Co-Ni Cyanide Bi-Metal Catalysts: Copolymerization of Carbon Dioxide with Propylene Oxide and Chain Transfer Agents

 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
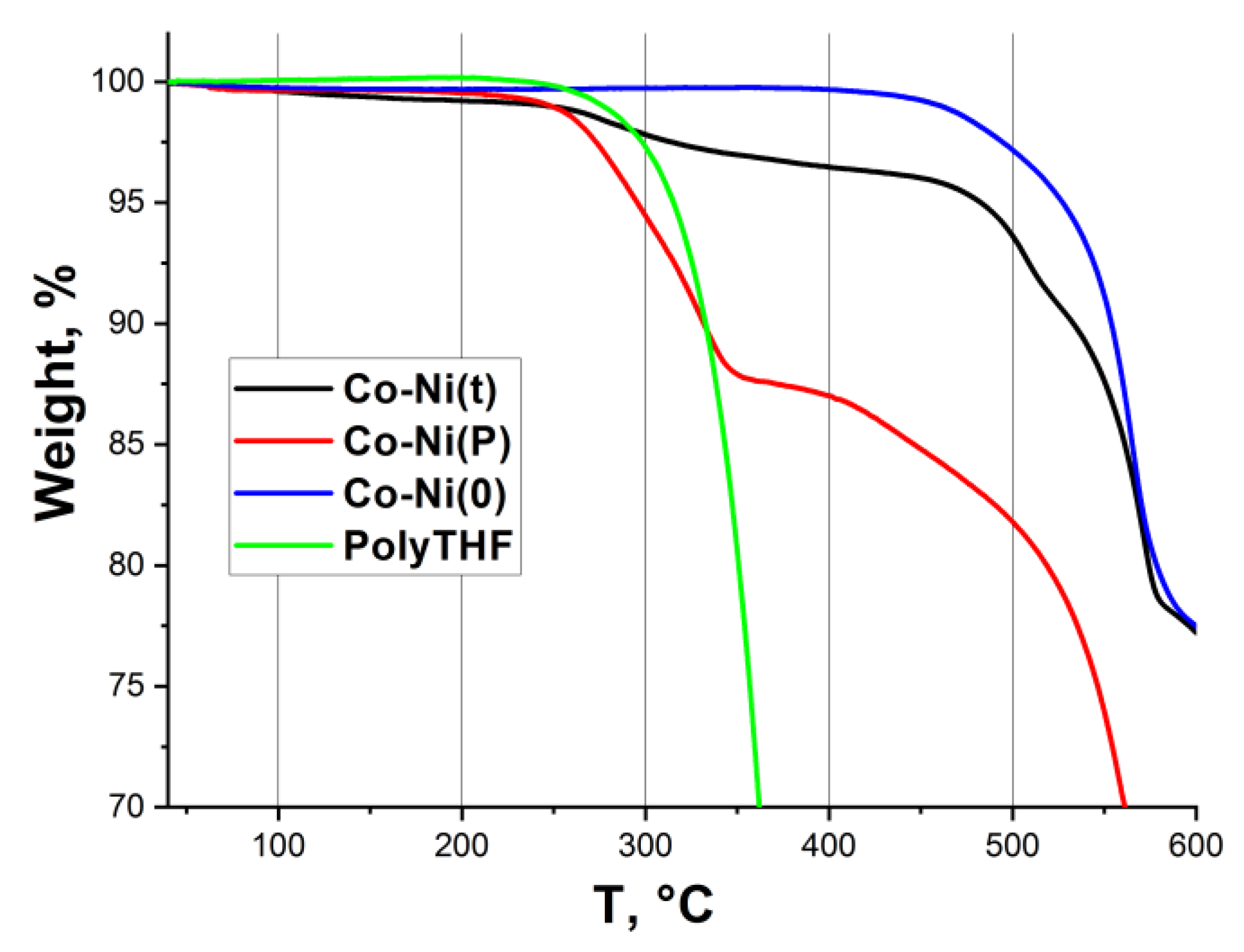
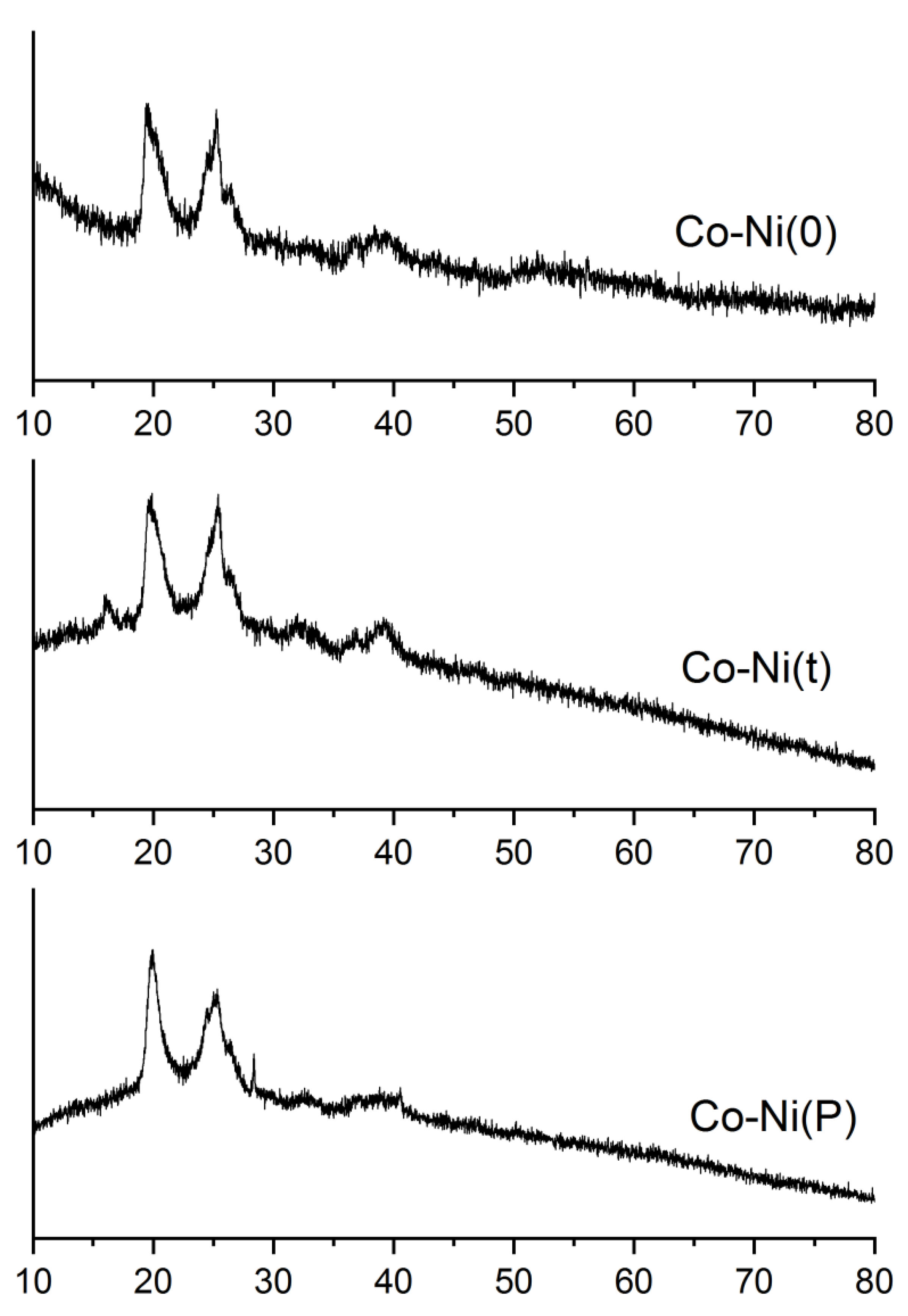
2.1. Catalyst Characterization
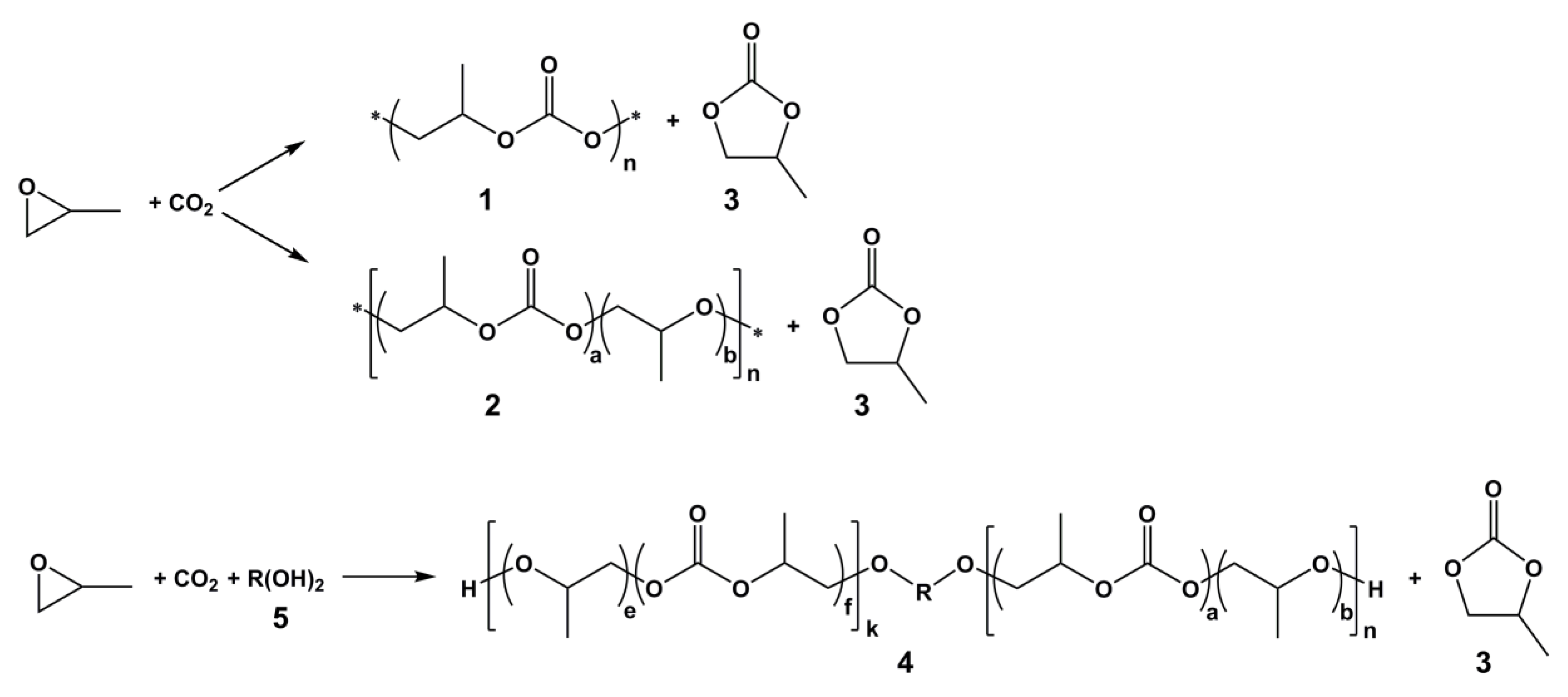
2.2. CO2/PO Copolymerization
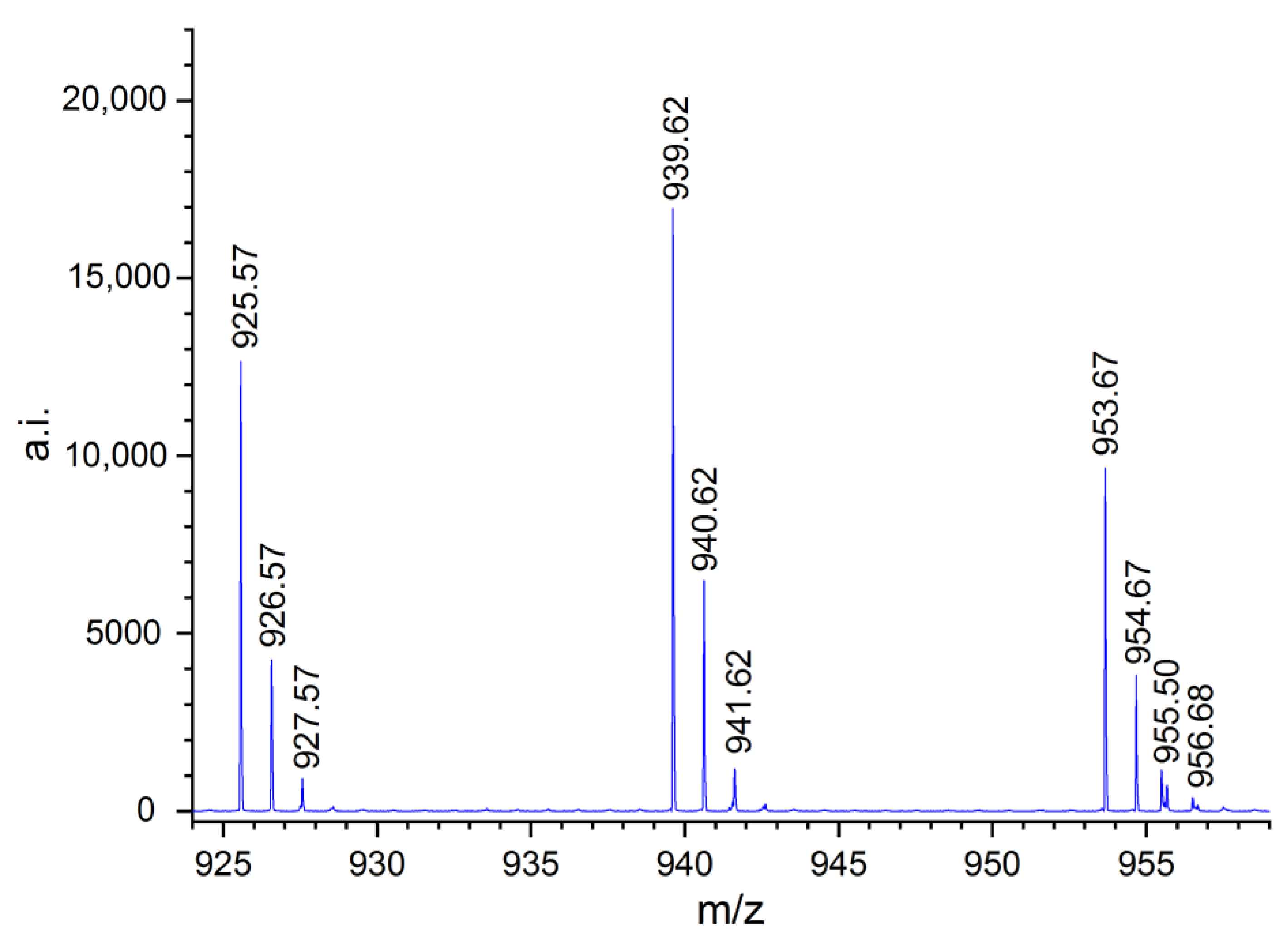
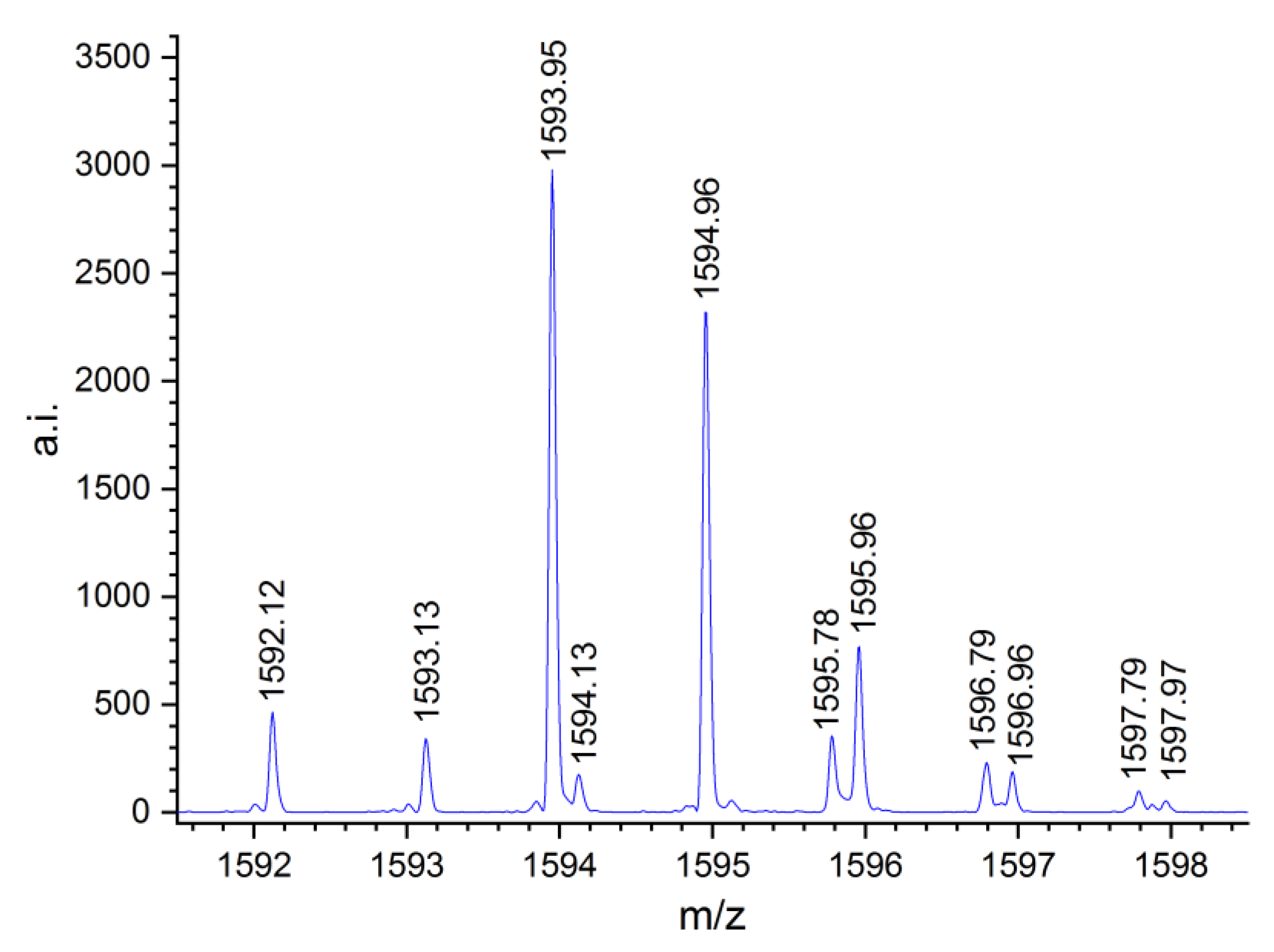
2.3. MALDI-TOF MS Analysis of the Oligo(Ethercarbonate) Synthesized in the Presence of HD
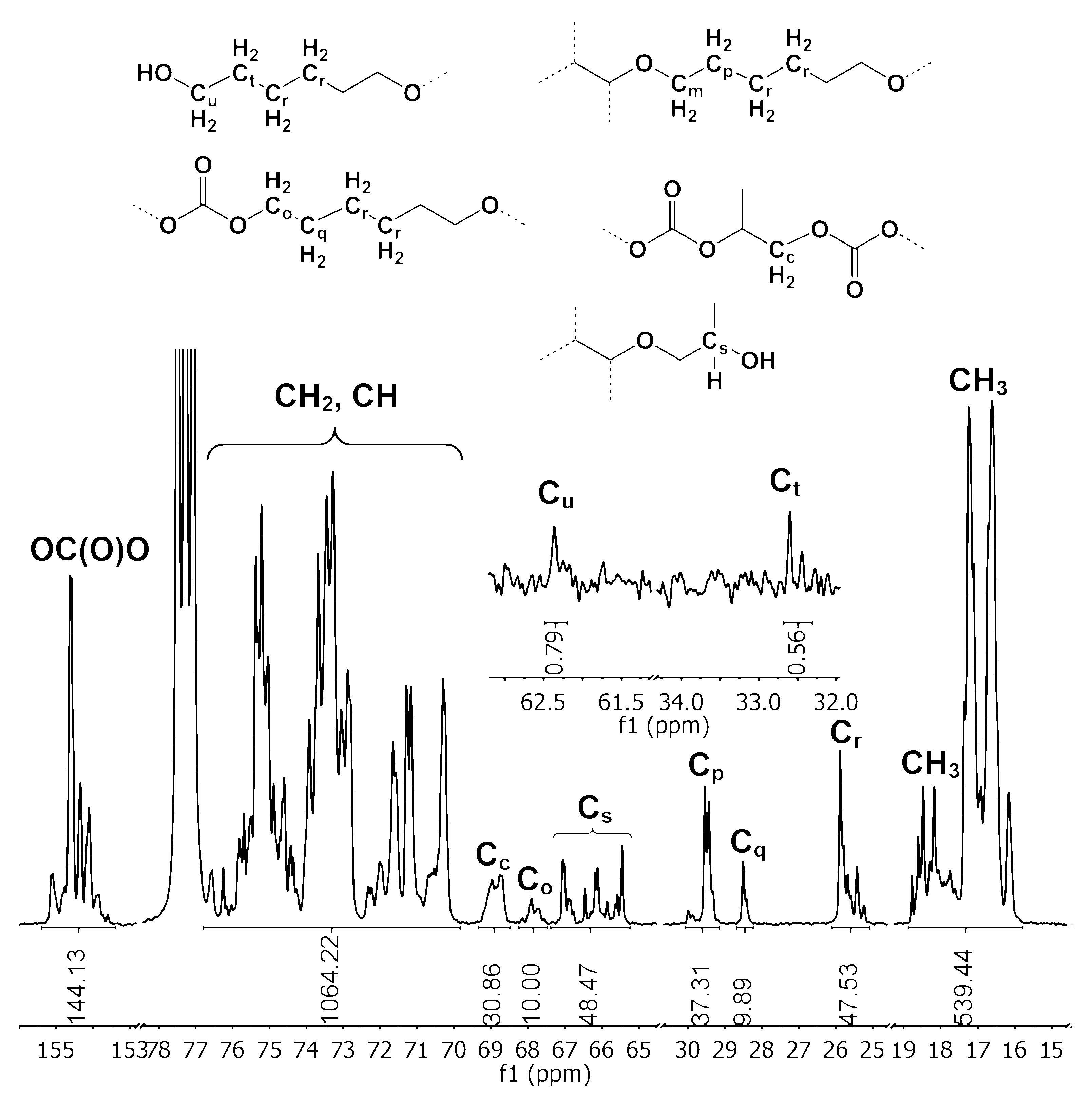
2.4. NMR Analysis of the Oligo(Ethercarbonate) Synthesized in the Presence of HD
2.5. Comparison of the Performance of Co-Ni(0) with the Published Data
3. Materials and Methods
3.1. Materials
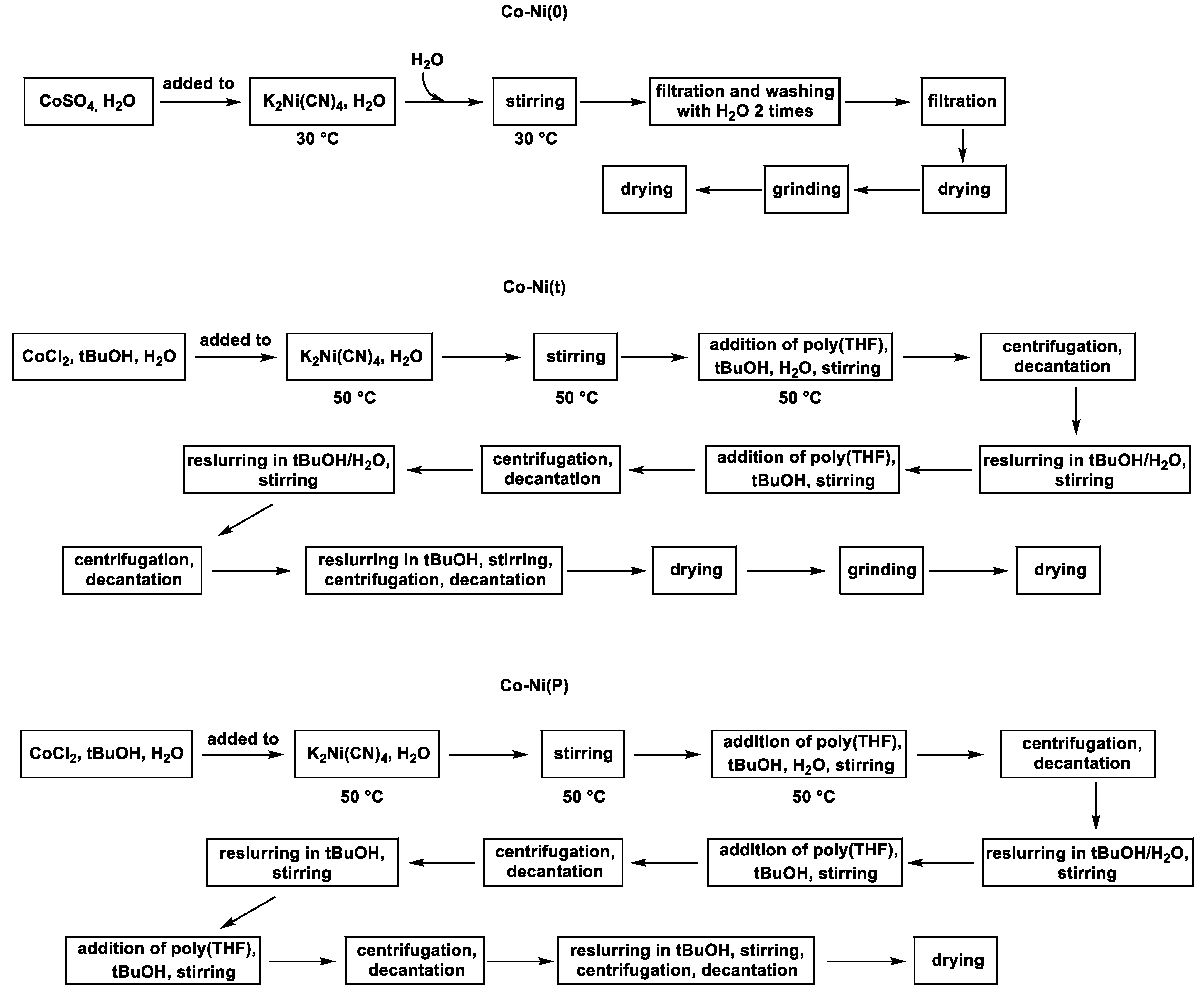
3.2. Catalyst Synthesis
3.3. Copolymerization Procedure
3.4. Characterization of Polymers and Catalysts
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Artz, J.; Müller, T.E.; Thenert, K.; Kleinekorte, J.; Meys, R.; Sternberg, A.; Bardow, A.; Leitner, W. Sustainable Conversion of Carbon Dioxide: An Integrated Review of Catalysis and Life Cycle Assessment. Chem. Rev. 2018, 118, 434–504. [Google Scholar] [CrossRef] [PubMed]
- Muthuraj, R.; Mekonnen, T. Recent progress in carbon dioxide (CO2) as feedstock for sustainable materials development: Co-polymers and polymer blends. Polymer 2018, 145, 348–373. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Y.Y.; Hu, L.F.; Zhang, X.H.; Du, B.Y.; Xu, J.T. Carbon dioxide-based copolymers with various architectures. Prog. Polym. Sci. 2018, 82, 120–157. [Google Scholar] [CrossRef]
- Xu, Y.; Lin, L.; Xiao, M.; Wang, S.; Smith, A.T.; Sun, L.; Meng, Y. Synthesis and properties of CO2-based plastics: Environmentally-friendly, energy-saving and biomedical polymeric materials. Prog. Polym. Sci. 2018, 80, 163–182. [Google Scholar] [CrossRef]
- Dai, Y.; Zhang, X. Recent development of functional aliphatic polycarbonates for the construction of amphiphilic polymers. Polym. Chem. 2017, 8, 7429–7437. [Google Scholar] [CrossRef]
- Kamphuis, A.J.; Picchioni, F.; Pescarmona, P.P. CO2-fixation into cyclic and polymeric carbonates: Principles and applications. Green Chem. 2019, 21, 406–448. [Google Scholar] [CrossRef]
- Poland, S.J.; Darensbourg, D.J. A quest for polycarbonates provided: Via sustainable epoxide/CO2 copolymerization processes. Green Chem. 2017, 19, 4990–5011. [Google Scholar] [CrossRef]
- Kozak, C.M.; Ambrose, K.; Anderson, T.S. Copolymerization of carbon dioxide and epoxides by metal coordination complexes. Coord. Chem. Rev. 2018, 376, 565–587. [Google Scholar] [CrossRef]
- Wang, Y.; Darensbourg, D.J. Carbon dioxide-based functional polycarbonates: Metal catalyzed copolymerization of CO2 and epoxides. Coord. Chem. Rev. 2018, 372, 85–100. [Google Scholar] [CrossRef]
- Bobbink, F.D.; Van Muyden, A.P.; Dyson, P.J. En route to CO2-containing renewable materials: Catalytic synthesis of polycarbonates and non-isocyanate polyhydroxyurethanes derived from cyclic carbonates. Chem. Commun. 2019, 55, 1360–1373. [Google Scholar] [CrossRef]
- Qin, Y.; Sheng, X.; Liu, S.; Ren, G.; Wang, X.; Wang, F. Recent advances in carbon dioxide based copolymers. J. CO2 Util. 2015, 11, 3–9. [Google Scholar] [CrossRef]
- Luo, W.; Qin, J.; Xiao, M.; Han, D.; Wang, S.; Meng, Y. Synthesis of Aliphatic Carbonate Macrodiols and Their Application as Sustainable Feedstock for Polyurethane. ACS Omega 2017, 2, 3205–3213. [Google Scholar] [CrossRef]
- Alagi, P.; Ghorpade, R.; Choi, Y.J.; Patil, U.; Kim, I.; Baik, J.H.; Hong, S.C. Carbon Dioxide-Based Polyols as Sustainable Feedstock of Thermoplastic Polyurethane for Corrosion-Resistant Metal Coating. ACS Sustain. Chem. Eng. 2017, 5, 3871–3881. [Google Scholar] [CrossRef]
- Cyriac, A.; Lee, S.H.; Varghese, J.K.; Park, J.H.; Jeon, J.Y.; Kim, S.J.; Lee, B.Y. Preparation of flame-retarding poly (propylene carbonate). Green Chem. 2011, 13, 3469–3475. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, H.; Miao, Y.; Qiao, L.; Wang, X.; Wang, F. Waterborne polyurethanes from CO2 based polyols with comprehensive hydrolysis/oxidation resistance. Green Chem. 2016, 18, 524–530. [Google Scholar] [CrossRef]
- Ying, Z.; Wu, C.; Jiang, S.; Shi, R.; Zhang, B.; Zhang, C.; Zhao, F. Synthesis of polyurethane-urea from double CO2-route oligomers. Green Chem. 2016, 18, 3614–3619. [Google Scholar] [CrossRef]
- DeBolt, M.; Kiziltas, A.; Mielewski, D.; Waddington, S.; Nagridge, M.J. Flexible polyurethane foams formulated with polyols derived from waste carbon dioxide. J. Appl. Polym. Sci. 2016, 133. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Z.; Han, S.; Han, J.; Jiang, D. Poly (propylene carbonate) polyurethane self-polishing coating for marine antifouling application. J. Appl. Polym. Sci. 2016, 133. [Google Scholar] [CrossRef]
- Xian, W.; Song, L.; Liu, B.; Ding, H.; Li, Z.; Cheng, M.; Ma, L. Rheological and mechanical properties of thermoplastic polyurethane elastomer derived from CO2 copolymer diol. J. Appl. Polym. Sci. 2018, 135, 45974. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, H.; Miao, Y.; Qiao, L.; Wang, X.; Wang, F. UV-curable waterborne polyurethane from CO2-polyol with high hydrolysis resistance. Polymer 2016, 100, 219–226. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, H.; Miao, Y.; Qiao, L.; Wang, X.; Wang, F. Microphase separation idea to toughen CO2-based waterborne polyurethane. Polymer 2018, 138, 211–217. [Google Scholar] [CrossRef]
- Langanke, J.; Wolf, A.; Hofmann, J.; Böhm, K.; Subhani, M.A.; Müller, T.E.; Leitner, W.; Gürtler, C. Carbon dioxide (CO2) as sustainable feedstock for polyurethane production. Green Chem. 2014, 16, 1865–1870. [Google Scholar] [CrossRef]
- Jang, J.H.; Ha, J.H.; Kim, I.; Baik, J.H.; Hong, S.C. Facile Room-Temperature Preparation of Flexible Polyurethane Foams from Carbon Dioxide Based Poly (ether carbonate) Polyols with a Reduced Generation of Acetaldehyde. ACS Omega 2019, 4, 7944–7952. [Google Scholar] [CrossRef]
- Liu, S.; Qin, Y.; Chen, X.; Wang, X.; Wang, F. One-pot controllable synthesis of oligo (carbonate-ether) triol using a Zn-Co-DMC catalyst: The special role of trimesic acid as an initiation-transfer agent. Polym. Chem. 2014, 5, 6171–6179. [Google Scholar] [CrossRef]
- Langanke, J.; Hofmann, J.; Gürtler, C.; Wolf, A. Facile synthesis of formaldehyde-based polyether (-carbonate) polyols. J. Polym. Sci. Part A Polym. Chem. 2015, 53, 2071–2074. [Google Scholar] [CrossRef]
- Gao, Y.; Qin, Y.; Zhao, X.; Wang, F.; Wang, X. Selective synthesis of oligo (carbonate-ether) diols from copolymerization of CO2 and propylene oxide under zinc-cobalt double metal cyanide complex. J. Polym. Res. 2012, 19, 9878. [Google Scholar] [CrossRef]
- Ma, K.; Bai, Q.; Zhang, L.; Liu, B. Synthesis of flame-retarding oligo (carbonate-ether) diols: Via double metal cyanide complex-catalyzed copolymerization of PO and CO2 using bisphenol A as a chain transfer agent. RSC Adv. 2016, 6, 48405–48410. [Google Scholar] [CrossRef]
- Luo, M.; Li, Y.; Zhang, Y.Y.; Zhang, X.H. Using carbon dioxide and its sulfur analogues as monomers in polymer synthesis. Polymer 2016, 82, 406–431. [Google Scholar] [CrossRef]
- Subhani, M.A.; Gürtler, C.; Leitner, W.; Müller, T.E. Nanoparticulate TiO2-Supported Double Metal Cyanide Catalyst for the Copolymerization of CO2 with Propylene Oxide. Eur. J. Inorg. Chem. 2016, 2016, 1944–1949. [Google Scholar] [CrossRef]
- Varghese, J.K.; Park, D.S.; Jeon, J.Y.; Lee, B.Y. Double metal cyanide catalyst prepared using H3Co (CN)6 for high carbonate fraction and molecular weight control in carbon dioxide/propylene oxide copolymerization. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 4811–4818. [Google Scholar] [CrossRef]
- Liu, S.; Qin, Y.; Qiao, L.; Miao, Y.; Wang, X.; Wang, F. Cheap and fast: Oxalic acid initiated CO2-based polyols synthesized by a novel preactivation approach. Polym. Chem. 2016, 7, 146–152. [Google Scholar] [CrossRef]
- Liu, S.; Miao, Y.; Qiao, L.; Qin, Y.; Wang, X.; Chen, X.; Wang, F. Controllable synthesis of a narrow polydispersity CO2-based oligo (carbonate-ether) tetraol. Polym. Chem. 2015, 6, 7580–7585. [Google Scholar] [CrossRef]
- Gao, Y.; Gu, L.; Qin, Y.; Wang, X.; Wang, F. Dicarboxylic acid promoted immortal copolymerization for controllable synthesis of low-molecular weight oligo (carbonate-ether) diols with tunable carbonate unit content. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 5177–5184. [Google Scholar] [CrossRef]
- An, N.; Li, Q.; Yin, N.; Kang, M.; Wang, J. Effects of addition mode on Zn–Co double metal cyanide catalyst for synthesis of oligo (propylene-carbonate) diols. Appl. Organomet. Chem. 2018, 32, e4509. [Google Scholar] [CrossRef]
- Qiang, L.; Zhifang, G.; Lisha, P.; Xue, X. Zn-Cr double metal cyanide catalysts synthesized by ball milling for the copolymerization of CO2/propylene oxide, phthalic anhydride/propylene oxide, and CO2/propylene oxide/phthalic anhydride. Catal. Commun. 2015, 64, 114–118. [Google Scholar] [CrossRef]
- Zhang, X.H.; Chen, S.; Wu, X.M.; Sun, X.K.; Liu, F.; Qi, G.R. Highly active double metal cyanide complexes: Effect of central metal and ligand on reaction of epoxide/CO2. Chin. Chem. Lett. 2007, 18, 887–890. [Google Scholar] [CrossRef]
- Zhang, W.; Lin, Q.; Cheng, Y.; Lu, L.; Lin, B.; Pan, L.; Xu, N. Double Metal Cyanide Complexes Synthesized by Solvent-Free Grinding Method for Copolymerization of CO2 and Propylene Oxide. J. Appl. Polym. Sci. 2012, 123, 977–985. [Google Scholar] [CrossRef]
- Guo, Z.; Lin, Q. Coupling reaction of CO2 and propylene oxide catalyzed by DMC with co-complexing agents incorporated via ball milling. J. Mol. Catal. A Chem. 2014, 390, 63–68. [Google Scholar] [CrossRef]
- Dai, C.; Zhu, Q.; Pang, H.; Zhu, L.; Lin, Q. Rapid copolymerization of carbon dioxide and propylene oxide catalyzed by double metal cyanide complexes in an ultrasonic field. Mater. Lett. 2016, 180, 89–92. [Google Scholar] [CrossRef]
- Shi, J.; Shi, Z.; Yan, H.; Wang, X.; Zhang, X.; Lin, Q.; Zhu, L. Synthesis of Zn-Fe double metal cyanide complexes with imidazolium-based ionic liquid cocatalysts: Via ball milling for copolymerization of CO2 and propylene oxide. RSC Adv. 2018, 8, 6565–6571. [Google Scholar] [CrossRef]
- Zhang, W.; Lu, L.; Cheng, Y.; Xu, N.; Pan, L.; Lin, Q.; Wang, Y. Clean and rapid synthesis of double metal cyanide complexes using mechanochemistry. Green Chem. 2011, 13, 2701–2703. [Google Scholar] [CrossRef]
- Lu, L.B.; Zhang, W.Y.; Lin, Q.; Pan, L.S.; Pang, S.J.; Xu, N. Copolymerization of Carbon Dioxide and Propylene Oxide with Multi-Metal Cyanide Complexes as Catalyst. Adv. Mater. Res. 2013, 646, 3–7. [Google Scholar] [CrossRef]
- Guo, Z.; Lin, Q.; Zhu, L.; Wang, X.; Niu, Y.; Yu, C.; Fang, T. Nanolamellar Zn–Ni/Co–Ni Catalysts Introduced by Ball Milling for the Copolymerization of CO2 with Propylene Oxide. Nanosci. Nanotechnol. Lett. 2014, 6, 353–356. [Google Scholar] [CrossRef]
- Chen, S.; Xiao, Z.; Ma, M. Copolymerization of carbon dioxide and epoxides with a novel effective Zn-Ni double-metal cyanide complex. J. Appl. Polym. Sci. 2008, 107, 3871–3877. [Google Scholar] [CrossRef]
- Robertson, N.J.; Qin, Z.; Dallinger, G.C.; Lobkovsky, E.B.; Lee, S.; Coates, G.W. Two-dimensional double metal cyanide complexes: Highly active catalysts for the homopolymerization of propylene oxide and copolymerization of propylene oxide and carbon dioxide. Dalton Trans. 2006, 5390. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.J.; Liu, Y.; Byeon, S.J.; Park, D.W.; Kim, I. Ring-opening polymerization of propylene oxide by double metal cyanide catalysts prepared by reacting CoCl2 with various metal cyanide salts. Catal. Today 2014, 232, 75–81. [Google Scholar] [CrossRef]
- Niu, T.; Crisci, G.; Lu, J.; Jacobson, A.J. Diaquacobalt Tetracyanonickelate Tetrahydrate. Acta Crystallogr. Sect. C 1998, 54, 565–567. [Google Scholar] [CrossRef]
- Patil, N.G.; Boopathi, S.K.; Alagi, P.; Hadjichristidis, N.; Gnanou, Y.; Feng, X. Carboxylate Salts as Ideal Initiators for the Metal-Free Copolymerization of CO2 with Epoxides: Synthesis of Well-Defined Polycarbonates Diols and Polyols. Macromolecules 2019, 52, 2431–2438. [Google Scholar] [CrossRef]
- Grobelny, Z.; Matlengiewicz, M.; Jurek-Suliga, J.; Golba, S.; Skrzeczyna, K. The influence of initiator and macrocyclic ligand on unsaturation and molar mass of poly(propylene oxide)s prepared with various anionic system. Polym. Bull. 2018, 75, 1101–1121. [Google Scholar] [CrossRef]
- Brocas, A.L.; Mantzaridis, C.; Tunc, D.; Carlotti, S. Polyether synthesis: From activated or metal-free anionic ring-opening polymerization of epoxides to functionalization. Prog. Polym. Sci. 2013, 38, 845–873. [Google Scholar] [CrossRef]
- Herzberger, J.; Niederer, K.; Pohlit, H.; Seiwert, J.; Worm, M.; Wurm, F.R.; Frey, H. Polymerization of ethylene oxide, propylene oxide, and other alkylene oxides: Synthesis, novel polymer architectures, and bioconjugation. Chem. Rev. 2016, 116, 2170–2243. [Google Scholar] [CrossRef] [PubMed]
- Carr, R.H.; Hernalsteen, J.; Devos, J. Determination of functionality and functionality distribution of polyether polyols by quantitative 13C NMR spectroscopy. J. Appl. Polym. Sci. 1994, 52, 1015–1022. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Co Source | Complexing Agents | Co/Ni (EDX) | BET, m2/g |
|---|---|---|---|---|
| Co-Ni(t) | CoCl2 | tBuOH, poly(THF) | 1.32 | 33.3 |
| Co-Ni(P) | CoCl2 | tBuOH, poly(THF) | 1.01 | 19.6 |
| Co-Ni(0) | CoSO4 | - | 1.01 | 23.3 |
| Run 1 | Catalyst | Prod. 2, g/g | w(PC) 2, wt.% | ν(POCO2) 2, mol.% | Mn, g/mol | PDI 2 | conv(PO) 2, % |
|---|---|---|---|---|---|---|---|
| 1 | Co-Ni(t) | 269 | 0.1 | 30 | 51,650 | 6.6 | 17 |
| 2 | Co-Ni(0) | 233 | 0.0 | 30 | 48,300 | 6.8 | 14 |
| 3 | Co-Ni(P) | inactive |
| Run 1 | CTA | CTA/cat, mol/g | Time, h | Prod. 2, g/g | w(PC) 2, wt.% | ν(POCO2) 2, mol.% | ν(POCO2) * 2, mol.% | Mn, g/mol | PDI 2 | Conv(PO) 2, % |
|---|---|---|---|---|---|---|---|---|---|---|
| 4 | no | 0 | 3 | 376 | 0 | 33 | 33 | 543,910 | 58.1 | 23 |
| 5 | PPG-425 | 0.024 | 3 | 233 | 0 | 30 | 31 | 48,300 | 6.8 | 14 |
| 6 | PPG-425 | 0.24 | 3 | 44 | 0.5 | 12 | 48 | 28,960 | 3.7 | 2 |
| 7 | AdipA | 0.024 | 3 | 176 | 0.2 | 36 | 37 | 25,490 | 6.1 | 10 |
| 8 | AdipA | 0.24 | 3 | 11 | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. |
| 9 | PPG-425 | 0.24 | 24 | 377 | 0.6 | 22 | 30 | 28,950 | 4.8 | 23 |
| 10 3 | PPG-425 | 0.24 | 24 | 407 | 19.5 | 19 | 27 | 26,170 (1,844,790) | 2.6 (1.2) | 27 |
| 11 4 | PPG-425 | 0.24 | 24 | traces | n.d. | - | - | n.d. | n.d. | n.d. |
| Run 1 | CTA/cat, mol/g | T, °C | Prod. 2, g/g | w(PC) 2, wt.% | ν(POCO2) 2, mol.% | ν(POCO2) * 2, % mol | Mn, g/mol | PDI 2 | Conv(PO) 2, % | Tg, °C |
|---|---|---|---|---|---|---|---|---|---|---|
| 12 | 0.24 | 90 | 1116 | 1.1 | 35 | 37 | 8360 | 9.2 | 66 | n.d. |
| 13 | 0.47 | 90 | 700 | 3.7 | 36 | 40 | 1970 | 6.0 | 41 | n.d. |
| 14 3 | 0.47 | 90 | 1172 | 3.4 | 33 | 35 | n.d. | n.d. | 70 | n.d. |
| 15 4 | 0.47 | 105 | 1163 | 3.9 | 31 | 33 | 3740 | 4.4 | 71 | −43.4 |
| 16 4 | 0.47 | 120 | 1642 | 3.6 | 23 | 24 | 4620 | 2.8 | 100 | −47.4 |
| 17 4 | 0.94 | 120 | 1528 | 6.5 | 25 | 26 | 2560 | 2.5 | 95 | −48.6 |
| Catalyst | T, °C | P, MPa | Time, h | PO/Cat, g/g | Prod. 1, g/gcat | CTA | ν(POCO2) 1, mol.% | w(PC) 1, wt.% | Mn, g/mol | PDI 1 | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Co-Ni | 120 | 4.3 | 24 | 1330 | 1528 | HD | 25 | 6.5 | 2560 | 2.5 | - |
| Zn-Co | 60 | 4.0 | 24 | 1037 | 1300 | PPG-350 | 50.5 | 4.8 | 3800 | 1.3 | [26] |
| Zn-Co | 80 | 4 | 5 | 1660 | 2440 | sebacic acid | 45.2 | 8.2 | 2500 | 1.35 | [33] |
| Zn-Fe | 60 | 3.8 | 40 | 138 | 774 g/gZn | - | 91 | n.a. | 98 | 1.9 | [37] |
| Zn-Cr | 70 | 4 | 24 | 83 | n.a. | - | 82.5 | n.a. | 68,600 | 1.68 | [35] |
| Zn-Fe | 60 | 4 | 40 | n.a. | 85 | - | 64.7 | n.a. | 2200 | 2.44 | [38] |
| Zn-Ni | 130 | 5.0 | 20 | 553 | 483 | - | 52 | 9.1 | 21,660 | 3.2 | [44] |
| Zn-Ni | 130 | 5.0 | 10 | 4610 | 483 | PPG-400 | 32 | 9.1 | Mw = 35,390 | n.a. | [36] |
| Zn-Fe | 55 | n.a. | 40 | 138 | 916 g/gZn | - | 92 | n.a. | 83,200 | 1.87 | [41] |
| Zn-Fe | 60 | 3 | 24 | 49.8 | 4.4 g/gZn | - | 48.3 | n.a. | 3354 | 1.01 | [40] |
| Zn-Ni-Co | 100 | 3 | 40 | 415 | 2480 g/gZn | - | 43.6 | n.a. | 10,000 | 4.3 | [42] |
| Zn-Ni | 60 | 4 | 24 | 83 | 259 g/gZn | - | 83.5 | n.a. | 10,344 | 1.45 | [43] |
| Co-Ni | 60 | 4 | 24 | 83 | 183 g/gCo | - | 73.7 | n.a. | 8478 | 1.44 | [43] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alferov, K.; Wang, S.; Li, T.; Xiao, M.; Guan, S.; Meng, Y. Co-Ni Cyanide Bi-Metal Catalysts: Copolymerization of Carbon Dioxide with Propylene Oxide and Chain Transfer Agents. Catalysts 2019, 9, 632. https://doi.org/10.3390/catal9080632
Alferov K, Wang S, Li T, Xiao M, Guan S, Meng Y. Co-Ni Cyanide Bi-Metal Catalysts: Copolymerization of Carbon Dioxide with Propylene Oxide and Chain Transfer Agents. Catalysts. 2019; 9(8):632. https://doi.org/10.3390/catal9080632
Chicago/Turabian StyleAlferov, Kirill, Shuanjin Wang, Tianhao Li, Min Xiao, Shanyue Guan, and Yuezhong Meng. 2019. "Co-Ni Cyanide Bi-Metal Catalysts: Copolymerization of Carbon Dioxide with Propylene Oxide and Chain Transfer Agents" Catalysts 9, no. 8: 632. https://doi.org/10.3390/catal9080632
APA StyleAlferov, K., Wang, S., Li, T., Xiao, M., Guan, S., & Meng, Y. (2019). Co-Ni Cyanide Bi-Metal Catalysts: Copolymerization of Carbon Dioxide with Propylene Oxide and Chain Transfer Agents. Catalysts, 9(8), 632. https://doi.org/10.3390/catal9080632