Ex-Situ Synthesis and Study of Nanosized Mo-Containing Catalyst for Petroleum Residue Hydro-Conversion



Abstract
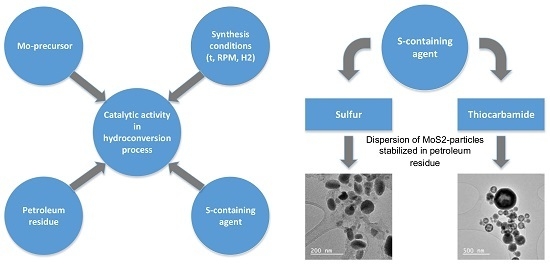
1. Introduction
2. Objects and Research Methods
2.1. Agents and Synthesis Process
2.2. Research of Catalytic Activity in the Hydro-Conversion Process
2.3. Catalyst Characterization Methods
3. Results and Discussion
3.1. Study of Ex-Situ Catalyst Characteristics
3.1.1. Dynamic Light Scattering (DLS)
3.1.2. Transmission Electron Microscopy (TEM)
3.1.3. Scanning Transmission Electron Microscopy and Energy Dispersive Analysis (HAADF STEM), Elemental Mapping
3.2. Elemental Analysis
3.2.1. Raman Spectroscopy
3.2.2. X-ray Photoelectron Spectrometry
3.2.3. X-ray Diffraction Analysis
3.3. Activity of Ex-Situ Catalysts in Hydro-Conversion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Khadzhiev, S.N.; Kadiev, K.M.; Kadieva, M.K. Synthesis and properties of nanosized systems as efficient catalysts for hydroconversion of heavy petroleum feedstock. Pet. Chem. 2014, 54, 323–346. [Google Scholar] [CrossRef]
- Rana, M.S.; Sámano, V.; Ancheyta, J.; Díaz, J. A review of recent advances on process technologies for upgrading of heavy oils and residua. Fuel 2007, 86, 1216–1231. [Google Scholar] [CrossRef]
- Bellussi, G.; Rispoli, G.; Landoni, A.; Millini, R.; Molinari, D.; Montanari, E.; Moscotti, D.; Pollesel, P. Hydroconversion of heavy residues in slurry reactors: Developments and perspectives. J. Catal. 2013, 308, 189–200. [Google Scholar] [CrossRef]
- Ortiz-Moreno, H.; Ramírez, J.; Sanchez-Minero, F.; Cuevas, R.; Ancheyta, J. Hydrocracking of Maya crude oil in a slurry-phase batch reactor. II. Effect of catalyst load. Fuel 2014, 130, 263–272. [Google Scholar] [CrossRef]
- Angeles, M.; Leyva, C.; Ancheyta, J.; Ramírez, S. A review of experimental procedures for heavy oil hydrocracking with dispersed catalyst. Catal. Today 2014, 220, 274–294. [Google Scholar] [CrossRef]
- AlKhaldi, S.; Husein, M.M. Hydrocracking of Heavy Oil by Means of In Situ Prepared Ultradispersed Nickel Nanocatalyst. Energy Fuels 2013, 28, 643–649. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, D.; Deng, W.; Que, G. A Review of Slurry-Phase Hydrocracking Heavy Oil Technology. Energy Fuels 2007, 21, 3057–3062. [Google Scholar] [CrossRef]
- Kadiev, K.H.; Khadzhiev, S.; Wadsworth, D.M. An efficient solution to the conversion of heavy hydrocarbon residue. In Proceedings of the 21st World Petroleum Congress, Moscow, Russia, 15–19 June 2014; Curran Associates Inc.: Red Hook, NY, USA, 2014; p. 3190. [Google Scholar]
- Liu, Y.; Gao, L.; Wen, L.; Zong, B. Recent Patents. Chem. Eng. 2009, 2, 22. [Google Scholar]
- Ramirez-Corredores, M.M. The Science and Technology of Unconventional Oils: Finding Refining Opportunities; Academic Press: Cambridge, MA, USA, 2017. [Google Scholar]
- Panariti, N.; Del Bianco, A.; Del Piero, G.; Marchionna, M. Petroleum residue upgrading with dispersed catalysts. Appl. Catal. A Gen. 2000, 204, 203–213. [Google Scholar] [CrossRef]
- Panariti, N.; Del Bianco, A.; Del Piero, G.; Marchionna, M.; Carniti, P. Petroleum residue upgrading with dispersed catalysts. Appl. Catal. A Gen. 2000, 204, 215–222. [Google Scholar] [CrossRef]
- Kadiev, K.M.; Khadzhiev, S.N.; Kadieva, M.K. Synthesis and use of polyfunctional catalyst nanoparticles for hydroconversion of natural bitumen. Pet. Chem. 2013, 53, 298–308. [Google Scholar] [CrossRef]
- Rezaei, H.; Ardakani, S.J.; Smith, K.J. Comparison of MoS 2 Catalysts Prepared from Mo-Micelle and Mo-Octoate Precursors for Hydroconversion of Cold Lake Vacuum Residue: Catalyst Activity, Coke Properties and Catalyst Recycle. Energy Fuels 2012, 26, 2768–2778. [Google Scholar] [CrossRef]
- Kadiev, K.M.; Gyul’Maliev, A.M.; Zekel’, L.A.; Kadieva, M.K. Hydrofining of Oil Shale Pyrolysis Tar in the Presence of Ultradispersed Catalysts. Solid Fuel Chem. 2018, 52, 336–342. [Google Scholar] [CrossRef]
- Khadzhiev, S.; Kadiev, K.; Kadieva, M. Formation and properties of nanosized particles of heavy feedstock conversion. Catal. Ind. 2014, 6, 312–319. [Google Scholar] [CrossRef]
- Nassar, N.N.; Husein, M.M. Ultradispersed particles in heavy oil: Part I, preparation and stabilization of iron oxide/hydroxide. Fuel Process. Technol. 2010, 91, 164–168. [Google Scholar] [CrossRef]
- Kadiev, K.M.; Khadzhiev, S.N.; Kadieva, M.K.; Dogova, E.S. Ex situ synthesis of sulfided molybdenum-containing ultrafine hydroconversion catalysts. Pet. Chem. 2017, 57, 608–617. [Google Scholar] [CrossRef]
- Khadzhiev, S.N.; Kadiev, K.M.; Gul’Maliev, A.M.; Kadieva, M.K. Properties and Structure of Nanosized Catalyst Systems Based on Molybdenum Sulfides. Pet. Chem. 2017, 57, 1277–1286. [Google Scholar] [CrossRef]
- Khadzhiev, S.N.; Kadiev, K.M.; Yampolskaya, G.P.; Kadieva, M.K. Trends in the synthesis of metal oxide nanoparticles through reverse microemulsions in hydrocarbon media. Adv. Colloid Interface Sci. 2013, 197, 132–145. [Google Scholar] [CrossRef]
- Eckert, B.; Janssen, A.J.H.; de Keizer, A.; Kleinjan, W.E.; Krossing, I.; Steudel, R.; Steudel, Y.; Wong, M.W. Elemental Sulfur and Sulfur-Rich Compounds I; Steudel, R., Ed.; Springer: Heidelberg, Germany, 2003; Volume 230, p. 202. [Google Scholar]
- Liu, Y.; Wu, K.; Guo, X.-L.; Wang, W.-Y.; Yang, Y.-Q. A comparison of MoS 2 catalysts hydrothermally synthesized from different sulfur precursors in their morphology and hydrodeoxygenation activity. J. Fuel Chem. Technol. 2018, 46, 535–542. [Google Scholar] [CrossRef]
- Peng, Y.; Meng, Z.; Zhong, C.; Lu, J.; Yu, W.; Yang, Z.; Qian, Y. Hydrothermal Synthesis of MoS2 and Its Pressure-Related Crystallization. J. Solid State Chem. 2001, 159, 170–173. [Google Scholar] [CrossRef]
- Vattikuti, S.V.P.; Byon, C. Synthesis and Characterization of Molybdenum Disulfide Nanoflowers and Nanosheets: Nanotribology. J. Nanomater. 2015, 2015, 1–11. [Google Scholar] [CrossRef]
- Han, J.H.; Kwak, M.; Kim, Y.; Cheon, J. Recent Advances in the Solution-Based Preparation of Two-Dimensional Layered Transition Metal Chalcogenide Nanostructures. Chem. Rev. 2018, 118, 6151–6188. [Google Scholar] [CrossRef]
- Nasirov, R.K.; Dianova, S.A.; Koval’Chuk, N.A.; Nasirov, I.R. Presulfiding of hydrotreating catalyst by elemental sulfur outside of the reactor. Chem. Technol. Fuels Oils 1998, 34, 344–348. [Google Scholar] [CrossRef]
- US4943547A—Method of Presulfiding a Hydrotreating Catalyst. Available online: https://patents.google.com/patent/US4943547A/en (accessed on 3 December 2018).
- Timchenko, V.P.; Novozhilov, A.L.; Slepysheva, O.A. Kinetics of Thermal Decomposition of Thiourea. Russ. J. Gen. Chem. 2004, 74, 1046–1050. [Google Scholar] [CrossRef]
- Morozov, E.V.; Trukhan, S.N.; Martyanov, O.N.; Larichev, Y.V.; Kazarian, S.G. The stability and evolution of oil systems studied via advanced methods in situ. Russ. Chem. Rev. 2017, 86, 999–1023. [Google Scholar]
- Yi, M.; Zhang, C. The synthesis of MoS 2 particles with different morphologies for tribological applications. Tribol. Int. 2017, 116, 285–294. [Google Scholar] [CrossRef]
- Stevens, G.C.; Edmonds, T. Preparation and Properties of Thiomolybdate Graphite Catalysts. Stud. Surf. Sci. Catal. 1979, 3, 507–517. [Google Scholar]
- Frey, G.L.; Tenne, R.; Matthews, M.J.; Dresselhaus, M.S.; Dresselhaus, G. Raman and resonance Raman investigation of MoS2 nanoparticles. Phys. Rev. B 1999, 60, 2883–2892. [Google Scholar] [CrossRef]
- Ma, L.; Chen, W.-X.; Xu, Z.-D.; Xia, J.-B.; Li, X. Carbon nanotubes coated with tubular MoS2 layers prepared by hydrothermal reaction. Nanotechnology 2006, 17, 571. [Google Scholar] [CrossRef]
- Ferrari, A.C. Raman spectroscopy of graphene and graphite: Disorder, electron–phonon coupling, doping and nonadiabatic effects. Solid State Commun. 2007, 143, 47–57. [Google Scholar] [CrossRef]
- Dresselhaus, M.S.; Jorio, A.; Souza Filho, A.G.; Saito, R. Defect characterization in graphene and carbon nanotubes using Raman spectroscopy. Philos. Trans. R. Soc. A: Math. Phys. Eng. Sci. 2010, 368, 5355–5377. [Google Scholar] [CrossRef]
- Benoist, L.; Gonbeau, D.; Pfister-Guillouzo, G.; Schmidt, E.; Meunier, G.; Levasseur, A. X-ray photoelectron spectroscopy characterization of amorphous molybdenum oxysulfide thin films. Thin Solid Films 1995, 258, 110–114. [Google Scholar] [CrossRef]
- Moulder, J.F.; Stickle, W.F.; Sobol, P.E.; Bomben, K.D. Handbook of X-ray Photoelectron Spectroscopy, Physical Electronics; Chastain, J., Ed.; Perkin-Elmer Corporation: Eden Prairie, MN, USA, 1992. [Google Scholar]
- McIntyre, N.S.; Johnston, D.D.; Coatsworth, L.L.; Davidson, R.D.; Brown, J.R. X-ray photoelectron spectroscopic studies of thin film oxides of cobalt and molybdenum. Surf. Interface Anal. 1990, 15, 265–272. [Google Scholar] [CrossRef]
- Latha, G.; Rajendran, N.; Rajeswari, S. Influence of alloying elements on the corrosion performance of alloy 33 and alloy 24 in seawater. J. Mater. Eng. Perform. 1997, 6, 743–748. [Google Scholar] [CrossRef]
- Briggs, D.; Sikh, M. Surface Analysis by Auger and X-Ray Photoelectron Spectroscopy; Mir: Moscow, Russia, 1987. [Google Scholar]
- Barr, T.L. An ESCA study of the termination of the passivation of elemental metals. J. Phys. Chem. 1978, 82, 1801–1810. [Google Scholar] [CrossRef]
- Anwar, M.; Hogarth, C.A.; Bulpett, R. Effect of substrate temperature and film thickness on the surface structure of some thin amorphous films of MoO3 studied by X-ray photoelectron spectroscopy (ESCA). J. Mater. Sci. 1989, 24, 3087–3090. [Google Scholar] [CrossRef]
- Hopfengärtner, G.; Borgmann, D.; Rademacher, I.; Wedler, G.; Hums, E.; Spitznagel, G. XPS studies of oxidic model catalysts: Internal standards and oxidation numbers. J. Electron. Spectrosc. Relat. Phenom. 1993, 63, 91–116. [Google Scholar] [CrossRef]
- Wang, H.; Skeldon, P.; Thompson, G. XPS studies of MoS2 formation from ammonium tetrathiomolybdate solutions. Surf. Coatings Technol. 1997, 91, 200–207. [Google Scholar] [CrossRef]
- Pourabbas, B.; Jamshidi, B. Preparation of MoS2 nanoparticles by a modified hydrothermal method and the photo-catalytic activity of MoS2/TiO2 hybrids in photo-oxidation of phenol. Chem. Eng. J. 2008, 138, 55–62. [Google Scholar] [CrossRef]
- Yi, Y.; Zhang, B.; Jin, X.; Wang, L.; Williams, C.T.; Xiong, G.; Su, D.; Liang, C. Unsupported NiMoW sulfide catalysts for hydrodesulfurization of dibenzothiophene by thermal decomposition of thiosalts. J. Mol. Catal. A: Chem. 2011, 351, 120–127. [Google Scholar] [CrossRef]
- Daage, M.; Chianelli, R. Structure-Function Relations in Molybdenum Sulfide Catalysts: The “Rim-Edge” Model. J. Catal. 1994, 149, 414–427. [Google Scholar] [CrossRef]
- Kadiev, K.M.; Zekel’, L.A.; Kadieva, M.K.; Khadzhiev, S.N. Formation of Polycondensation Products in Heavy Oil Feedstock Hydroconversion in the Presence of Ultrafine Catalyst: Physicochemical Study. Pet. Chem. 2018, 58, 519–527. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value |
|---|---|
| Density (20 °C), kg/m3 | 1030.0 |
| Fraction boiling above 500 °C, wt % | 95.0 |
| V, ppm | 150 |
| Ni, ppm | 38 |
| S, wt % | 3.1 |
| Asphaltenes, wt % | 12.5 |
| Resins, wt % | 22.0 |
| Parameter | Cat (S) | Cat (TC) |
|---|---|---|
| VR-stabilized dispersion of Mo-particles | ||
| D (DLS), nm | 209 ± 10 | 223 ± 11 |
| PI (DLS) | 0.51 | 0.60 |
| Mo, wt % | 6.0 | 10.2 |
| V, wt % | 0.0186 | 0.0174 |
| Ni, wt % | 0.0061 | 0.0062 |
| Fe, wt % | 0.0102 | 0.0045 |
| Co, wt % | 0.0034 | 0.0027 |
| Toluene insoluble components | ||
| N, wt % | 0.3 | 1.0 |
| C, wt % | 18.7 | 14.6 |
| H, wt % | 1.4 | 1.2 |
| S, wt % | 25.9 | 31.4 |
| O, wt % | 15.0 | 5.2 |
| Mo, wt % | 38.6 | 46.6 |
| S/Mo, molar | 1.9 | 2.1 |
| O/Mo, molar | 3.2 | 0.6 |
| Sample | Elemental Content, at. % | Ratio of Elemental Content | |||||
|---|---|---|---|---|---|---|---|
| C | O | S | Mo | S/Mo | O/Mo | C/Mo | |
| Cat (S)-TI | 37.6 | 43.7 | 9.7 | 9.1 | 1.1 | 4.8 | 4.2 |
| Cat (TC)-TI | 55.9 | 24.9 | 12.4 | 7.0 | 1.8 | 3.6 | 8.0 |
| Sample | Parameter | Mo3d | S2p | O1s | C1s | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1-1′ MoS2 | 2-2′ MoOySz | 3-3′ Mo5+ | 4-4′ Mo6+ | 1-1′ MoS2 | 2-2′ S22-/ MoOySz | 3-3′ S0 | 4-4′ SO42- | ||||
| Cat (S)-TI | E, eV | 228.9 | 229.5 | 231.7 | 233.0 | 161.8 | 162.9 | 163.9 | 168.6 | 531.6 | 284.1 |
| I, % | 9 | 6 | 21 | 64 | 14 | 7 | 5 | 75 | 100 | 100 | |
| Cat (TC)-TI | E, eV | 228.9 | 229.5 | 231.8 | 233.0 | 161.8 | 163.0 | 164.5 | 168.5 | 531.6 | 284.1 |
| I, % | 52 | 14 | 16 | 18 | 50 | 13 | 1 | 36 | 100 | 100 | |
| Test # | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| Catalyst | Cat (S) | Cat (TC) | In-situ | ||
| Composition of the dispersed phase (XRD, elemental analysis) | MoS2, Mo3S4 and | MoS2, Mo3S4 and | Mo sulfide | ||
| C-containing amorphous matrix (XRD), | C-containing amorphous matrix (XRD), | ||||
| residual precursor (S/Mo = 1.9, O/Mo = 3.2 (mol.)) | residual precursor (S/Mo = 2.1, O/Mo = 0.6 (mol.)) | ||||
| Particle size | 40–920 nm (DLS) | 40–1140 nm (DLS) | 10–50 nm [1] | ||
| 80 nm (TEM) | 120 m (TEM) | ||||
| d = 3.8 nm, N = 6 (XRD) | d = 1.7 nm, N = 3 (XRD) | ||||
| Particle structure | Spherical and elongated rounded particles, mono-layered slabs (length 8 nm) | Solid and hollow nanostructured spheres, multilayered “packets” (number of layers 3–12, length 8–40 nm) | [1] | ||
| Feed space velocity, hr−1 | 0.4 | 1.4 | 0.4 | 1.4 | 0.4 |
| Yield of products, wt % | |||||
| Gaseous products (without H2) | 2.6 | 1.7 | 2.1 | 1.8 | 2.3 |
| Liquid hydrogenation product | 97.2 | 98.2 | 94.5 | 98.1 | 97.6 |
| Fractional composition of liquid product, wt % | |||||
| IBP-180 °C fraction | 7.9 | 9.3 | 13.7 | 4.0 | 8.6 |
| 180–350 °C fraction | 30.2 | 15.6 | 32.6 | 18.8 | 25.6 |
| 350–500 °C fraction | 18.6 | 11.0 | 21.0 | 10.0 | 16.7 |
| 500 °C + fraction | 40.5 | 62.3 | 27.2 | 65.3 | 46.7 |
| Coke yield, wt % | 0.2 | 0.1 | 3.4 | 0.1 | <0.1 |
| Conversion of 500 °C+ fraction, wt % | 55.5 | 31.4 | 70.0 | 28.0 | 48.6 |
| Product properties: | |||||
| Liquid hydrogenation product | |||||
| S, wt % | 1 | 1.18 | 0.87 | 1.2 | 1.03 |
| ρ(20 °C), kg/m3 | 928 | 961 | 908 | 965 | 930 |
| IBP-180 °C fraction | |||||
| S, wt % | 0.34 | 0.48 | 0.33 | 0.49 | 0.38 |
| Olefins, wt % | 22.1 | 21.5 | 27 | 29.2 | 23.3 |
| 180–350 °C fraction | |||||
| S, wt % | 0.68 | 0.75 | 0.55 | 0.7 | 0.73 |
| Olefins, wt % | 25.4 | 20.3 | 29.4 | 22.3 | 19.4 |
| 350–500 °C fraction | |||||
| S, wt % | 1.01 | 1.17 | 0.96 | 0.16 | 1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kadieva, M.K.; Maximov, A.L.; Kadiev, K.M. Ex-Situ Synthesis and Study of Nanosized Mo-Containing Catalyst for Petroleum Residue Hydro-Conversion. Catalysts 2019, 9, 649. https://doi.org/10.3390/catal9080649
Kadieva MK, Maximov AL, Kadiev KM. Ex-Situ Synthesis and Study of Nanosized Mo-Containing Catalyst for Petroleum Residue Hydro-Conversion. Catalysts. 2019; 9(8):649. https://doi.org/10.3390/catal9080649
Chicago/Turabian StyleKadieva, Malkan Kh., Anton L. Maximov, and Khusain M. Kadiev. 2019. "Ex-Situ Synthesis and Study of Nanosized Mo-Containing Catalyst for Petroleum Residue Hydro-Conversion" Catalysts 9, no. 8: 649. https://doi.org/10.3390/catal9080649
APA StyleKadieva, M. K., Maximov, A. L., & Kadiev, K. M. (2019). Ex-Situ Synthesis and Study of Nanosized Mo-Containing Catalyst for Petroleum Residue Hydro-Conversion. Catalysts, 9(8), 649. https://doi.org/10.3390/catal9080649