Perovskite Structure Associated with Precious Metals: Influence on Heterogenous Catalytic Process


Abstract
:1. Introduction
2. General Considerations on Photocatalysis
2.1. Heterogeneous Photocatalysis: TiO2 vs. Perovskite-based Materials
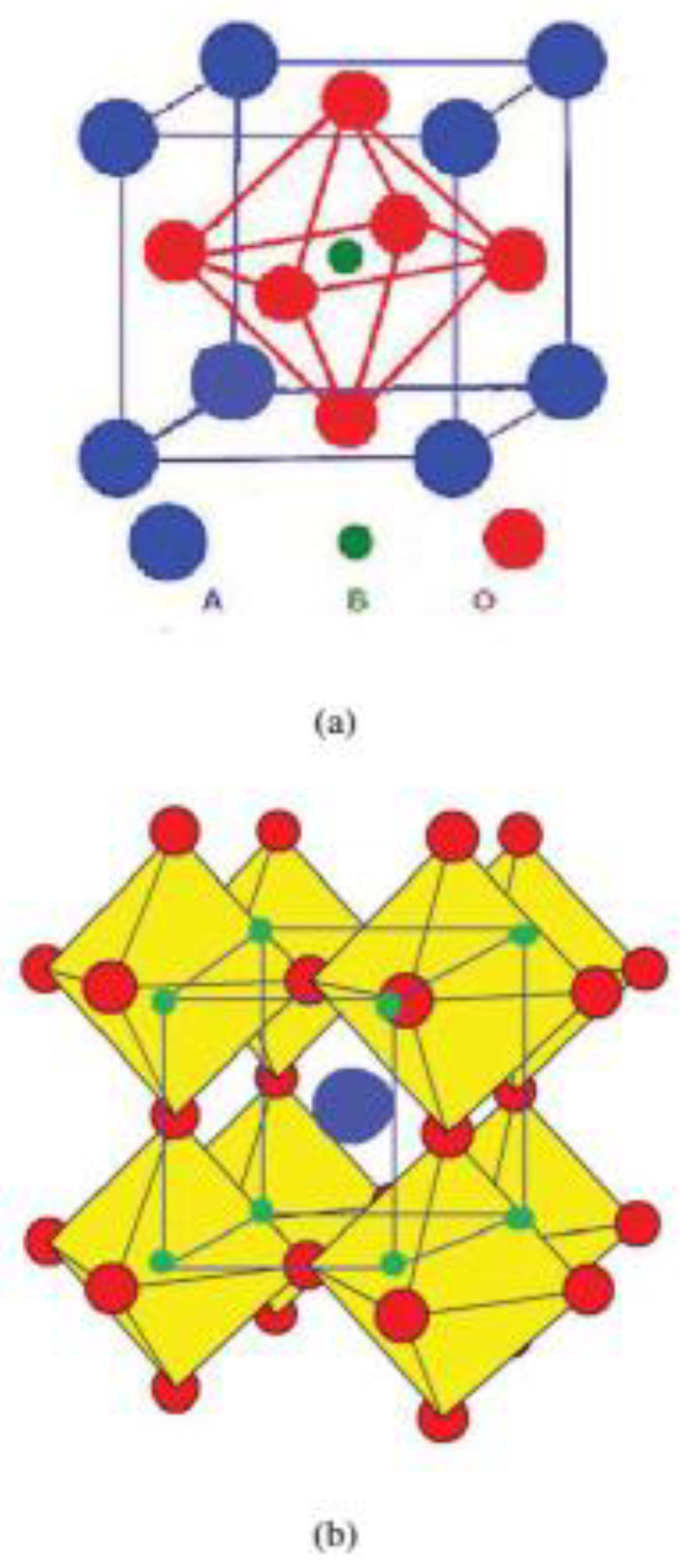
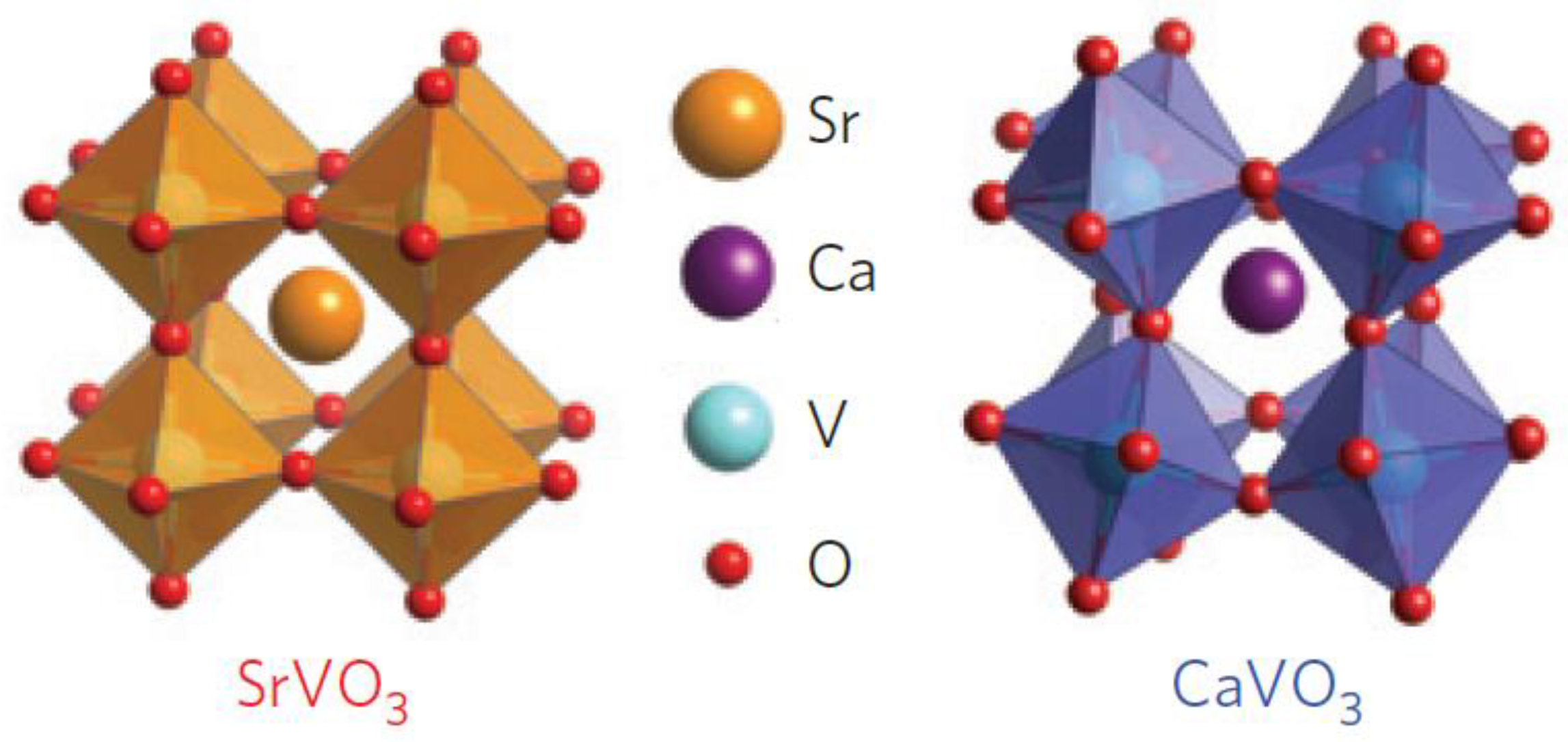
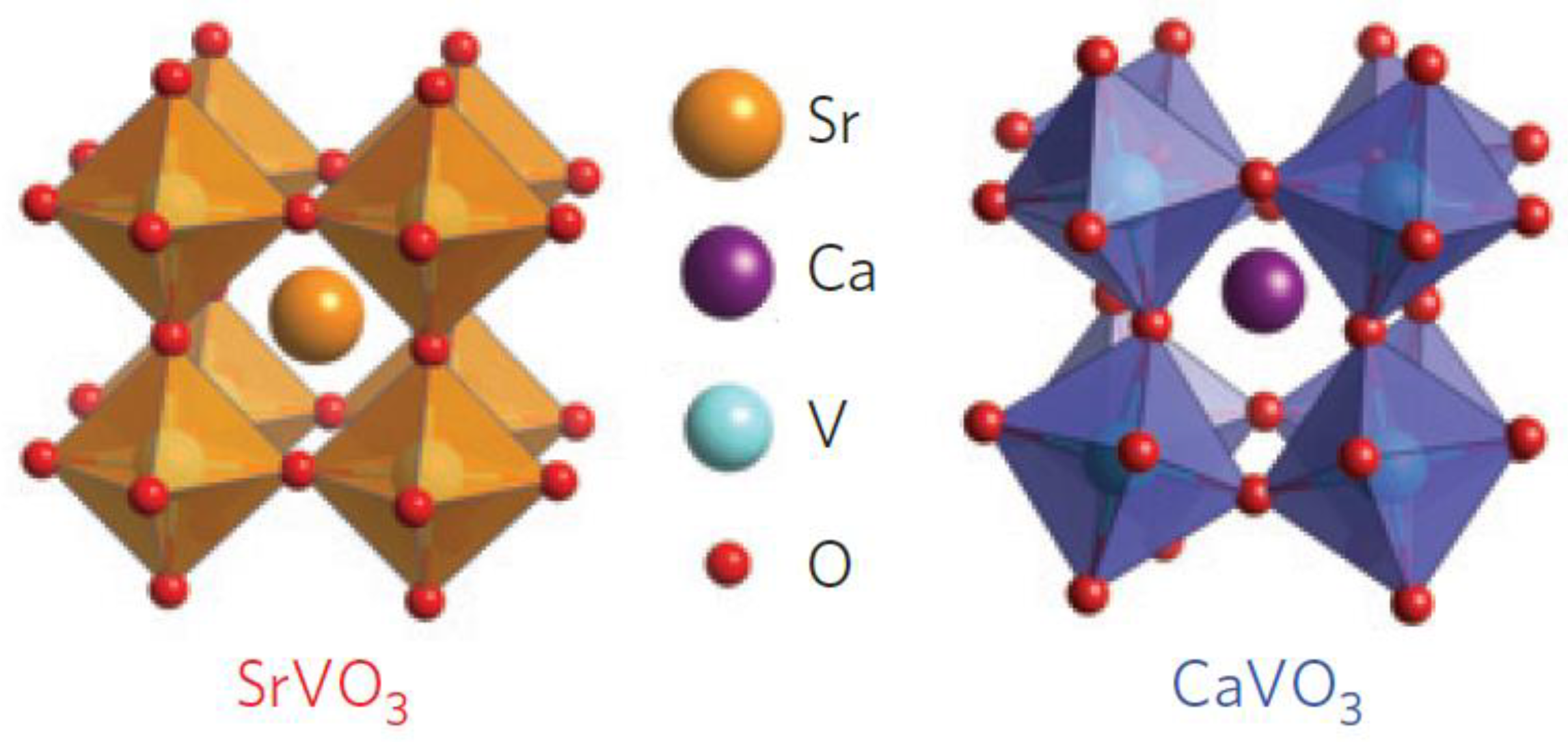
2.2. Perovskite Structure
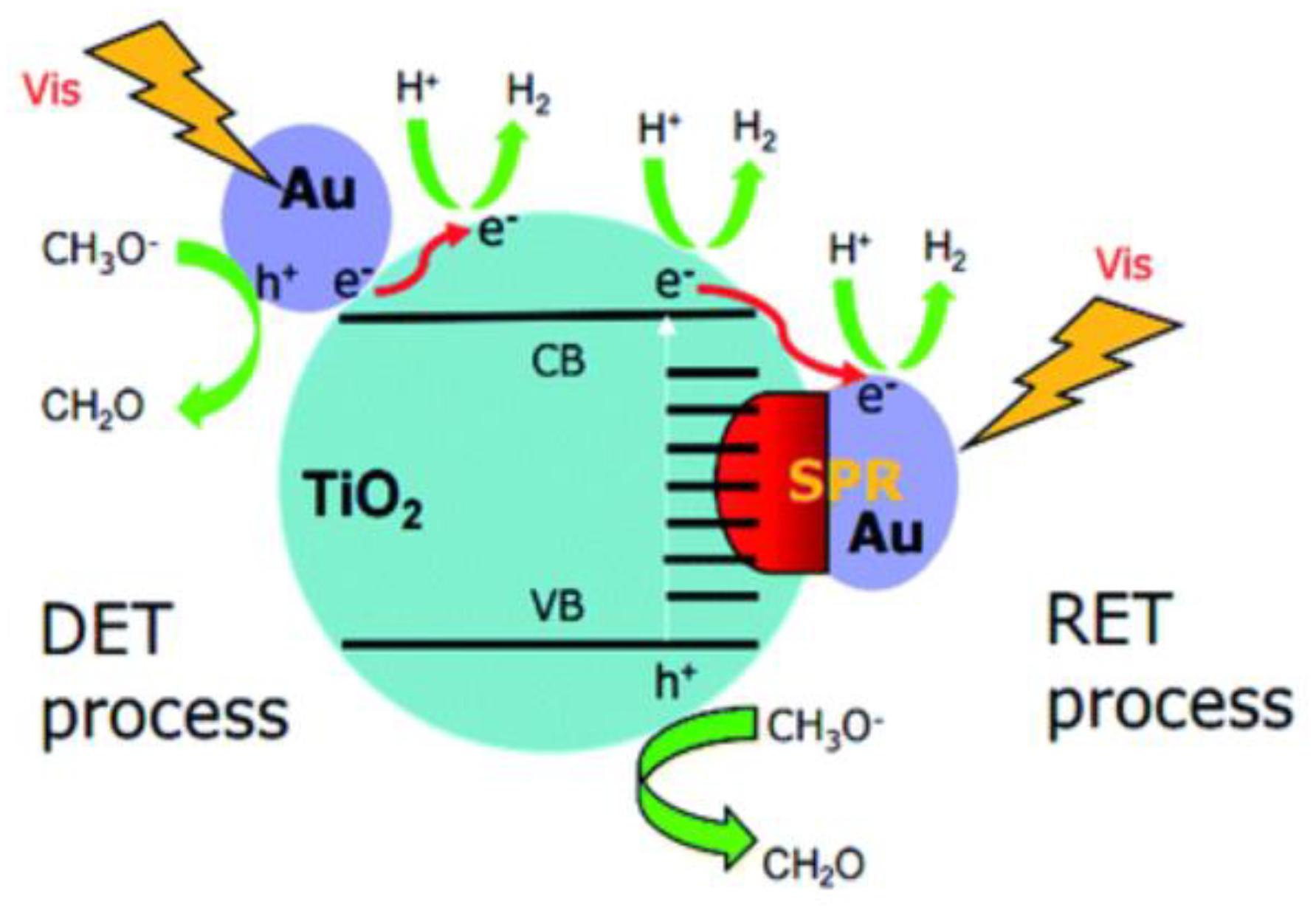
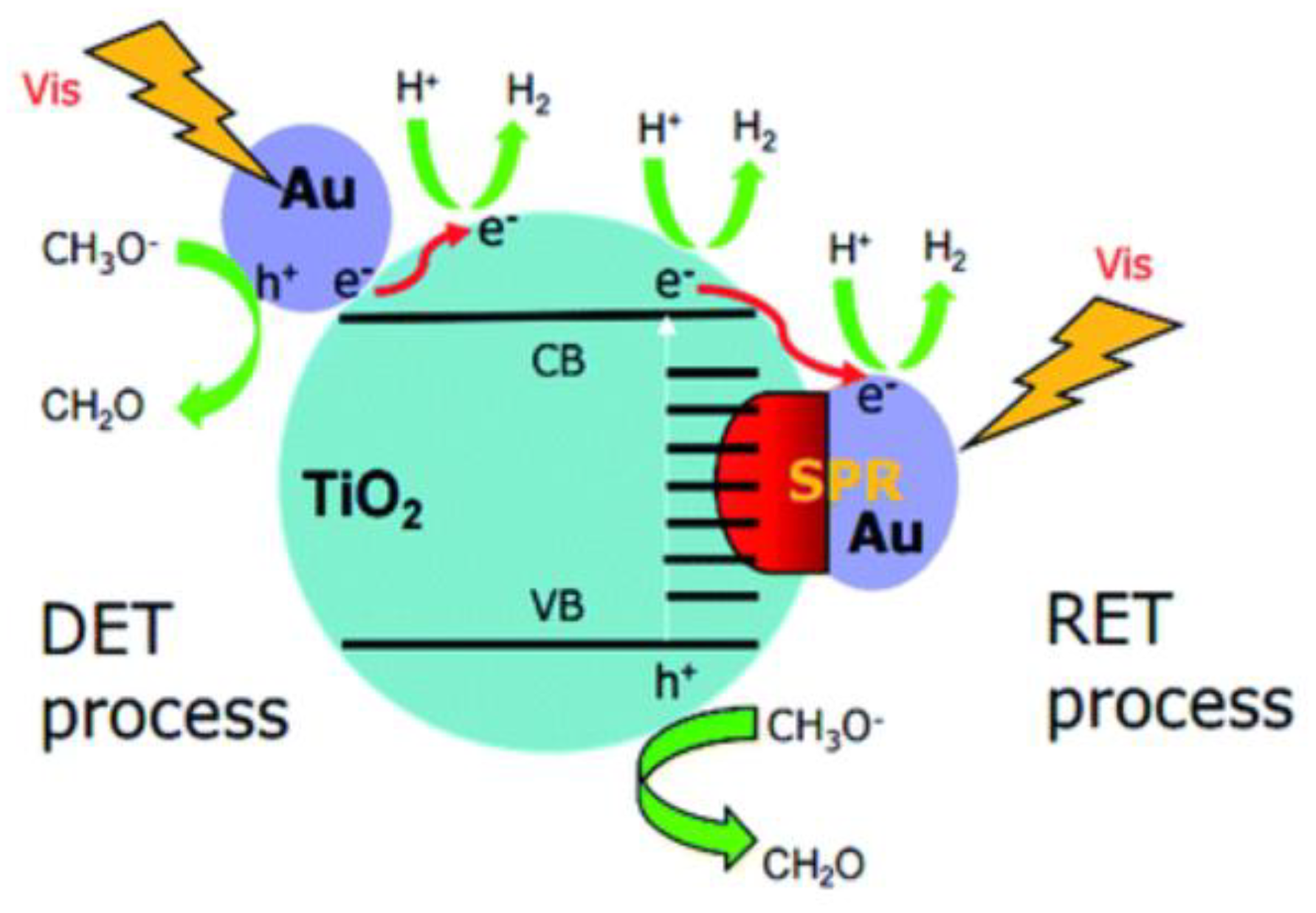
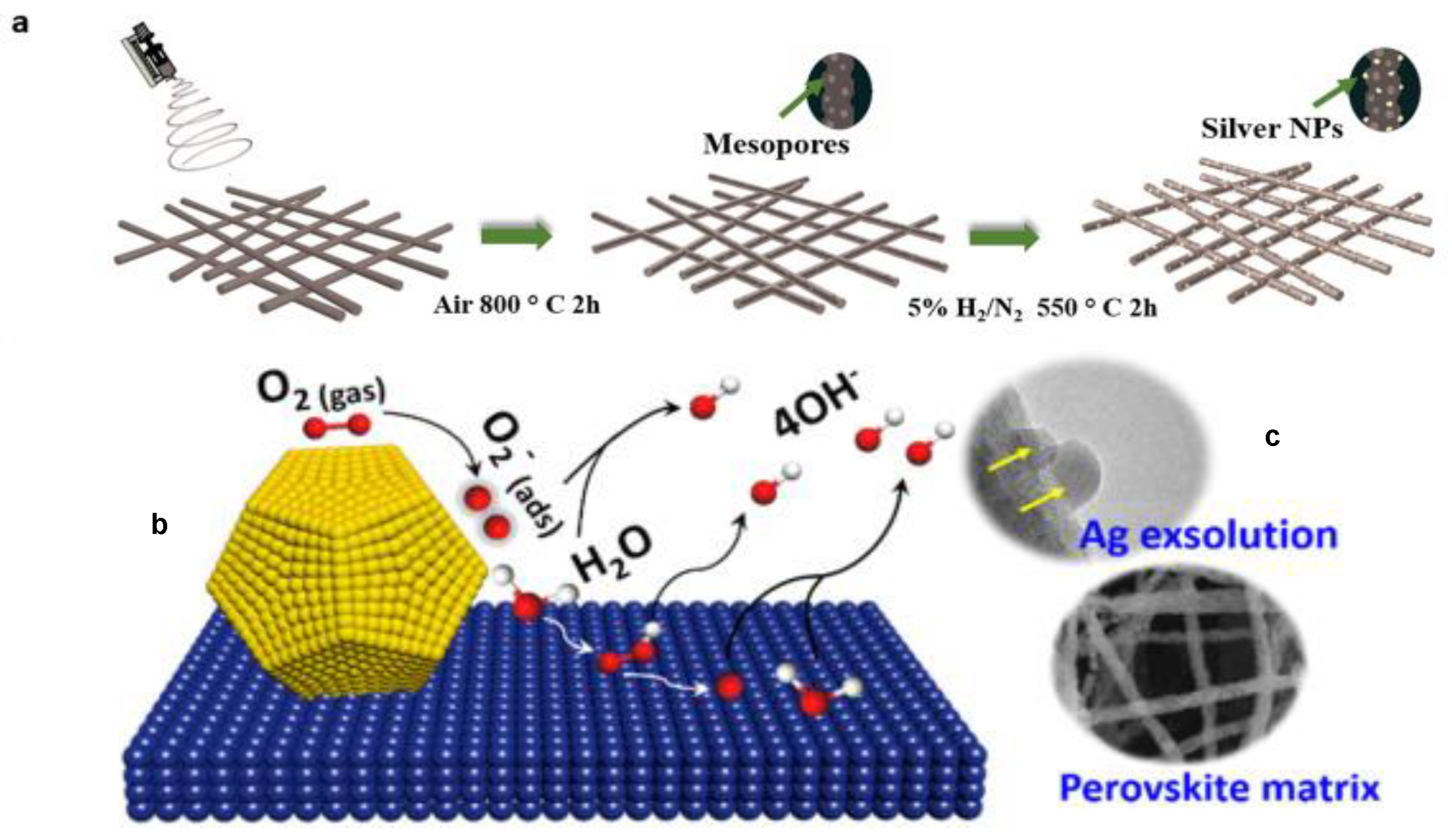
2.3. Perovskite-based Photocatalysts Supported by Precious Metals: A Synergistic Effect on Photocatalytic Performance
3. Chemical Synthesis for Perovskite Photocatalyst Obtaining
3.1. Solid-State Reaction
3.2. Sol–Gel Method
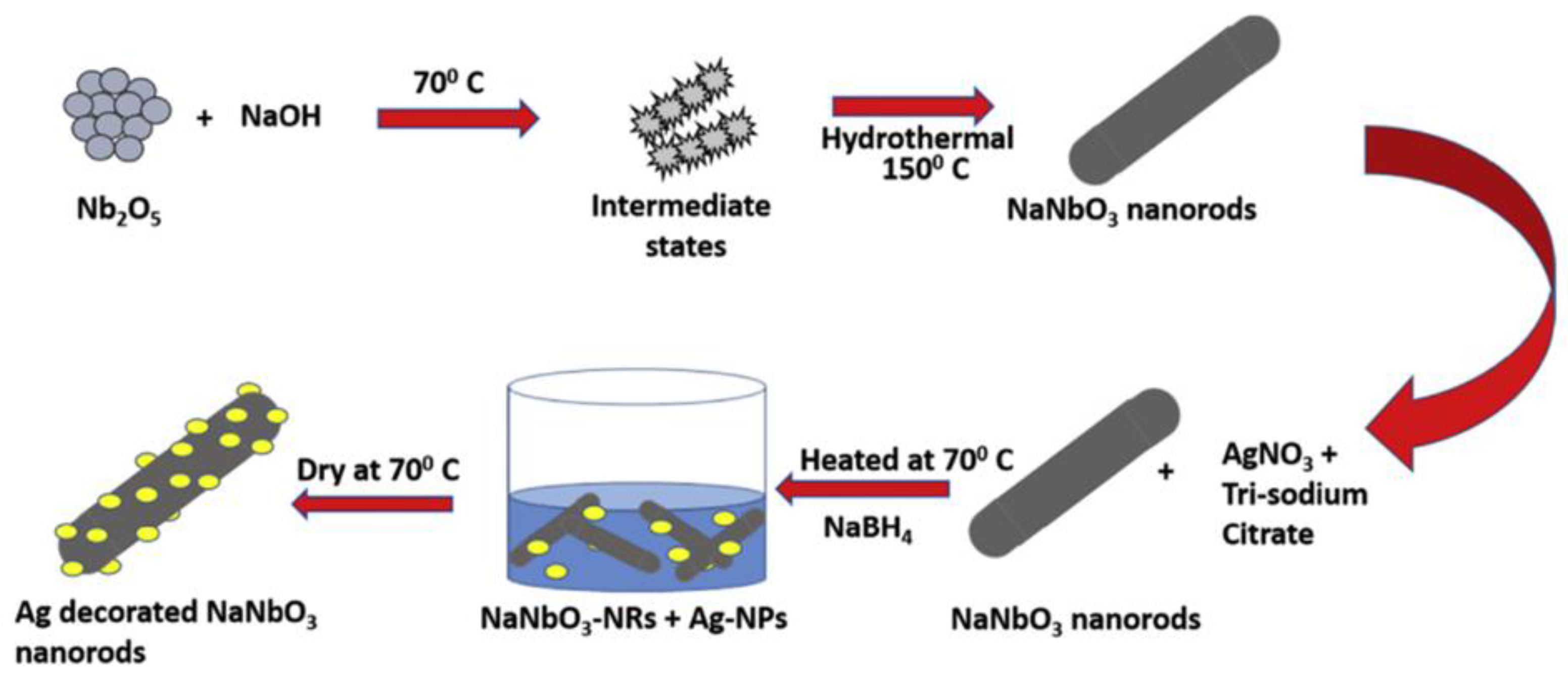
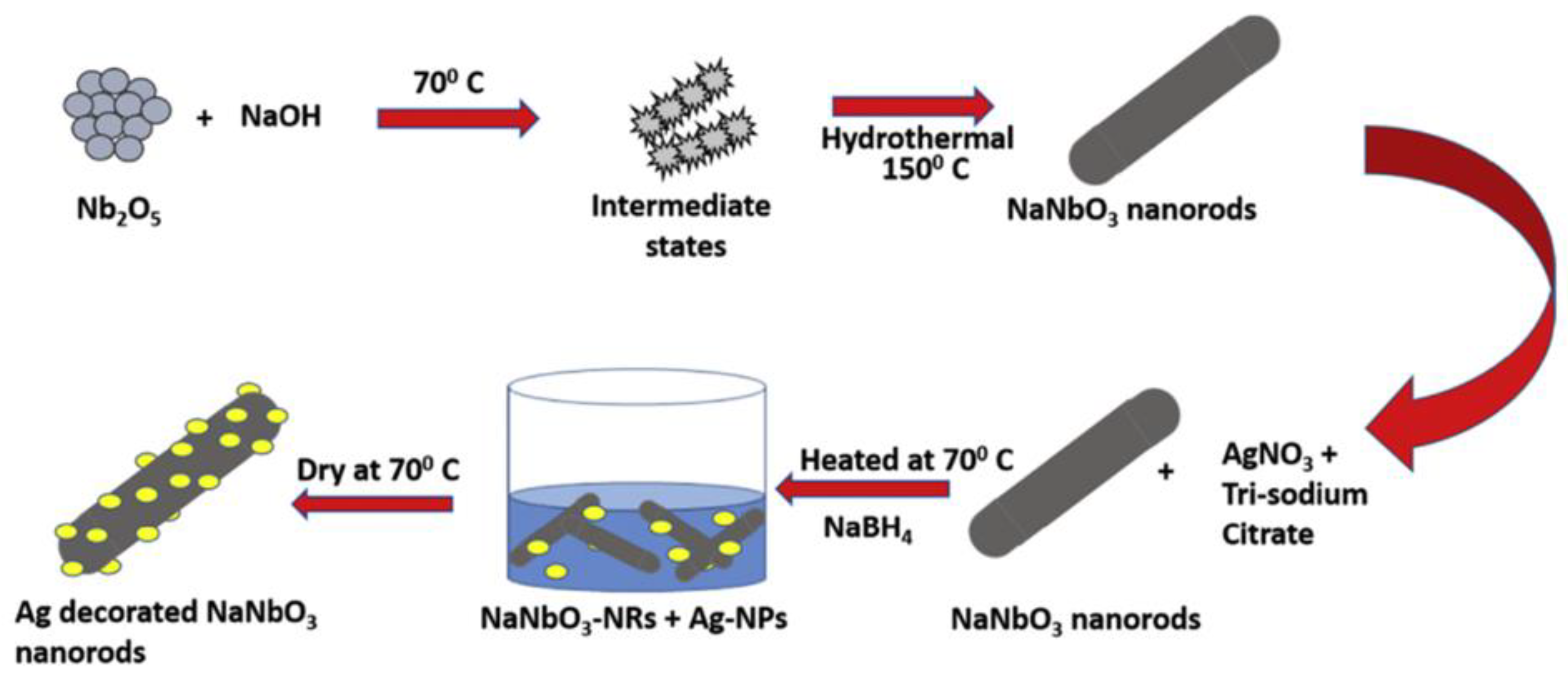
3.3. Hydrothermal Method
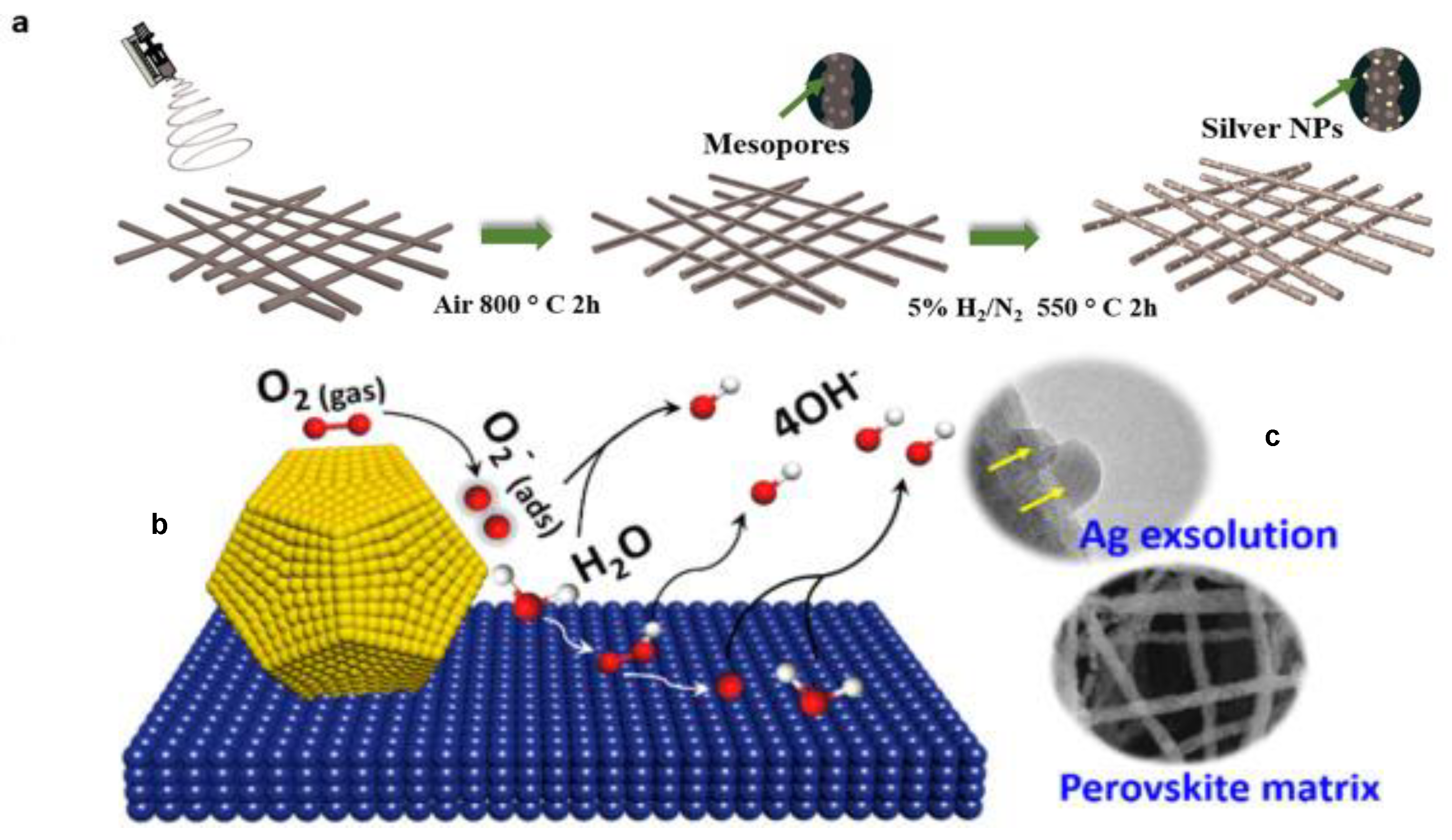
4. Application of Perovskites in Electrocatalysis
| ORR in alkaline medium | |
| O2 + 2H2O + 4e− → 4OH− (4-electrons process) | −1 |
| O2 + H2O + 2e− → HO2− + OH− | (2a) |
| H2O + HO2− + 2e− → 3OH− (2-electrons process) | (2b) |
| ORR in acid medium | |
| O2 + 4H+ + 4e− → 2H2O (4-electrons process) | −3 |
| O2 + 2H+ + 2e− → H2O2 | (4a) |
| H2O2 + 2H+ + 2e− → 2H2O (2-electrons process) | (4b) |
5. Final Considerations
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- IUPAC. Compendium of chemical terminology. In The Gold Book, 2nd ed.; McNaught, A.D., Wilkinson, A., Eds.; Blackwell Scientific Publications: Oxford, UK, 1997. [Google Scholar]
- Deng, Y.; Zhao, R. Advanced Oxidation Processes (AOPs) in Wastewater Treatment. Curr. Pollut. Rep. 2015, 1, 167–176. [Google Scholar] [CrossRef]
- Yang, L.; Yang, L.; Ding, L.; Deng, F.; Luo, X.-B.; Luo, S.-L. Principle for the application of nanomaterials in environmental pollution control and resource reutilization. In Nanomaterial for the Removal of Pollutants and Resource Reutilization; Luo, X., Deng, F., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 1–23. [Google Scholar]
- Glaze, W.H.; Kang, J.-W.; Chapin, D.H. The chemistry of water treatment processes involving ozone, hydrogen peroxide and ultraviolet radiation. Ozone Sci. Eng. 1987, 9, 335–352. [Google Scholar] [CrossRef]
- Parsons, S. Advanced Oxidation Processes for Water and Wastewater Treatment; IWA Publishing: London, UK, 2004; pp. 1–347. [Google Scholar]
- Fan, X.; Hao, H.; Shen, X.; Chen, F.; Zhang, J. Removal and degradation pathway study of sulfasalazine with Fenton-like reaction. J. Hazard. Mater. 2001, 190, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S.; Ullah, R.; Butt, A.M.; Gohar, N.D. Strategies of making TiO2 and ZnO visible light active. J. Hazard. Mater. 2009, 170, 560–569. [Google Scholar]
- WHO. Fluoride in drinking-water. In Guidelines for Drinking-Water Quality, 3rd ed.; WHO Press: Geneva, Switzerland, 2006; Volume 1, pp. 375–377. [Google Scholar]
- Ibrahim, M.; Asimrasheed, M.; Sumalatha, M.; Prabhakar, P. Effects of fluoride contents in ground water: A review. Inter. J. Pharm. Appl. 2011, 2, 128–134. [Google Scholar]
- Singh, J.; Singh, P.; Singh, A. Fluoride ions vs. removal technologies: A study. Arab. J. Chem. 2016, 9, 815–824. [Google Scholar] [CrossRef]
- Modi, S.; Soni, R. Merits and Demerits of different technologies of defluoridation for drinking water. IOSR J. Environ. Sci. Toxicol. Food. Technol. 2013, 3, 24–27. [Google Scholar] [CrossRef]
- Gupta, V.K.; Ali, I.; Saleh, T.A.; Nayak, A.; Agarwal, S. Chemical treatment technologies for waste-water recycling—An overview. RSC Adv. 2012, 2, 6380–6388. [Google Scholar] [CrossRef]
- Gogate, P.R.; Pandit, A.B. A review of imperative technologies for wastewater treatment II: Hybrid methods. Adv. Environ. Res. 2004, 8, 553–597. [Google Scholar] [CrossRef]
- Oller, I.; Malato, S.; Sanchez-Perez, J.A. Combination of advanced oxidation processes and biological treatments for wastewater decontamination—A review. Sci. Total Environ. 2011, 409, 4141–4166. [Google Scholar] [CrossRef]
- Esplugas, S.; Bila, D.M.; Krause, L.G.T.; Dezotti, M. Ozonation and advanced oxidation technologies to remove endocrine disrupting chemicals (EDCs) and pharmaceuticals and personal care products (PPCPs) in water effluents. J. Hazard. Mater. 2007, 149, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Saravanan, R.; Gracia, F.; Stephen, A. Basic principles, mechanism, and challenges of photocatalysis. In Nanocomposites for Visible Light-Induced Photocatalysis; Khan, M.M., Pradhan, D., Shon, Y., Eds.; Springer: Cham, Switzerland, 2018; pp. 19–40. [Google Scholar]
- Serpone, N.; Horikoshi, S.; Emeline, A.V. Microwaves in advanced oxidation processes for environmental applications. A brief review. J. Photochem. Photobiol. C 2010, 11, 114–131. [Google Scholar] [CrossRef]
- Legrini, O.; Oliveros, E.; Braun, A.M. Photochemical processes for water treatment. Chem. Rev. 1993, 93, 671–698. [Google Scholar] [CrossRef]
- Huang, C.P.; Dong, C.; Tang, Z. Advanced chemical oxidation: Its present role and potential future in hazardous waste treatment. Waste Manag. 1993, 13, 361–377. [Google Scholar] [CrossRef]
- Pignatello, J.J.; Oliveros, S.E.; Mackay, A. Advanced oxidation processes of organic contaminant destruction based of the Fenton reaction and related chemistry. Crit. Rev. Environ. Sci. Technol. 2006, 36, 1–84. [Google Scholar] [CrossRef]
- Carraway, E.R.; Hoffman, A.J.; Hoffmann, M.R. Photocatalytic oxidation of organic acids on quantum-sized semiconductor colloids. Environ. Sci. Technol. 1994, 28, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Neppolian, B.; Choi, H.S.; Sakthivel, S.; Arabindoo, B.; Murugesan, V. Solar light induced and TiO2 assisted degradation of textile dye reactive blue 4. Chemosphere 2002, 46, 1173–1181. [Google Scholar] [CrossRef]
- Reza, K.M.; Kurny, A.S.; Gulshan, F. Parameters affecting the photocatalytic degradation of dyes using TiO2: A review. Appl. Water Sci. 2015, 4, 1569–1578. [Google Scholar] [CrossRef]
- Lim, M.; Son, Y.; Khim, J. Frequency effects on the sonochemical degradation of chlorinated compounds. Ultrason. Sonochem. 2011, 8, 460–465. [Google Scholar] [CrossRef]
- Doodeve, C.F.; Kitchener, J.A. Photosensitisation by titanium dioxide. Trans. Faraday Soc. 1938, 34, 570–579. [Google Scholar]
- Hashimoto, K.; Irie, H.; Fujishima, A. TiO2 photocatalysis: A historical overview and future prospects. Jpn. J. Appl. Phys. 2005, 44, 8269–8285. [Google Scholar] [CrossRef]
- Ameta, S.C.; Ameta, R. Introduction. In Advanced Oxidation Processes for Wastewater Treatment; Ameta, S.C., Ameta, R., Eds.; Academic Press: Oxford, UK, 2018; pp. 1–12. [Google Scholar]
- Gaya, U.I. Principles of heterogeneous photocatalysis. In Heterogeneous Photocatalysis Using Inorganic Semiconductor Solids; Gaya, U.I., Ed.; Springer Science: Dordrecht, The Netherlands, 2014; pp. 1–34. [Google Scholar]
- Gratzel, M. Photoelectrochemical cells. Nature 2001, 414, 338–344. [Google Scholar] [CrossRef]
- Fujishima, A.; Honda, K. Electrochemical photolysis of water at a semiconductor electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef]
- Cowan, A.J.; Durrant, J. Long-lived charge separated states in nanostructured semiconductor photoelectrodes for the production of solar fuels. Chem. Soc. Rev. 2013, 42, 2281–2293. [Google Scholar] [CrossRef]
- Al-Ekabi, H.; Serpone, M. Kinetic studies in heterogeneous photocatalysis. 1. Photocatalytic degradation of chlorinated phenols in aerated aqueous solutions over TiO2 supported on a glass matrix. J. Phys. Chem. 1988, 92, 5726–5731. [Google Scholar] [CrossRef]
- Serpone, N. Brief introductory remarks on heterogeneous photocatalysis. Sol. Energy Mater. Sol. Cells 1995, 38, 369–379. [Google Scholar] [CrossRef]
- Paramasivam, I.; Jha, H.; Liu, N.; Schmuki, P. A review of photocatalysis using self-organized TiO2 nanotubes and other ordered oxide nanostructures. Small 2012, 8, 3073–3103. [Google Scholar] [CrossRef]
- Shen, Y.; Guo, X.; Bo, X.; Wang, T.; Guo, X.; Xie, M.; Guo, X. Effect of template-induced surface species on electronic structure and photocatalytic activity of g-C3N4. Appl. Surf. Sci. 2017, 396, 933–938. [Google Scholar] [CrossRef]
- Ravelli, D.; Dondi, D.; Fagnoni, M.; Albini, A. Photocatalysis. A multi-faceted concept for green chemistry. Chem. Soc. Rev. 2009, 38, 1999–2011. [Google Scholar] [CrossRef]
- Liu, B.; Zhao, X.; Terashima, C.; Fujishima, A.; Nakata, K. Thermodynamic and kinetic analysis of heterogeneous photocatalysis for semiconductor systems. Phys. Chem. Chem. Phys. 2014, 16, 8751–8760. [Google Scholar] [CrossRef]
- Henderson, M.A. A surface science perspective on TiO2 photocatalysis. Surf. Sci. Rep. 2011, 66, 185–297. [Google Scholar] [CrossRef]
- Grabowska, E. Selected perovskite oxides: Characterization, preparation and photocatalytic properties—A review. Appl. Catal. B Environ. 2016, 186, 97–126. [Google Scholar] [CrossRef]
- Zhang, G.; Liu, G.; Wang, L.; Irvine, J.T.S. Inorganic perovskite photocatalysts for solar energy utilization. Chem. Soc. Rev. 2016, 45, 5951–5984. [Google Scholar] [CrossRef] [Green Version]
- Rojas-Cervantes, M.L.; Castillejos, E. Perovskites as catalysts in advanced oxidation processes for wastewater treatment. Catalysts 2019, 9, 230. [Google Scholar] [CrossRef]
- Shi, J.; Guo, L. ABO3-based photocatalysts for water splitting. Prog. Nat. Sci. Mater. Inter. 2012, 22, 592–615. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, R.H. Perovskites: Modern and Ancient; Almaz Press: Thunder Bay, ON, Canada, 2002; pp. 1–318. [Google Scholar]
- Glazer, A.M. The classification of tilted octahedra in perovskites. Acta Crystallogr. Sect. B 1972, 28, 3384–3392. [Google Scholar] [CrossRef]
- Tilley, R.J.D. Perovskites. Structure-Property Relationships; John Wiley & Sons Ltd.: Chichester, UK, 2016; pp. 1–315. [Google Scholar]
- Mishra, A.; Prasad, R. Preparation and application of perovskite catalyst for diesel soot emission control: An overview. Catal. Rev. 2014, 56, 57–81. [Google Scholar] [CrossRef]
- Knight, K.S. Structural phase transitions, oxygen vacancy ordering and protonation in doped BaCeO3: Results from time-of-flight neutron powder diffraction investigations. Solid State Ion. 2001, 145, 275–294. [Google Scholar] [CrossRef]
- Woodward, P.M. Octahedral tilting in perovskites. I. geometrical considerations. Acta Crystallogr. Sect. B Struct. Sci. 1997, 53, 32–43. [Google Scholar] [CrossRef]
- Goldschmidt, V.M. Die Gesetze der Krystallochemie. Naturwissenschaften 1926, 14, 477–485. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomie distances in halides and chaleogenides. Acta Crystallogr. Sect. A Cryst. Phys. Diffr. Theor. Gen. Crystallogr. 1976, A32, 751–767. [Google Scholar] [CrossRef]
- Fan, Z.; Sun, K.; Wang, J. Perovskites for photovoltaics: A combined review of organic–inorganic halide perovskites and ferroelectric oxide perovskites. J. Mater. Chem. A 2015, 3, 18809–18828. [Google Scholar] [CrossRef]
- Yamada, I.; Takamatsu, A.; Ikeno, H. Complementary evaluation of structure stability of perovskite oxides using bond-valence and density functional-theory calculations. Sci. Technol. Adv. Mater. 2018, 19, 102–107. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, Y.; Guo, L.; Zhao, W.W.; Barnes, A.; Zhang, H.-T.; Eatom, C.; Zheng, Y.; Brahlek, M.; Haneef, H.F.; et al. Correlated metals as transparent conductor. Nat. Mater. 2016, 15, 204–210. [Google Scholar] [CrossRef]
- Imran, Z.; Rafiq, M.A.; Hasan, M.M. Charge carrier transport mechanisms in perovskite CdTiO3 fibers. AIP Adv. 2014, 4, 067137. [Google Scholar] [CrossRef]
- Nikonov, A.V.; Kuterbekov, K.A.; Bekmyrza, K.Z.; Pavzderin, N.B. A brief review of conductivity and thermal expansion of perovskite-related oxides for SOFC cathode. Eurasian J. Phys. Funct. Mater. 2018, 2, 274–292. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Yang, L.; Ai, C.; Xie, P.; Lin, S.; Wang, C.-Z.; Lu, X. Tailoring band gap of perovskite BaTiO3 by Transition metals co-doping for visible-light photoelectrical applications: A first-principles study. Nanomaterials 2018, 8, 455. [Google Scholar] [CrossRef]
- Hwang, S.W.; Noh, T.H.; Cho, I.S. Optical properties, electronic structures, and photocatalytic performances of bandgap-tailored SrBi2Nb2−xVxO9 compounds. Catalysts 2019, 9, 393. [Google Scholar] [CrossRef]
- Linic, S.; Christopher, P.; Ingram, D.B. Plasmonic-metal nanostructures for efficient conversion of solar to chemical energy. Nat. Mater. 2001, 10, 911–921. [Google Scholar] [CrossRef]
- Linic, S.; Aslam, U.; Boerigter, C.; Morabito, M. Photochemical transformations on plasmonic metal nanoparticles. Nat. Mater. 2015, 14, 567–576. [Google Scholar] [CrossRef]
- Nycenga, M.; Cobley, C.M.; Zeng, J.; Li, W.; Moran, C.H.; Zhang, Q.; Qin, D.; Xia, Y. Controlling the synthesis and assembly of silver nanostructures for plasmonic applications. Chem. Rev. 2011, 111, 3669–3712. [Google Scholar] [CrossRef]
- Wang, M.; Ye, M.; Iocozzia, J.; Lin, C.; Lin, Z. Plasmon-mediated solar energy conversion via photocatalysis in noble metal/semiconductor composites. Adv. Sci. 2016, 3, 1600024. [Google Scholar] [CrossRef]
- Hou, W.; Liu, Z.; Pavaskar, P.; Hung, W.H.; Cronin, S.B. Plasmonic enhancement of photocatalytic decomposition of methyl orange under visible light. J. Catal. 2011, 277, 149–153. [Google Scholar] [CrossRef]
- Nie, J.; Scheneider, J.; Sieland, F.; Zhou, L.; Xia, S.; Bahnemann. New insights into the surface plasmon resonance (SPR) driven photocatalytic H2 production of Au-TiO2. RSC Adv. 2018, 8, 25881–25887. [Google Scholar] [CrossRef]
- Liz-Marzán, L.M. Tailoring surface plasmons through the morphology and assembly of metal nanoparticles. Langmuir 2006, 22, 32–41. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, Y.; Yang, X.; Wang, S.; Shen, J.; Lin, B.; Li, C. Enhanced visible light photocatalytic activity of interlayer-isolated triplex Ag@SiO2@TiO2 core-shell nanoparticles. Nanoscale 2013, 5, 3359–3366. [Google Scholar] [CrossRef]
- Hou, W.; Cronin, S.B. A review of surface plasmon resonance-enhanced photocatalysis. Adv. Funct. Mater. 2013, 23, 1612–1619. [Google Scholar] [CrossRef]
- Wang, H.; Tam, F.; Grady, N.K.; Halas, N.J. Cu nanoshells: Effects of interband transitions on the nanoparticle plasmon resonance. J. Phys. Chem. B 2005, 109, 18218–18222. [Google Scholar] [CrossRef]
- Musialic-Piotrowwska, A.; Landmesser, H. Noble metal-doped perovskites for the oxidation of organic air pollutants. Catal. Today 2008, 137, 357–361. [Google Scholar] [CrossRef]
- Smart, L.E.; Moore, E.A. Solid State Chemistry, 3rd ed.; Taylor & Francis: Abingdon, UK, 2005; pp. 148–177. [Google Scholar]
- West, A.R. Solid State Chemistry and Its Applications, 2nd ed.; Wiley: Chichester, UK, 2014; pp. 187–228. [Google Scholar]
- Iwashina, K.; Kudo, A. Rh-doped SrTiO3 photocatalyst electrode showing cathodic photocurrent for water splitting under visible-light irradiation. J. Am. Chem. Soc. 2011, 133, 13272–13275. [Google Scholar] [CrossRef]
- Irie, H.; Maruyama, Y.; Hashimoto, K. Ag+ and Pb2+ doped SrTiO3 Photocatalysts. A correlation between band structure and photocatalytic activity. J. Phys. Chem. C 2007, 111, 1847–1852. [Google Scholar] [CrossRef]
- Saadetnejad, D.; Yıldırım, R. Photocatalytic hydrogen production by water splitting over Au/Al-SrTiO3. Int. J. Hydrog. Energy 2018, 43, 1116–1122. [Google Scholar] [CrossRef]
- Wang, F.; Wang, T.; Lang, J.; Su, Y.; Wang, X. Improved photocatalytic activity and durability of AgTaO3/AgBr heterojunction: The relevance of phase and electronic structure. J. Mol. Catal. A Chem. 2017, 426, 52–59. [Google Scholar] [CrossRef]
- Konta, R.; Ishii, T.; Kato, H.; Kudo, A. Photocatalytic activities of noble metal ion doped SrTiO3 under visible light irradiation. J. Phys. Chem. B 2004, 108, 8992–8995. [Google Scholar] [CrossRef]
- Sajjadi, S.P. Sol-gel process and its application in Nanotechnology. J. Polym. Eng. Technol. 2005, 13, 38–41. [Google Scholar]
- Rao, B.G.; Mukherjee, D.; Reddy, B.M. Nanostructures for novel therapy novel approaches for preparation of nanoparticles. In Nanostructures for Novel Therapy; Fricai, D., Grumezescu, A., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 1–36. [Google Scholar]
- Nayak, A.K.; Das, B. Introduction to polymeric gels. In Polymeric Gels. Characterization, Properties and Biomedical Applications; Pal, K., Banerjee, I., Eds.; Elsevier: Duxford, UK, 2018; pp. 3–27. [Google Scholar]
- Zhang, H.; Chen, G.; He, X.; Xu, J. Electronic structure and photocatalytic properties of Ag-La codoped CaTiO3. J. Alloy. Compd. 2012, 516, 91–95. [Google Scholar] [CrossRef]
- Reddy, K.H.; Martha, S.; Parida, K.M. Erratic charge transfer dynamics of Au/ZnTiO3 nanocomposites under UV and visible light irradiation and their related photocatalytic activities. Nanoscale 2018, 10, 18540–18554. [Google Scholar] [CrossRef]
- Li, Y.; Li, J.; Chen, L.; Sun, H.; Zhang, H.; Guo, H.; Feng, L. In situ synthesis of au-induced hierarchical nanofibers/nanoflakes structured BiFeO3 homojunction photocatalyst with enhanced photocatalytic activity. Front. Chem. 2019, 6, 649–657. [Google Scholar] [CrossRef]
- Feng, S.; Li, G. Hydrothermal and solvothermal syntheses. In Modern Inorganic Synthetic Chemistry, 2nd ed.; Xu, R., Xu, Y., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 73–104. [Google Scholar]
- Yan, Y.; Yang, H.; Yi, Z.; Li, R.; Wang, X. Enhanced photocatalytic performance and mechanism of Au@CaTiO3 composites with au nanoparticles assembled on CaTiO3 nanocuboids. Micromachines 2019, 10, 254. [Google Scholar] [CrossRef]
- Malkhasian, A.Y.S. Synthesis and characterization of Pt/AgVO3 nanowires for degradation of atrazine using visible light irradiation. J. Alloy. Compd. 2015, 649, 394–399. [Google Scholar] [CrossRef]
- Zielińska, B.; Borowiak-Palen, E.; Kalenczuk, R.J. Preparation, characterization and photocatalytic activity of metal-loaded NaNbO3. J. Phys. Chem. Solids 2011, 72, 117–123. [Google Scholar] [CrossRef]
- Chen, W.; Hu, Y.; Ba, M. Surface interaction between cubic phase NaNbO3 nanoflowers and Ru nanoparticles for enhancing visible-light driven photosensitized photocatalysis. Appl. Surf. Sci. 2018, 435, 483–493. [Google Scholar] [CrossRef]
- Zhang, T.; Lei, W.; Liu, P.; Rodriguez, J.A.; Yu, J.; Qi, Y.; Liu, G.; Liu, M. Organic Pollutant Photodecomposition by Ag/KNbO3 Nanocomposites: A combined experimental and theoretical study. J. Phys. Chem. C 2016, 120, 2777–2786. [Google Scholar] [CrossRef]
- Xing, P.; Wu, S.; Chen, Y.; Chen, P.; Hu, X.; Lin, H.; Zhao, L.; He, Y. New Application and Excellent Performance of Ag/KNbO3 Nanocomposite in Photocatalytic NH3 Synthesis. ACS Sustain. Chem. Eng. 2019, 7, 12408–124118. [Google Scholar] [CrossRef]
- Xu, D.; Yang, S.; Jin, Y.; Chen, M.; Fan, W.; Luo, B.; Shi, W. Ag-decorated ATaO3 (A = K, Na) nanocube plasmonic photocatalysts with enhanced photocatalytic water-splitting properties. Langmuir 2015, 31, 9694–9699. [Google Scholar] [CrossRef]
- Jayabal, P.; Sasirekha, V.; Mayandi, J.; Jeganathan, K.; Ramakrishnan, V. A facile hydrothermal synthesis of SrTiO3 for dye sensitized solar cell application. J. Alloy. Compd. 2014, 586, 456–461. [Google Scholar] [CrossRef]
- Gao, H.; Yang, H.; Wang, S. Hydrothermal synthesis, growth mechanism, optical properties and photocatalytic activity of cubic SrTiO3 particles for the degradation of cationic and anionic dyes. Optik 2018, 175, 237–249. [Google Scholar] [CrossRef]
- Zhang, Q.; Huang, Y.; Xu, L.; Cao, J.; Ho, W.; Lee, S.C. Visible-light-active plasmonic Ag-SrTiO3 nanocomposites for the degradation of NO in air with high selectivity. ACS Appl. Mater. Interfaces 2016, 8, 4165–4178. [Google Scholar] [CrossRef]
- Kalyani, V.; Vasile, B.S.; Ianculescu, A.; Testino, A.; Carino, A.; Buscaglia, M.T.; Buscaglia, V.; Nanni, P. Hydrothermal synthesis of SrTiO3: Role of interfaces. Cryst. Growth Des. 2015, 15, 5712–5725. [Google Scholar] [CrossRef]
- Kumar, D.; Singh, S.; Khare, N. Plasmonic Ag nanoparticles decorated NaNbO3 nanorods for efficient photoelectrochemical water splitting. Int. J. Hydrog. Energy 2018, 43, 1–8. [Google Scholar] [CrossRef]
- Jeong, S.Y.; Shin, H.-M.; Jo, Y.-R.; Kin, Y.J.; Kim, S.; Lee, W.-J.; Lee, G.J.; Song, J.; Moon, B.J.; Seo, S.; et al. Plasmonic Silver nanoparticle-impregnated nanocomposite BiVO4 photoanode for plasmon-enhanced photocatalytic water splitting. J. Phys. Chem. C 2018, 122, 7088–7093. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, B.-P.; Li, S.; Huang, Z.; Yang, C.; Wang, H. Enhanced photocatalytic activity in Ag-nanoparticle-dispersed BaTiO3 composite thin films: Role of charge transfer. J. Adv. Ceram. 2017, 6, 1–10. [Google Scholar] [CrossRef]
- Gpel, W.; Schierbaum, K.D.; Vayenas, C.G.; Yentekakis, I.V.; Gerischer, H.; Vielstich, W.; Savage, P.E.; Moyes, R.B.; Bond, G.; Suslick, K.S. Special Catalytic Systems. In Handbook of Heterogeneous Catalysis; Ertl, G., Knözinger, H., Weitkamp, J., Eds.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 1997; pp. 1283–1357. [Google Scholar]
- Wang, Y.-J.; Zhao, N.; Fang, B.; Li, H.; Bi, X.T.; Wang, H. Carbon-Supported Pt-Based Alloy Electrocatalysts for the Oxygen Reduction Reaction in Polymer Electrolyte Membrane Fuel Cells: Particle Size, Shape, and Composition Manipulation and Their Impact to Activity. Chem. Rev. 2015, 115, 3433–3467. [Google Scholar] [CrossRef] [Green Version]
- Shan, S.; Wu, J.; Kang, N.; Cronk, H.; Zhao, Y.; Zhao, W.; Skeete, Z.; Joseph, P.; Trimm, B.; Luo, J.; et al. Nanoscale Alloying in Electrocatalysts. Catalysts 2015, 5, 1465–1478. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Zhang, P.; Dai, S. Recent Advances of Lanthanum-Based Perovskite Oxides for Catalysis. ACS Catal. 2015, 5, 6370–6385. [Google Scholar] [CrossRef]
- Ghosh, S.; Basu, R.N. Multifunctional nanostructured electrocatalysts for energy conversion and storage: Current status and perspectives. Nanoscale 2018, 10, 11241–11280. [Google Scholar] [CrossRef]
- Ghosh, S.K.; Rahaman, H. Noble metal-manganese oxide hybrid nanocatalysts. In Noble Metal-Metal Oxide Hybrid Nanoparticles; Elsevier: Amsterdam, The Netherlands, 2019; pp. 313–340. [Google Scholar]
- Zhang, J.; Xia, Z.; Dai, L. Carbon-based electrocatalysts for advanced energy conversion and storage. Sci. Adv. 2015, 1, e1500564. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, B. Recent advances in porous Pt-based nanostructures: Synthesis and electrochemical applications. Chem. Soc. Rev. 2014, 43, 2439–2444. [Google Scholar] [CrossRef]
- Shao-Horn, Y.; Hwang, J.; Yu, Y.; Katayama, Y.; Rao, R.R.; Giordano, L. Perovskites in catalysis and electrocatalysis. Science 2017, 358, 751–756. [Google Scholar] [Green Version]
- Zhang, Y.Q.; Tao, H.B.; Liu, J.; Sun, Y.F.; Chen, J.; Hua, B.; Thundat, T.; Luo, J.L. A rational design for enhanced oxygen reduction: Strongly coupled silver nanoparticles and engineered perovskite nanofibers. Nano Energy 2017, 38, 392–400. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhou, W.; Ran, R.; Chen, Y.; Shao, Z.; Liu, M. Promotion of Oxygen Reduction by Exsolved Silver Nanoparticles on a Perovskite Scaffold for Low-Temperature Solid Oxide Fuel Cells. Nano Lett. 2016, 16, 512–518. [Google Scholar] [CrossRef]
- Huang, K. An emerging platform for electrocatalysis: Perovskite exsolution. Sci. Bull. 2016, 61, 1783–1784. [Google Scholar] [CrossRef]
- Mitlitsky, F.; Myers, B.; Weisberg, A.H. Regenerative fuel cell systems. Energy Fuels 1998, 12, 56–71. [Google Scholar] [CrossRef]
- Retuerto, M.; Calle-Vallejo, F.; Pascual, L.; Lumbeeck, G.; Fernandez-Diaz, M.T.; Croft, M.; Gopalakrishnan, J.; Peña, M.A.; Hadermann, J.; Greenblatt, M. La1.5Sr0.5NiMn0.5Ru0.5O6 double perovskite with enhanced ORR/OER bifunctional catalytic activity. ACS Appl. Mater. Interfaces 2019, 11, 21454–21464. [Google Scholar] [CrossRef]
- Elumeeva, K.; Masa, J.; Tietz, F.; Yang, F.; Xia, W.; Muhler, M.; Schuhmann, W. A Simple Approach towards High-Performance Perovskite-Based Bifunctional Oxygen Electrocatalysts. ChemElectroChem 2016, 3, 138–143. [Google Scholar] [CrossRef]
- Rincón, R.A.; Masa, J.; Mehrpour, S.; Tietz, F.; Schuhmann, W. Activation of oxygen evolving perovskites for oxygen reduction by functionalization with Fe-Nx/C groups. Chem. Commun. 2014, 50, 14760–14762. [Google Scholar] [CrossRef]
- Elumeeva, K.; Masa, J.; Sierau, J.; Tietz, F.; Muhler, M.; Schuhmann, W. Perovskite-based bifunctional electrocatalysts for oxygen evolution and oxygen reduction in alkaline electrolytes. Electrochim. Acta 2016, 208, 25–32. [Google Scholar] [CrossRef]
- Zhang, D.; Song, Y.; Du, Z.; Wang, L.; Li, Y.; Goodenough, J.B. Active LaNi1−xFexO3 bifunctional catalysts for air cathodes in alkaline media. J. Mater. Chem. A 2015, 3, 9421–9426. [Google Scholar] [CrossRef]
- Du, Z.; Yang, P.; Wang, L.; Lu, Y.; Goodenough, J.B.; Zhang, J.; Zhang, D. Electrocatalytic performances of LaNi1−xMgxO3 perovskite oxides as bi-functional catalysts for lithium air batteries. J. Power Sources 2014, 265, 91–96. [Google Scholar] [CrossRef]
- Warwick, M.E.A.; Kaunisto, K.; Barreca, D.; Carraro, G.; Gasparotto, A.; Maccato, C.; Bontempi, E.; Sada, C.; Ruoko, T.-P.; Turner, S.; et al. Vapor Phase Processing of α-Fe2O3 Photoelectrodes for Water Splitting: An Insight into the Structure/Property Interplay. ACS Appl. Mater. Interfaces 2015, 7, 8667–8676. [Google Scholar] [CrossRef]
- Guerrero, A.; Bisquert, J. Perovskite semiconductors for photoelectrochemical water splitting applications. Curr. Opin. Electrochem. 2017, 2, 144–147. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| M | Eg/eV | Activity/µmol h−1 | |
|---|---|---|---|
| H2 b | O2 c | ||
| Mn | 2.7 | 0.2 | 2.7 |
| Ru | 1.9 | 1.7 | 3.9 |
| Rh | 1.7 | 17.2 | 0 |
| Ir | 2.3 | 8.6 | 0.4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teixeira, G.F.; Silva Junior, E.; Vilela, R.; Zaghete, M.A.; Colmati, F. Perovskite Structure Associated with Precious Metals: Influence on Heterogenous Catalytic Process. Catalysts 2019, 9, 721. https://doi.org/10.3390/catal9090721
Teixeira GF, Silva Junior E, Vilela R, Zaghete MA, Colmati F. Perovskite Structure Associated with Precious Metals: Influence on Heterogenous Catalytic Process. Catalysts. 2019; 9(9):721. https://doi.org/10.3390/catal9090721
Chicago/Turabian StyleTeixeira, Guilhermina Ferreira, Euripedes Silva Junior, Ramon Vilela, Maria Aparecida Zaghete, and Flávio Colmati. 2019. "Perovskite Structure Associated with Precious Metals: Influence on Heterogenous Catalytic Process" Catalysts 9, no. 9: 721. https://doi.org/10.3390/catal9090721
APA StyleTeixeira, G. F., Silva Junior, E., Vilela, R., Zaghete, M. A., & Colmati, F. (2019). Perovskite Structure Associated with Precious Metals: Influence on Heterogenous Catalytic Process. Catalysts, 9(9), 721. https://doi.org/10.3390/catal9090721