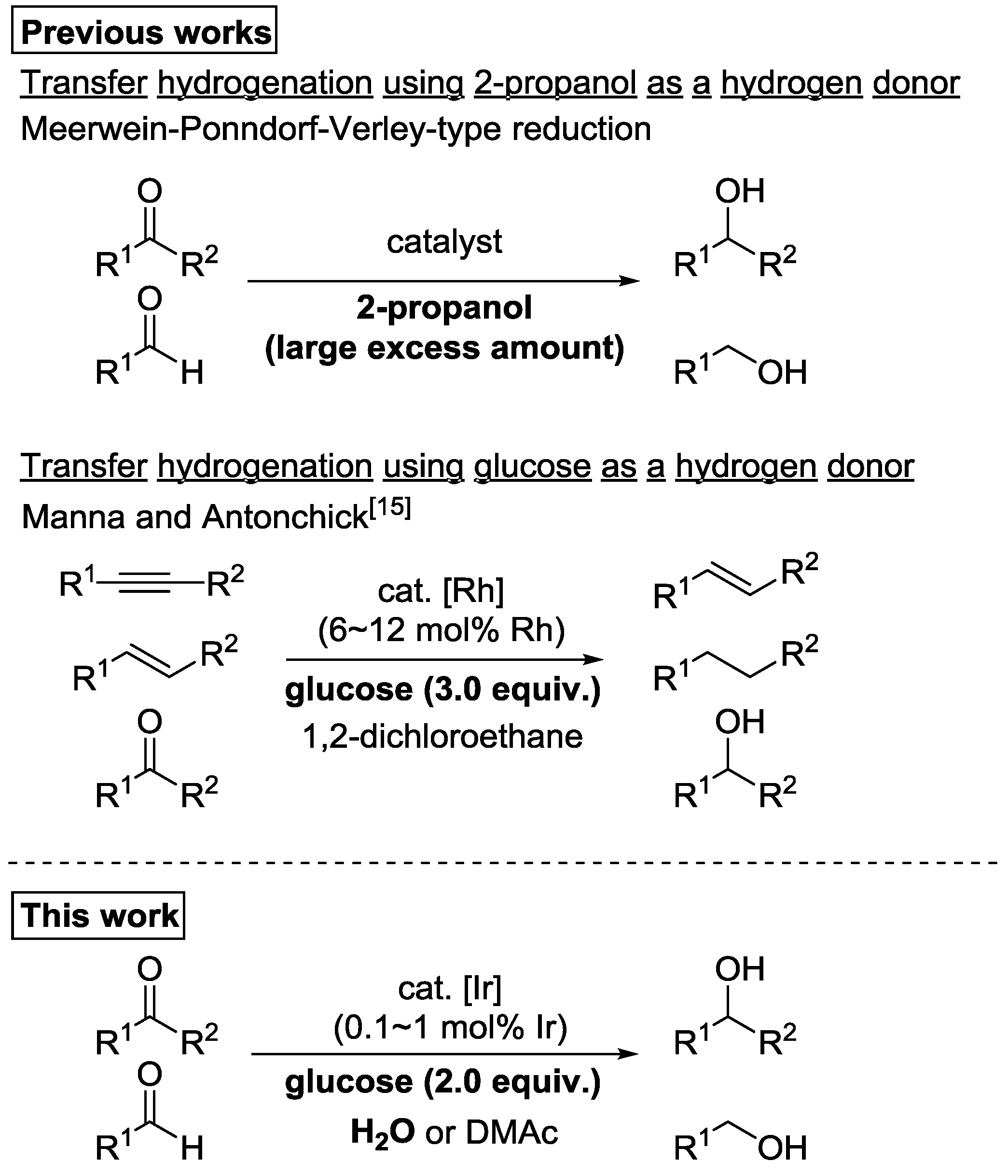
1. Introduction
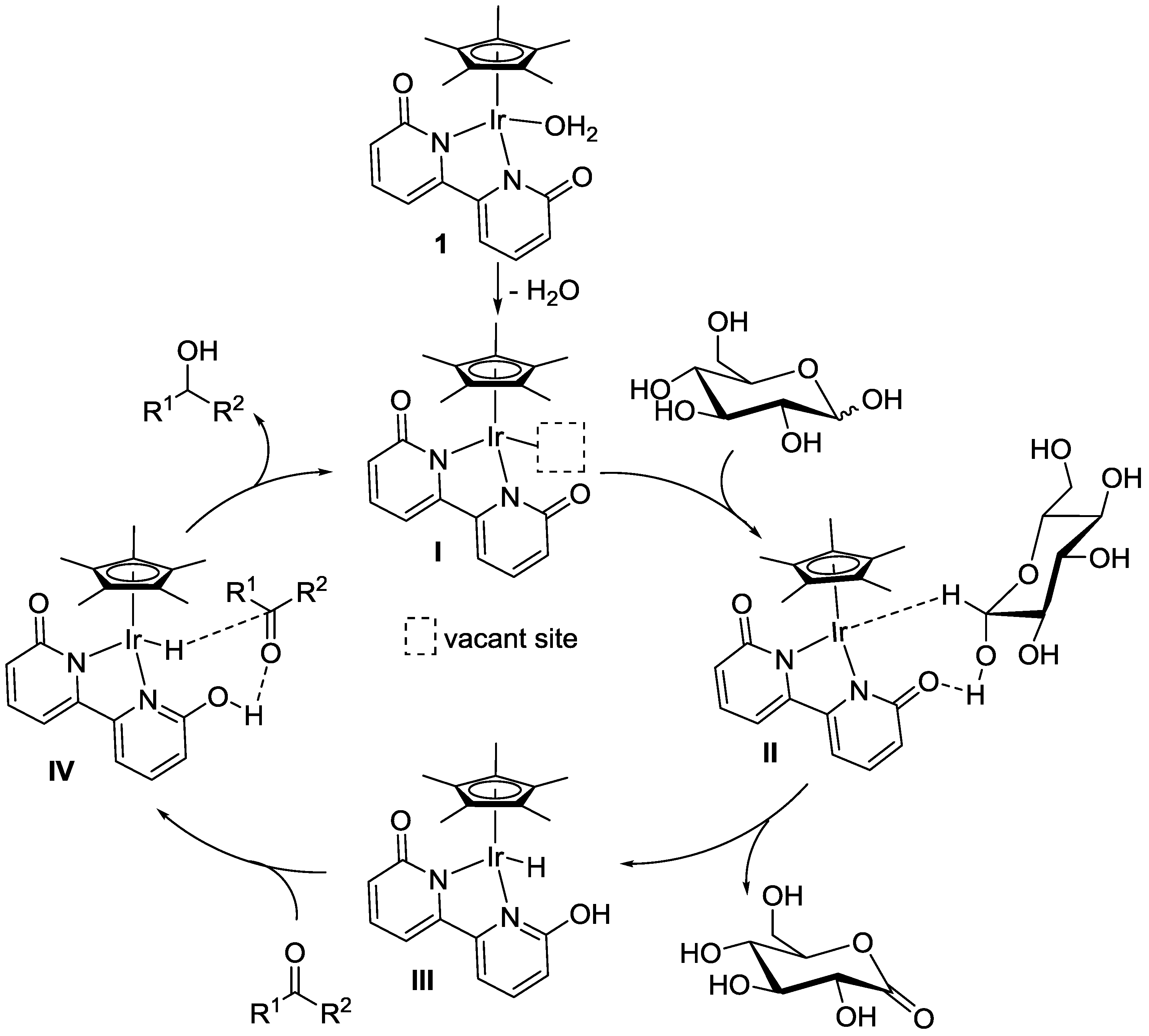
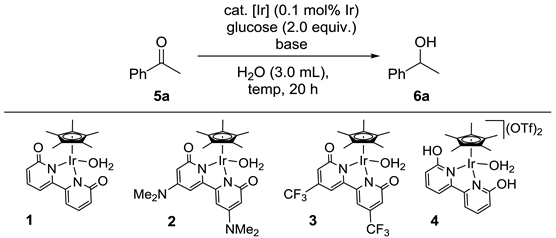
2. Results
3. Materials and Methods
3.1. General
3.2. General Procedure for Transfer Hydrogenation of Acetophenone to 1-phenylethanol Using Glucose (Table 1)
3.3. General Procedure for Transfer Hydrogenation of Ketones to the Corresponding Secondary Alcohols Using Glucose (Table 2)
3.3.1. Conditions A
3.3.2. Conditions B
3.4. General Procedure for Transfer Hydrogenation of Aldehydes to the Corresponding Alcohols Using Glucose (Table 3)
3.4.1. Conditions A
3.4.2. Conditions B
3.5. Preparation of 2,3,4,6-tetra-O-methyl-D-glucopyranose (10). (Equation 2)
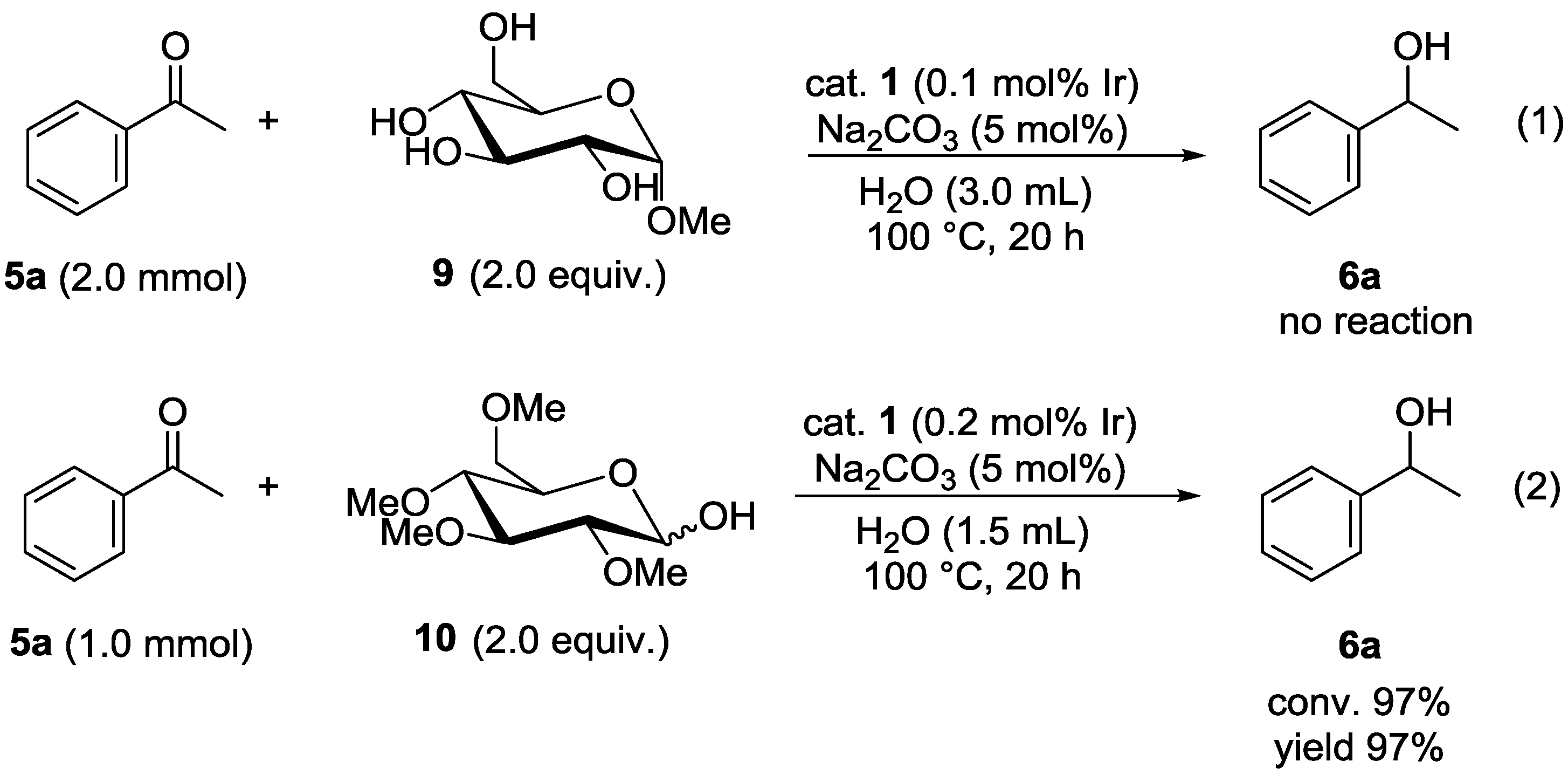
3.6. Reaction of Acetophenone Using α-D-glucopyranoside (9) (Equation 1)
3.7. Reaction of Acetophenone Using 2,3,4,6-tetra-O-methyl-D-glucopyranose (10) (Equation 2)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dupau, P. Ruthenium-Catalyzed Selective Hydrogenation for Flavor and Fragrance Applications. In Organometallics as Catalysts in the Fine Chemical Industry; Beller, M., Blaser, H.-U., Eds.; Topics in Organometallic Chemistry; Springer: Berlin/Heidelberg, Germany, 2012; pp. 47–63. ISBN 978-3-642-32833-6. [Google Scholar]
- Štefane, B.; Požgan, F. Metal-Catalysed Transfer Hydrogenation of Ketones. Top. Curr. Chem. 2016, 374, 1–67. [Google Scholar]
- Brieger, G.; Nestrick, T.J. Catalytic transfer hydrogenation. Chem. Rev. 1974, 74, 567–580. [Google Scholar] [CrossRef]
- Wang, D.; Astruc, D. The Golden Age of Transfer Hydrogenation. Chem. Rev. 2015, 115, 6621–6686. [Google Scholar] [CrossRef] [PubMed]
- Meerwein, H.; Schmidt, R. Ein neues Verfahren zur Reduktion von Aldehyden und Ketonen. Liebigs Ann. 1925, 444, 221–238. [Google Scholar] [CrossRef]
- Li, J.J. Meerwein–Ponndorf–Verley reduction. In Name Reactions: A Collection of Detailed Mechanisms and Synthetic Applications; Li, J.J., Ed.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 345–346. ISBN 978-3-642-01053-8. [Google Scholar]
- Saidi, O.; Williams, J.M.J. Iridium-Catalyzed Hydrogen Transfer Reactions. In Iridium Catalysis; Andersson, P.G., Ed.; Topics in Organometallic Chemistry; Springer: Berlin/Heidelberg, Germany, 2011; pp. 77–106. ISBN 978-3-642-15334-1. [Google Scholar]
- Wang, C.; Wu, X.; Xiao, J. Broader, Greener, and More Efficient: Recent Advances in Asymmetric Transfer Hydrogenation. Chem. Asian J. 2008, 3, 1750–1770. [Google Scholar] [CrossRef] [PubMed]
- Noyori, R.; Hashiguchi, S. Asymmetric Transfer Hydrogenation Catalyzed by Chiral Ruthenium Complexes. Acc. Chem. Res. 1997, 30, 97–102. [Google Scholar] [CrossRef]
- Ikariya, T.; Blacker, A.J. Asymmetric Transfer Hydrogenation of Ketones with Bifunctional Transition Metal-Based Molecular Catalysts. Acc. Chem. Res. 2007, 40, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Miecznikowski, J.R.; Crabtree, R.H. Hydrogen Transfer Reduction of Aldehydes with Alkali-Metal Carbonates and Iridium NHC Complexes. Organometallics 2004, 23, 629–631. [Google Scholar] [CrossRef]
- Wang, R.; Tang, Y.; Xu, M.; Meng, C.; Li, F. Transfer Hydrogenation of Aldehydes and Ketones with Isopropanol under Neutral Conditions Catalyzed by a Metal–Ligand Bifunctional Catalyst [Cp*Ir(2,2′-bpyO)(H2O)]. J. Org. Chem. 2018, 83, 2274–2281. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, L. Industrial Catalysts. In Handbook of Industrial Catalysts; Lloyd, L., Ed.; Fundamental and Applied Catalysis; Springer: Boston, MA, USA, 2011; pp. 1–22. ISBN 978-0-387-49962-8. [Google Scholar]
- Simon, M.-O.; Li, C.-J. Green chemistry oriented organic synthesis in water. Chem. Soc. Rev. 2012, 41, 1415–1427. [Google Scholar] [CrossRef]
- Manna, S.; Antonchick, A.P. Catalytic Transfer Hydrogenation Using Biomass as Hydrogen Source. ChemSusChem. in press. [CrossRef] [PubMed]
- Fujita, K.; Tanino, N.; Yamaguchi, R. Ligand-Promoted Dehydrogenation of Alcohols Catalyzed by Cp*Ir Complexes. A New Catalytic System for Oxidant-Free Oxidation of Alcohols. Org. Lett. 2007, 9, 109–111. [Google Scholar] [CrossRef]
- Fujita, K.; Yoshida, T.; Imori, Y.; Yamaguchi, R. Dehydrogenative Oxidation of Primary and Secondary Alcohols Catalyzed by a Cp*Ir Complex Having a Functional C,N-Chelate Ligand. Org. Lett. 2011, 13, 2278–2281. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, R.; Fujita, K.; Yamaguchi, R. Dehydrogenative Oxidation of Alcohols in Aqueous Media Using Water-Soluble and Reusable Cp*Ir Catalysts Bearing a Functional Bipyridine Ligand. J. Am. Chem. Soc. 2012, 134, 3643–3646. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, R.; Fujita, K.I.; Yamaguchi, R. Cooperative Catalysis by Iridium Complexes with a Bipyridonate Ligand: Versatile Dehydrogenative Oxidation of Alcohols and Reversible Dehydrogenation–Hydrogenation between 2-Propanol and Acetone. Angew. Chem. Int. Ed. 2012, 51, 12790–12794. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Uejima, T.; Yamaguchi, R. Hydrogen-transfer Oxidation of Primary Alcohols Catalyzed by Iridium Complexes Bearing a Functional Pyridonate Ligand Using Isopropenyl Acetate as a Hydrogen Acceptor. Chem. Lett. 2013, 42, 1496–1498. [Google Scholar] [CrossRef]
- Ball, R.G.; Graham, W.A.G.; Heinekey, D.M.; Hoyano, J.K.; McMaster, A.D.; Mattson, B.M.; Michel, S.T. Synthesis and structure of dicarbonylbis(η-pentamethylcyclopentadienyl)diiridium. Inorg. Chem. 1990, 29, 2023–2025. [Google Scholar] [CrossRef]
- Kuwahara, M.; Nishioka, M.; Yoshida, M.; Fujita, K. A Sustainable Method for the Synthesis of Acetic Acid Based on Dehydrogenation of an Ethanol–Water Solution Catalyzed by an Iridium Complex Bearing a Functional Bipyridonate Ligand. ChemCatChem 2018, 10, 3636–3640. [Google Scholar] [CrossRef]
- Fujita, K.; Wada, T.; Shiraishi, T. Reversible Interconversion between 2,5-Dimethylpyrazine and 2,5-Dimethylpiperazine by Iridium-Catalyzed Hydrogenation/Dehydrogenation for Efficient Hydrogen Storage. Angew. Chem. Int. Ed. 2017, 56, 10886–10889. [Google Scholar] [CrossRef]
- Nogueira Fernandes, J.L.; de Souza, M.C.; Brenelli, E.C.S.; Brenelli, J.A. Reduction of Acetophenones Using Borohydride Exchange Resins (BER) and a BER-Lithium Salt System. Synthesis 2009, 2009, 4058–4062. [Google Scholar]
- Yamamoto, Y.; Hasegawa, H.; Yamataka, H. Dynamic Path Bifurcation in the Beckmann Reaction: Support from Kinetic Analyses. J. Org. Chem. 2011, 76, 4652–4660. [Google Scholar] [CrossRef] [PubMed]
- Bastin, S.; Eaves, R.J.; Edwards, C.W.; Ichihara, O.; Whittaker, M.; Wills, M. A Soluble-Polymer System for the Asymmetric Transfer Hydrogenation of Ketones. J. Org. Chem. 2004, 69, 5405–5412. [Google Scholar] [CrossRef]
- Rahaim, R.J.; Maleczka, R.E. C−O Hydrogenolysis Catalyzed by Pd-PMHS Nanoparticles in the Company of Chloroarenes. Org. Lett. 2011, 13, 584–587. [Google Scholar] [CrossRef] [PubMed]
- Hevia, E.; Kennedy, A.R.; Klett, J.; Livingstone, Z.; McCall, M.D. New insights into addition reactions of dialkylzinc reagents to trifluoromethyl ketones: Structural authentication of a β-hydride elimination product containing a tetranuclear zinc chain. Dalton Trans. 2009, 39, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Maytum, H.C.; Francos, J.; Whatrup, D.J.; Williams, J.M.J. 1,4-Butanediol as a Reducing Agent in Transfer Hydrogenation Reactions. Chem. Asian J. 2010, 5, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Castro, L.C.M.; Bézier, D.; Sortais, J.-B.; Darcel, C. Iron Dihydride Complex as the Pre-catalyst for Efficient Hydrosilylation of Aldehydes and Ketones Under Visible Light Activation. Adv. Synth. Catal. 2011, 353, 1279–1284. [Google Scholar] [CrossRef]
- Azerraf, C.; Gelman, D. New Shapes of PC(sp3)P Pincer Complexes. Organometallics 2009, 28, 6578–6584. [Google Scholar] [CrossRef]
- Koren-Selfridge, L.; Londino, H.N.; Vellucci, J.K.; Simmons, B.J.; Casey, C.P.; Clark, T.B. A Boron-Substituted Analogue of the Shvo Hydrogenation Catalyst: Catalytic Hydroboration of Aldehydes, Imines, and Ketones. Organometallics 2009, 28, 2085–2090. [Google Scholar] [CrossRef]
- Basu, B.; Mandal, B.; Das, S.; Das, P.; Nanda, A.K. Chemoselective reduction of aldehydes by ruthenium trichloride and resin-bound formates. Beilstein J. Org. Chem. 2008, 4, 53. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, N.S.; Junge, K.; Beller, M. A Convenient and General Iron-Catalyzed Hydrosilylation of Aldehydes. Org. Lett. 2007, 9, 5429–5432. [Google Scholar] [CrossRef]
- Dieskau, A.P.; Begouin, J.-M.; Plietker, B. Bu4N[Fe(CO)3(NO)]-Catalyzed Hydrosilylation of Aldehydes and Ketones. Eur. J. Org. Chem. 2011, 2011, 5291–5296. [Google Scholar] [CrossRef]
- Pouchert, C.J.; Behnke, J. The Aldrich Library of 13C and 1H FT NMR Spectra, 1st ed.; Aldrich Chemical Company Inc.: St. Louis, MI, USA, 1993; Volume 2, p. 357B. [Google Scholar]
- Bhattacharya, P.; Krause, J.A.; Guan, H. Iron Hydride Complexes Bearing Phosphinite-Based Pincer Ligands: Synthesis, Reactivity, and Catalytic Application in Hydrosilylation Reactions. Organometallics 2011, 30, 4720–4729. [Google Scholar] [CrossRef]
- Xu, G.; Moeller, K.D. Anodic Coupling Reactions and the Synthesis of C-Glycosides. Org. Lett. 2010, 12, 2590–2593. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Cat. | Temp. (°C) | Base (mol%) | Conv. (%) a | Yield (%) a |
|---|---|---|---|---|---|
| 1 b | [Cp*IrCl2]2 | 100 | Na2CO3 (5.0) | <5 | 0 |
| 2 | 1 | 100 | Na2CO3 (5.0) | 86 | 85 |
| 3 | 2 | 100 | Na2CO3 (5.0) | 62 | 59 |
| 4 | 3 | 100 | Na2CO3 (5.0) | 2 | 2 |
| 5 | 4 | 100 | Na2CO3 (5.0) | 67 | 67 |
| 6 | 1 | 80 | Na2CO3 (5.0) | 48 | 47 |
| 7 | 1 | 120 | Na2CO3 (5.0) | 92 | 81 |
| 8 | 1 | 100 | K2CO3 (5.0) | 75 | 72 |
| 9 | 1 | 100 | NaOtBu (10.0) | 58 | 56 |
| 10 | 1 | 100 | KOtBu (10.0) | 62 | 56 |
| 11 c | 1 | 100 | Na2CO3 (5.0) | 54 | 53 |
| 12 d | 1 | 100 | Na2CO3 (5.0) | 94 | 89 |
| 13 b,e | 1 | 100 | Na2CO3 (5.0) | 85 | 80 |

| Conditions A: H2O was used as a solvent a | |||
 |  |  | 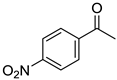 |
| 5a, 89(79) c, d | 5b, 61(56) | 5c, 60(56) e | 5d, 85(84) |
 | 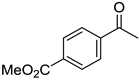 | 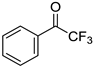 | 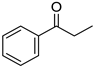 |
| 5e, 72(67) | 5f, 83(81) | 5g, 89(79) | 5h, 68(58) |
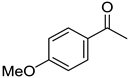
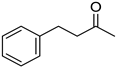 |  |  | |
| 5i, 45(45) | 5j, 86(77) d | 5k, 79(70) | |
| Conditions B: DMAc was used as a solvent b | |||
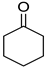
 |  | 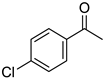 | 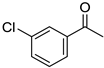 |
| 5l, 63(54) | 5m, 90(79) | 5n, 88(81) | 5o, 96(74) |
 |  | ||
| 5p, 84(82) | 5q, 92(89) | ||

| Conditions A: H2O was used as a solvent a | |||
 |  | 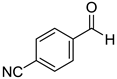 | 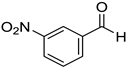 |
| 7a, 87(86) c, d | 7b, 72(58) | 7c, 54(56) | 7d, 63(54) |
 | 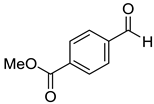 | ||
| 7e, 58(54) | 7f, 68(63) | ||
| Conditions B: DMAc was used as a solvent b | |||
 | 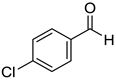 | 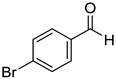 | 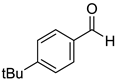 |
| 7g, 73(69) | 7h, 88(88) | 7i, 95(88) | 7j, 81(78) |
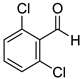 | 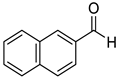 | 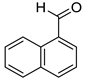 | |
| 7k, 94(87) | 7l, 97(91) | 7m, 88(83) | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoshida, M.; Hirahata, R.; Inoue, T.; Shimbayashi, T.; Fujita, K.-i. Iridium-Catalyzed Transfer Hydrogenation of Ketones and Aldehydes Using Glucose as a Sustainable Hydrogen Donor. Catalysts 2019, 9, 503. https://doi.org/10.3390/catal9060503
Yoshida M, Hirahata R, Inoue T, Shimbayashi T, Fujita K-i. Iridium-Catalyzed Transfer Hydrogenation of Ketones and Aldehydes Using Glucose as a Sustainable Hydrogen Donor. Catalysts. 2019; 9(6):503. https://doi.org/10.3390/catal9060503
Chicago/Turabian StyleYoshida, Masato, Ryota Hirahata, Takayoshi Inoue, Takuya Shimbayashi, and Ken-ichi Fujita. 2019. "Iridium-Catalyzed Transfer Hydrogenation of Ketones and Aldehydes Using Glucose as a Sustainable Hydrogen Donor" Catalysts 9, no. 6: 503. https://doi.org/10.3390/catal9060503