Structure-Activity Relationship of Manganese Oxide Catalysts for the Catalytic Oxidation of (chloro)-VOCs



Abstract
:1. Introduction
2. Results and Discussion
3. Experimental
3.1. Catalysts Preparation
3.2. Catalyst Characterizations
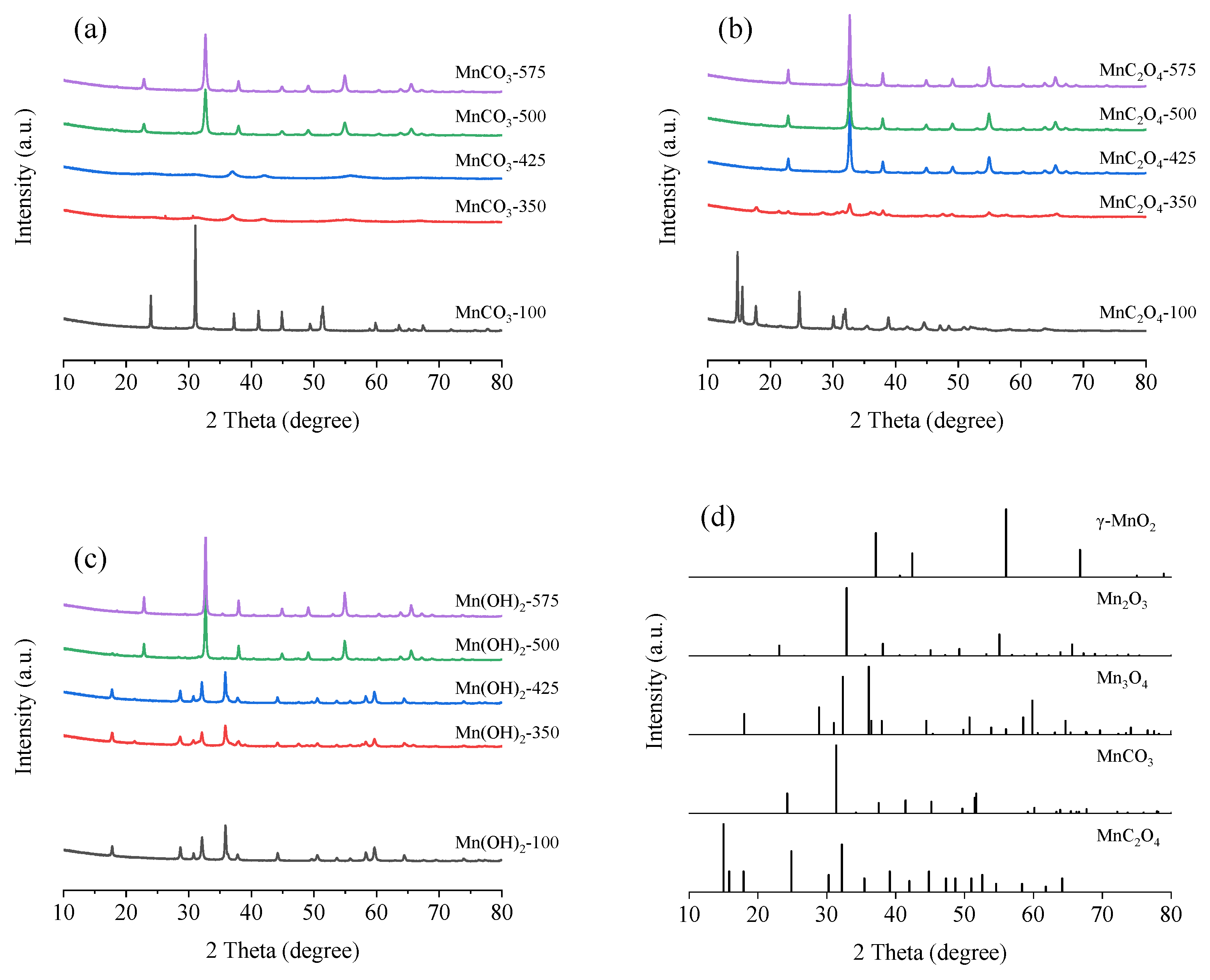
3.2.1. X-ray Diffraction (XRD)
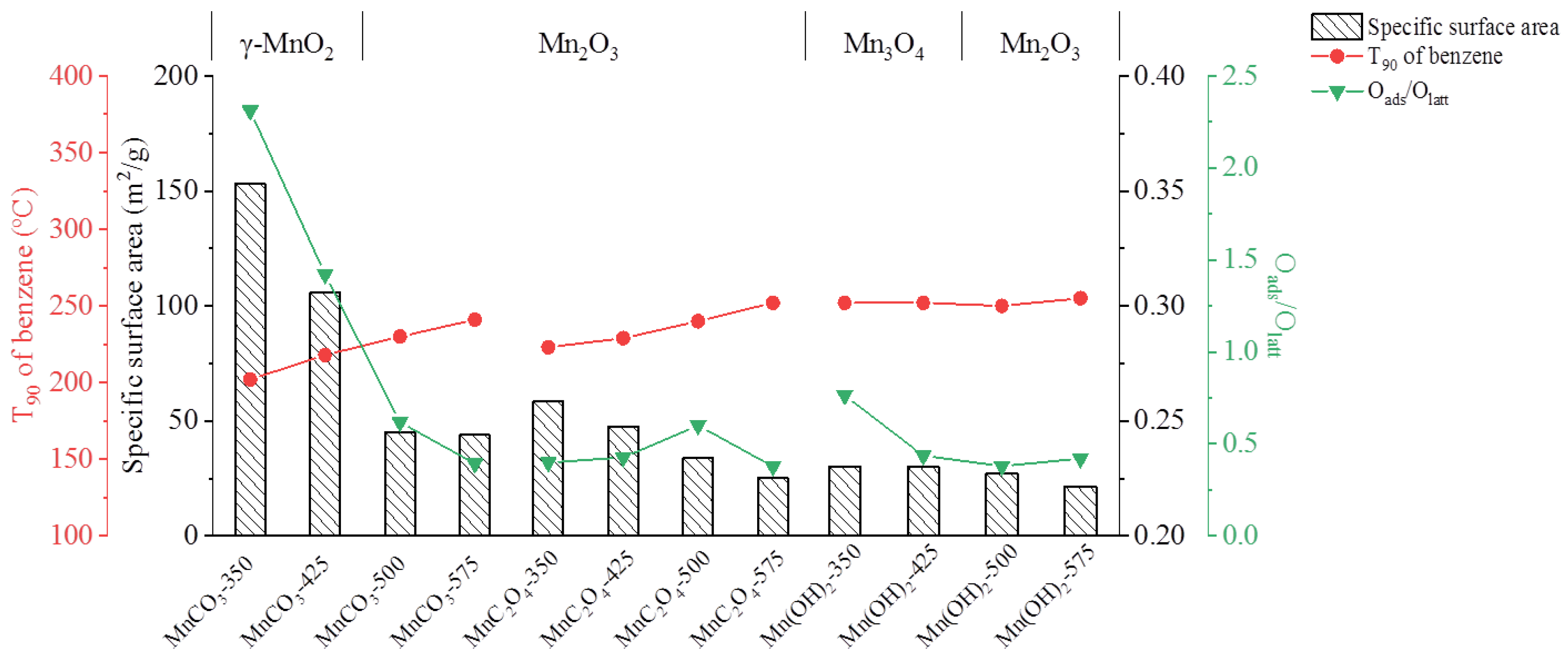
3.2.2. N2 Adsorption/Desorption
3.2.3. Scanning Electron Microscopy (SEM) and Transmission Electron Microscopy (TEM)
3.2.4. X-ray Photoelectron Spectroscopy (XPS)
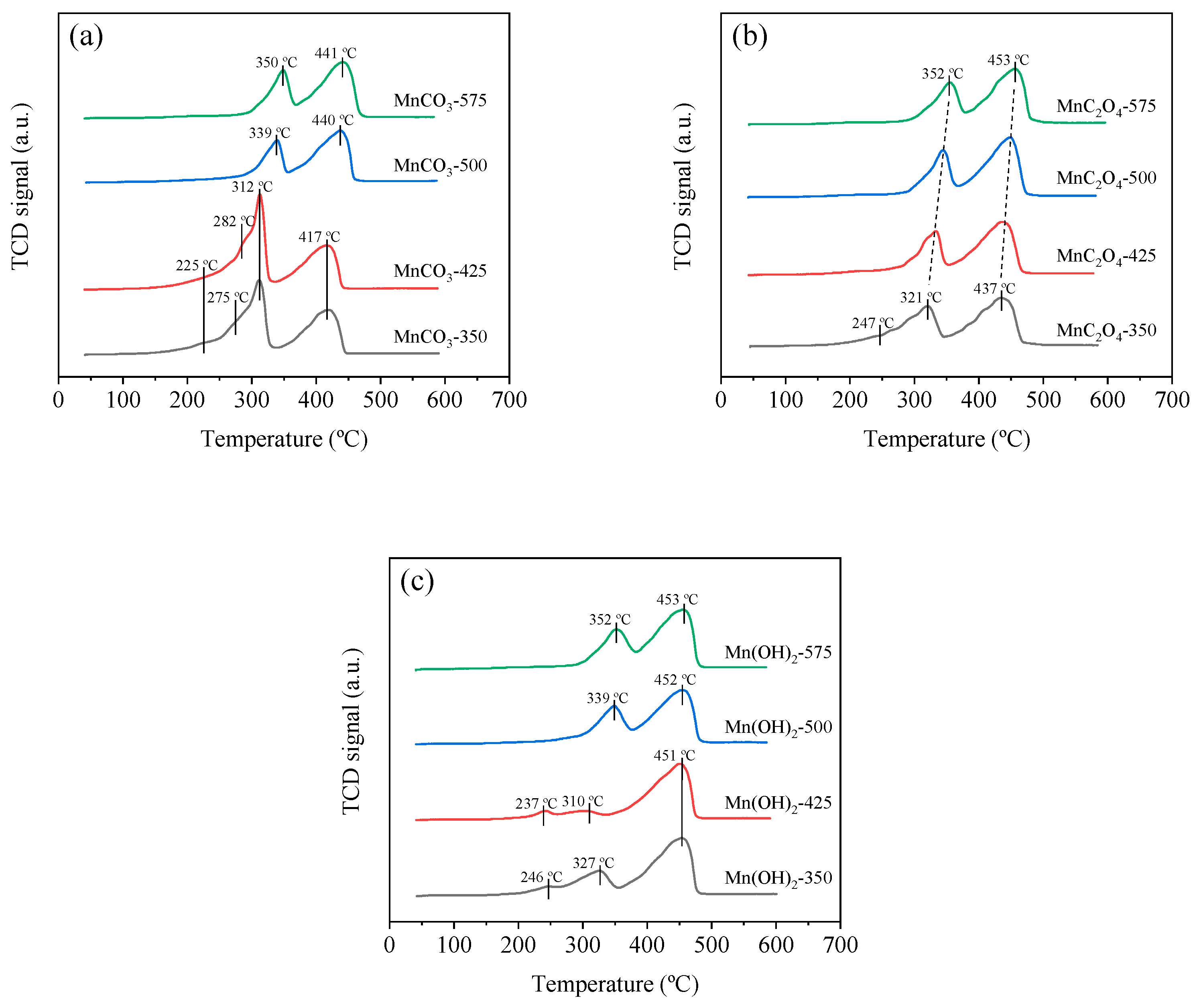
3.2.5. H2-Temperature-Programmed Reduction (H2-TPR)
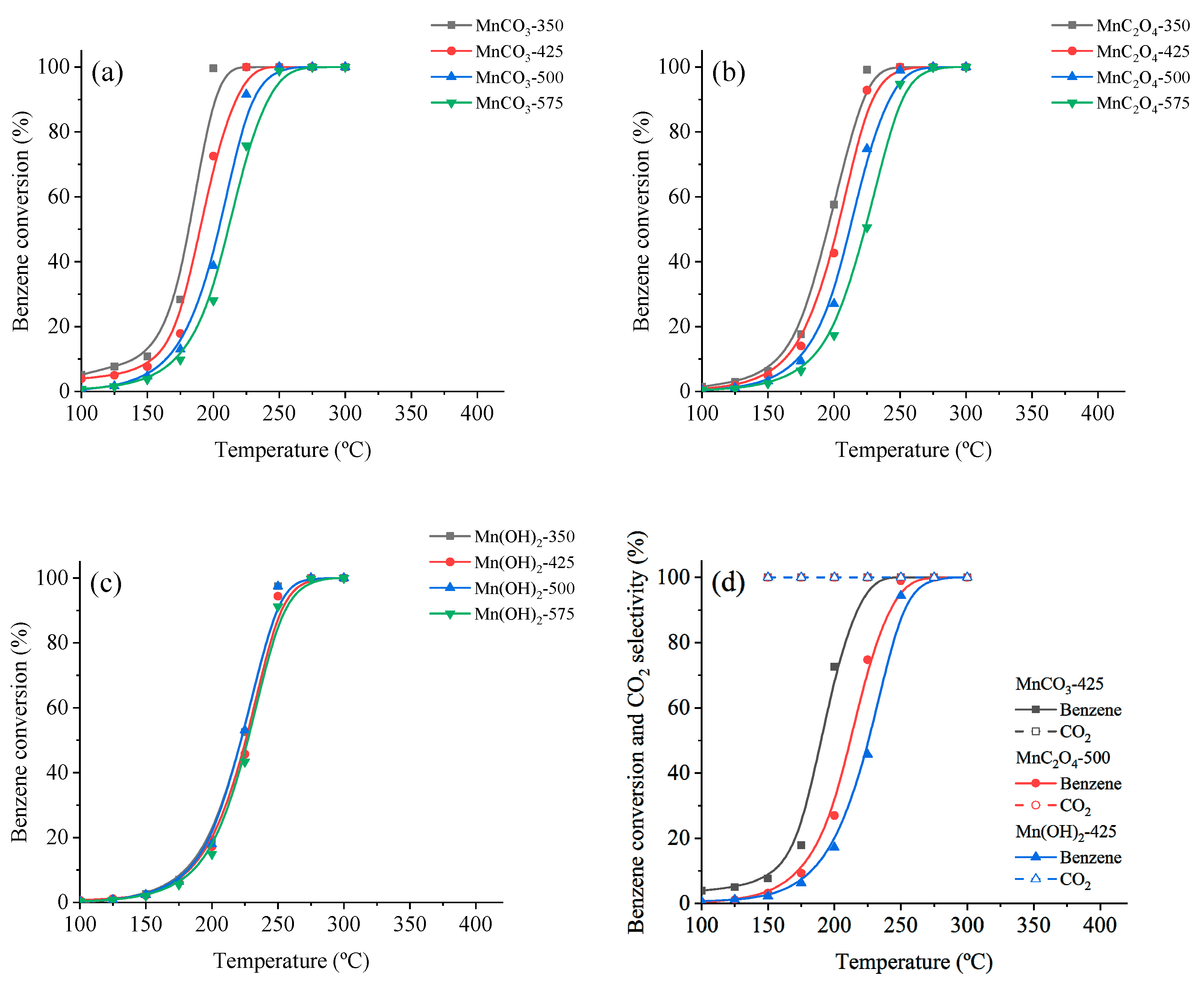
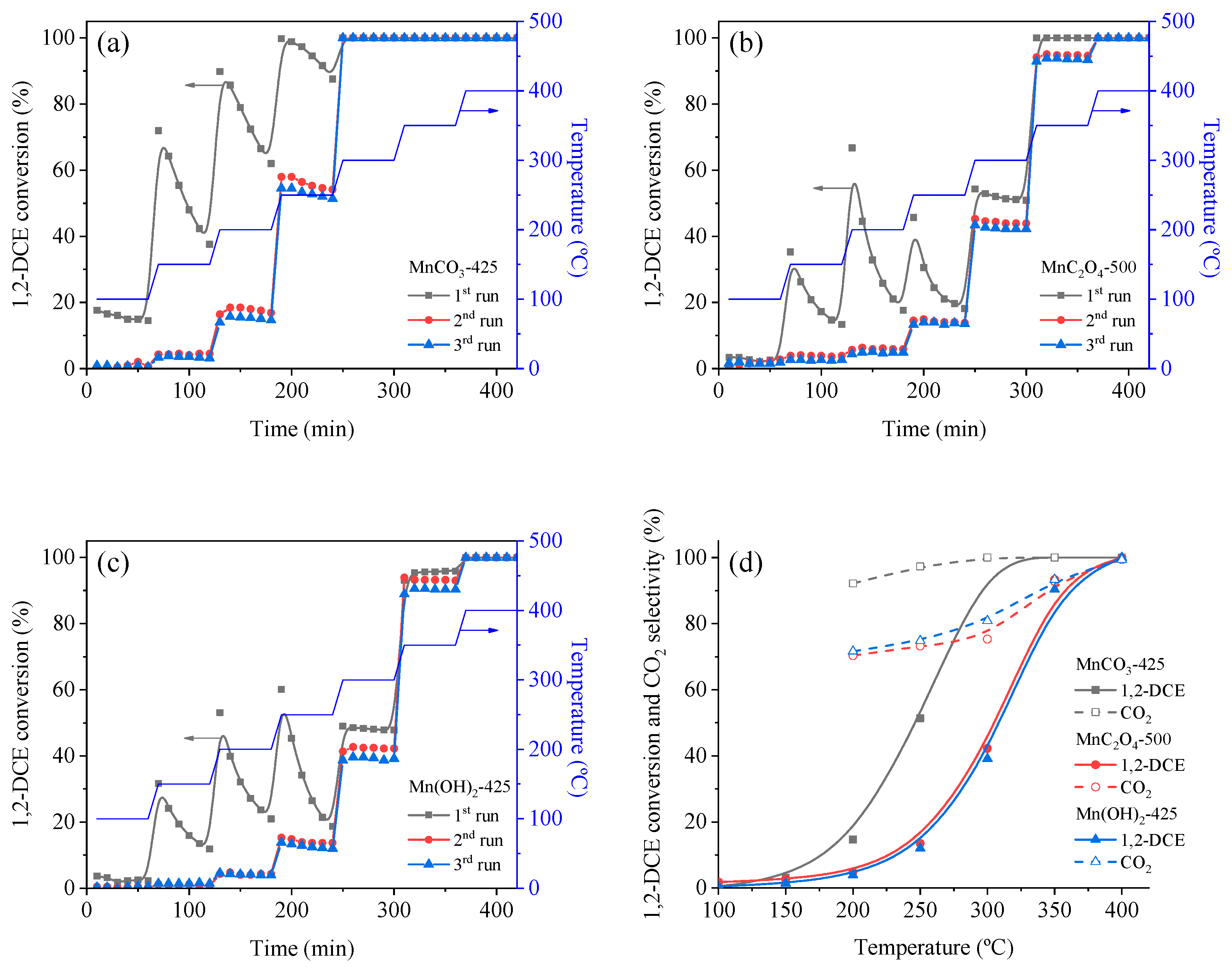
3.3. Catalytic Oxidation of VOCs
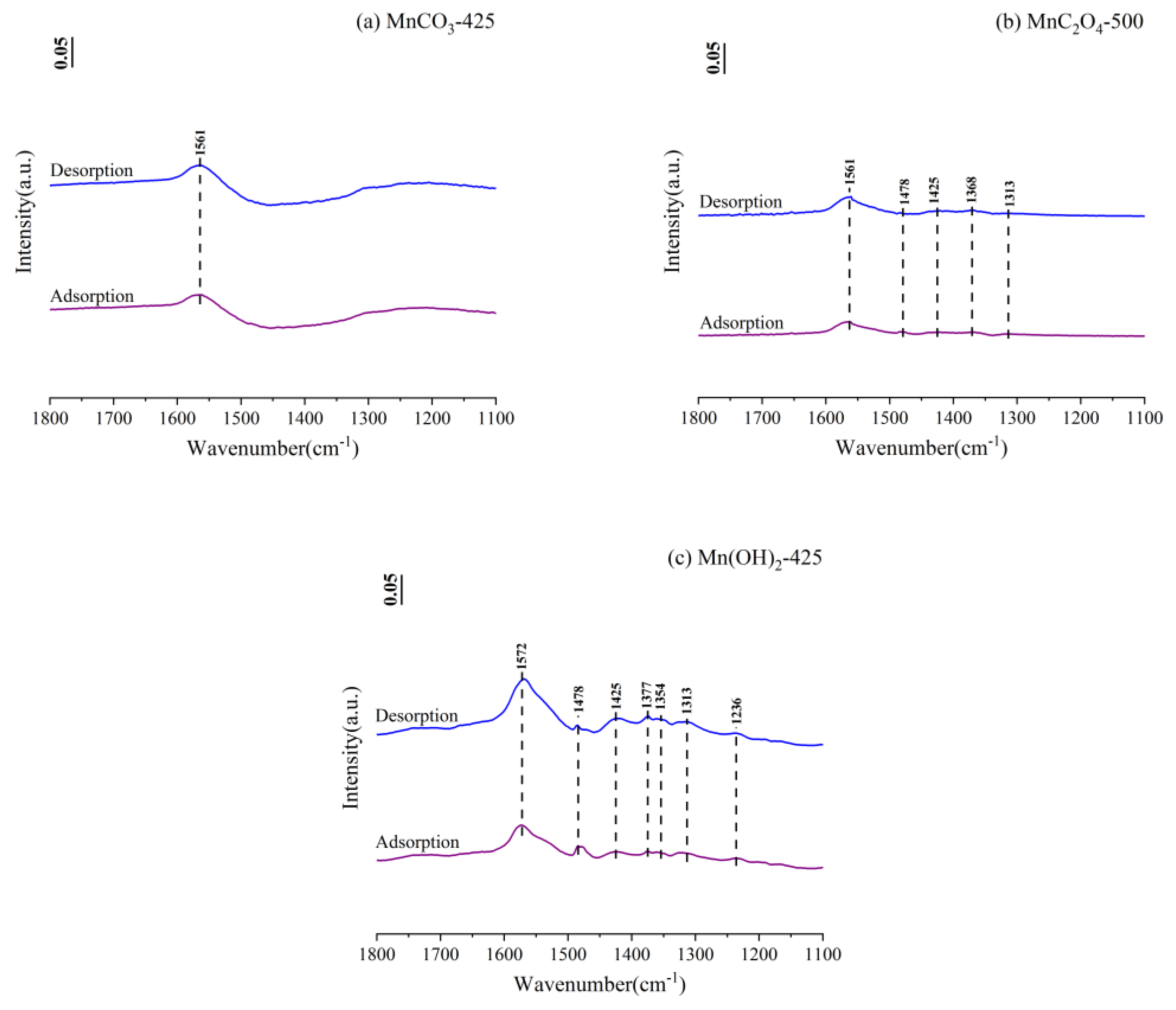
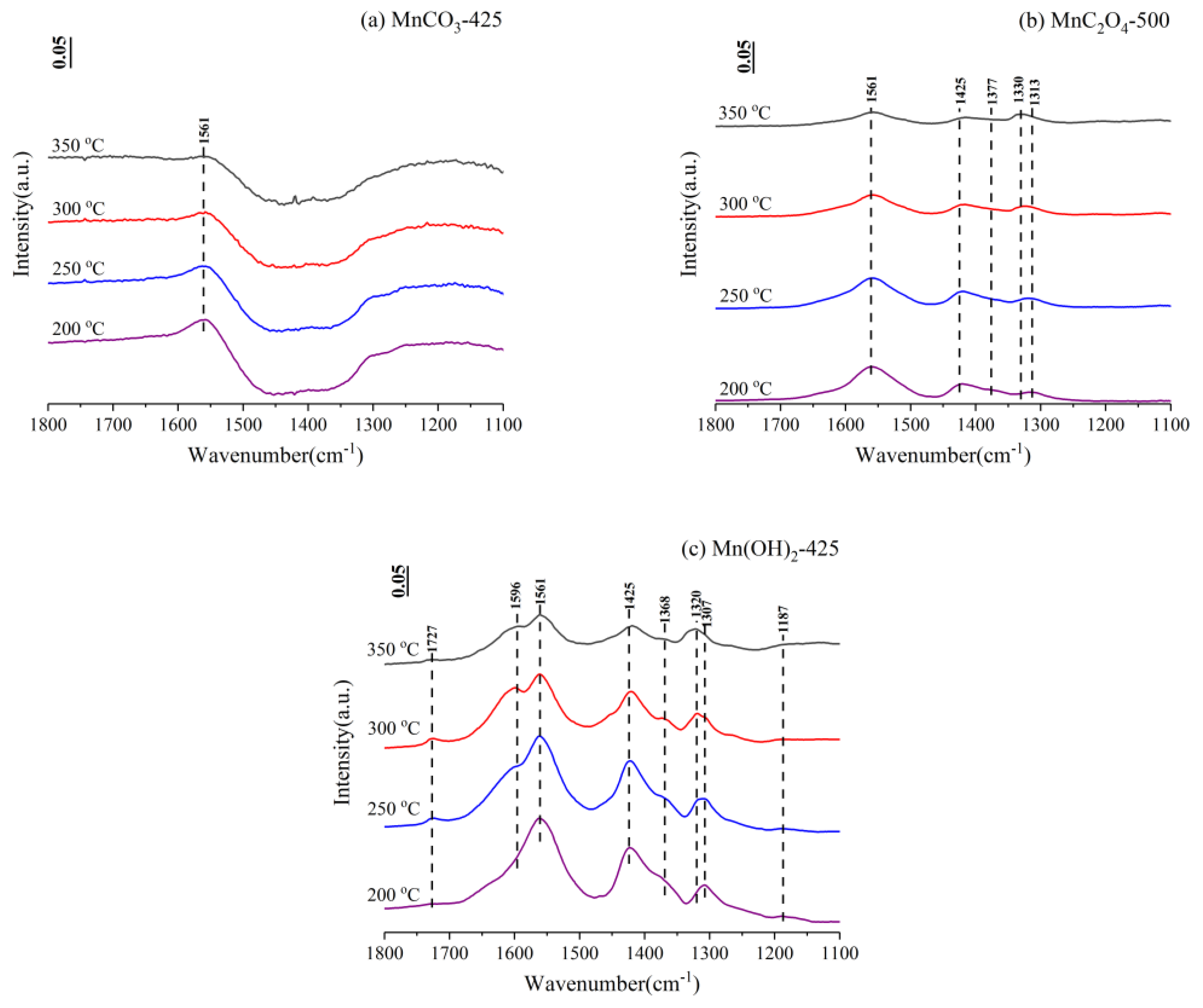
3.4. In-Situ Fourier Transform Infrared Spectroscopy (FTIR)
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kampa, M.; Castanas, E. Human health effects of air pollution. Environ. Pollut. 2008, 151, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Volkamer, R.; Jimenez, J.L.; Martini, F.S.; Dzepina, K.; Zhang, Q.; Salcedo, D.; Molina, L.T.; Worsnop, D.R.; Molina, M.J. Secondary organic aerosol formation from anthropogenic air pollution: Rapid and higher than expected. Geophys. Res. Lett. 2006, 33, L17811. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Su, H.; Zhong, L.J.; Cheng, Y.F.; Zeng, L.M.; Wang, X.S.; Xiang, Y.R.; Wang, J.L.; Gao, D.F.; Shao, M.; et al. Regional ozone pollution and observation-based approach for analyzing ozone-precursor relationship during the PRIDE-PRD2004 campaign. Atmos. Environ. 2008, 42, 6203–6218. [Google Scholar] [CrossRef]
- Shao, M.; Zhang, Y.; Zeng, L.; Tang, X.; Zhang, J.; Zhong, L.; Wang, B. Ground-level ozone in the Pearl River Delta and the roles of VOC and NOx in its production. J. Environ. Manag. 2009, 90, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Li, W.B.; Wang, J.X.; Gong, H. Catalytic combustion of VOCs on non-noble metal catalysts. Catal. Today 2009, 148, 81–87. [Google Scholar] [CrossRef]
- Liotta, L.F. Catalytic oxidation of volatile organic compounds on supported noble metals. Appl. Catal. B-Environ. 2010, 100, 403–412. [Google Scholar] [CrossRef]
- Zheng, J.; Yu, Y.; Mo, Z.; Zhang, Z.; Wang, X.; Yin, S.; Peng, K.; Yang, Y.; Feng, X.; Cai, H. Industrial sector-based volatile organic compound (VOC) source profiles measured in manufacturing facilities in the Pearl River Delta, China. Sci. Total Environ. 2013, 456, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.J.; Xiang, P.; Liang, S.W.; Chen, W.T.; Wang, M.; Lu, S.H.; Wang, Z.W. Sources profiles of volatile organic compounds (VOCs) measured in a typical industrial process in Wuhan, central China. Atmosphere 2018, 9, 297. [Google Scholar] [CrossRef]
- Burgos, N.; Paulis, M.; Antxustegi, M.M.; Montes, M. Deep oxidation of VOC mixtures with platinum supported on Al2O3/Al monoliths. Appl. Catal. B-Environ. 2002, 38, 251–258. [Google Scholar] [CrossRef]
- Yang, K.S.; Choi, J.S.; Lee, S.H.; Chung, J.S. Development of Al/Al2O3-coated wire-mesh honeycombs for catalytic combustion of volatile organic compounds in air. Ind. Eng. Chem. Res. 2004, 43, 907–912. [Google Scholar] [CrossRef]
- Aguilera, D.A.; Perez, A.; Molina, R.; Moreno, S. Cu-Mn and Co-Mn catalysts synthesized from hydrotalcites and their use in the oxidation of VOCs. Appl. Catal. B-Environ. 2011, 104, 144–150. [Google Scholar] [CrossRef]
- Tian, Z.-Y.; Ngamou, P.H.T.; Vannier, V.; Kohse-Hoeinghaus, K.; Bahlawane, N. Catalytic oxidation of VOCs over mixed Co-Mn oxides. Appl. Catal. B-Environ. 2012, 117, 125–134. [Google Scholar] [CrossRef]
- Gomez, D.M.; Gatica, J.M.; Hernandez-Garrido, J.C.; Cifredo, G.A.; Montes, M.; Sanz, O.; Rebled, J.M.; Vidal, H. A novel CoOx/La-modified-CeO2 formulation for powdered and washcoated onto cordierite honeycomb catalysts with application in VOCs oxidation. Appl. Catal. B-Environ. 2014, 144, 425–434. [Google Scholar] [CrossRef]
- Castano, M.H.; Molina, R.; Moreno, S. Cooperative effect of the Co-Mn mixed oxides for the catalytic oxidation of VOCs: Influence of the synthesis method. Appl. Catal. A-Gen. 2015, 492, 48–59. [Google Scholar] [CrossRef]
- Zhang, X.; Ye, J.; Yuan, J.; Cai, T.; Xiao, B.; Liu, Z.; Zhao, K.; Yang, L.; He, D. Excellent low-temperature catalytic performance of nanosheet Co-Mn oxides for total benzene oxidation. Appl. Catal. A-Gen. 2018, 566, 104–112. [Google Scholar] [CrossRef]
- Gandia, L.M.; Vicente, M.A.; Gil, A. Complete oxidation of acetone over manganese oxide catalysts supported on alumina- and zirconia-pillared clays. Appl. Catal. B-Environ. 2002, 38, 295–307. [Google Scholar] [CrossRef]
- Lamaita, L.; Peluso, M.A.; Sambeth, J.E.; Thomas, H.J. Synthesis and characterization of manganese oxides employed in VOCs abatement. Appl. Catal. B-Environ. 2005, 61, 114–119. [Google Scholar] [CrossRef]
- Gandhe, A.R.; Rebello, J.S.; Figueiredo, J.L.; Fernandes, J.B. Manganese oxide OMS-2 as an effective catalyst for total oxidation of ethyl acetate. Appl. Catal. B-Environ. 2007, 72, 129–135. [Google Scholar] [CrossRef]
- Peluso, M.A.; Gambaro, L.A.; Pronsato, E.; Gazzoli, D.; Thomas, H.J.; Sambeth, J.E. Synthesis and catalytic activity of manganese dioxide (type OMS-2) for the abatement of oxygenated VOCs. Catal. Today 2008, 133, 487–492. [Google Scholar] [CrossRef]
- Santos, V.P.; Pereira, M.F.R.; Orfao, J.J.M.; Figueiredo, J.L. The role of lattice oxygen on the activity of manganese oxides towards the oxidation of volatile organic compounds. Appl. Catal. B-Environ. 2010, 99, 353–363. [Google Scholar] [CrossRef]
- Kim, S.C.; Shim, W.G. Catalytic combustion of VOCs over a series of manganese oxide catalysts. Appl. Catal. B-Environ. 2010, 98, 180–185. [Google Scholar] [CrossRef]
- Hou, J.; Li, Y.; Liu, L.; Ren, L.; Zhao, X. Effect of giant oxygen vacancy defects on the catalytic oxidation of OMS-2 nanorods. J. Mater. Chem. A 2013, 1, 6736–6741. [Google Scholar] [CrossRef]
- Tang, W.X.; Wu, X.F.; Li, D.Y.; Wang, Z.; Liu, G.; Liu, H.D.; Chen, Y.F. Oxalate route for promoting activity of manganese oxide catalysts in total VOCs’ oxidation: Effect of calcination temperature and preparation method. J. Mater. Chem. A 2014, 2, 2544–2554. [Google Scholar] [CrossRef]
- Piumetti, M.; Fino, D.; Russo, N. Mesoporous manganese oxides prepared by solution combustion synthesis as catalysts for the total oxidation of VOCs. Appl. Catal. B-Environ. 2015, 163, 277–287. [Google Scholar] [CrossRef]
- Wang, J.L.; Li, J.E.; Jiang, C.J.; Zhou, P.; Zhang, P.Y.; Yu, J.G. The effect of manganese vacancy in birnessite-type MnO2 on room-temperature oxidation of formaldehyde in air. Appl. Catal. B-Environ. 2017, 204, 147–155. [Google Scholar] [CrossRef]
- Blanch-Raga, N.; Palomares, A.E.; Martinez-Triguero, J.; Valencia, S. Cu and Co modified beta zeolite catalysts for the trichloroethylene oxidation. Appl. Catal. B-Environ. 2016, 187, 90–97. [Google Scholar] [CrossRef]
- Wang, X.Y.; Kang, Q.; Li, D. Catalytic combustion of chlorobenzene over MnOx-CeO2 mixed oxide catalysts. Appl. Catal. B-Environ. 2009, 86, 166–175. [Google Scholar] [CrossRef]
- Zhang, K.; Han, X.; Hu, Z.; Zhang, X.; Tao, Z.; Chen, J. Nanostructured Mn-based oxides for electrochemical energy storage and conversion. Chem. Soc. Rev. 2015, 44, 699–728. [Google Scholar] [CrossRef]
- Stoerzinger, K.A.; Risch, M.; Han, B.; Shao-Horn, Y. Recent insights into manganese oxides in catalyzing oxygen reduction kinetics. ACS Catal. 2015, 5, 6021–6031. [Google Scholar] [CrossRef]
- Valyaev, D.A.; Lavigne, G.; Lugan, N. Manganese organometallic compounds in homogeneous catalysis: Past, present, and prospects. Coord. Chem. Rev. 2016, 308, 191–235. [Google Scholar] [CrossRef]
- Najafpour, M.M.; Renger, G.; Holynska, M.; Moghaddam, A.N.; Aro, E.-M.; Carpentier, R.; Nishihara, H.; Eaton-Rye, J.J.; Shen, J.-R.; Allakhverdiev, S.I. Manganese compounds as water-oxidizing catalysts: From the natural water-oxidizing complex to nanosized manganese oxide structures. Chem. Rev. 2016, 116, 2886–2936. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, X.; Xu, Z.; Xu, W.J.; Li, J.J.; Jia, H.P.; Chen, J. Syntheses of hierarchical MnO2 via H2O2 selectively reducing KMnO4 for catalytic combustion of toluene. Chemistryselect 2016, 1, 4052–4056. [Google Scholar] [CrossRef]
- Bertinchamps, F.; Poleunis, C.; Gregoire, C.; Eloy, P.; Bertrand, P.; Gaigneaux, E.M. Elucidation of deactivation or resistance mechanisms of CrOx, VOx, and MnOx supported phases in the total oxidation of chlorobenzene via ToF-SIMS and XPS analyses. Surf. Interface Anal. 2008, 40, 231–236. [Google Scholar] [CrossRef]
- Kapteijn, F.; Singoredjo, L.; Andreini, A.; Moulijn, J.A. Activity and selectivity of pure manganese oxides in the selective catalytic reduction of nitric-oxide with ammonia. Appl. Catal. B-Environ. 1994, 3, 173–189. [Google Scholar] [CrossRef]
- He, F.; Chen, Y.; Zhao, P.; Liu, S. Effect of calcination temperature on the structure and performance of CeOx-MnOx/TiO2 nanoparticles for the catalytic combustion of chlorobenzene. J. Nanoparticle Res. 2016, 18, 119. [Google Scholar] [CrossRef]
- Barrio, I.; Legorburu, I.; Montes, M.; Dominguez, M.I.; Centeno, M.A.; Odriozola, J.A. New redox deposition-precipitation method for preparation of supported manganese oxide catalysts. Catal. Lett. 2005, 101, 151–157. [Google Scholar] [CrossRef]
- Sun, Y.F.; Gao, S.; Lei, F.C.; Xie, Y. Atomically-thin two-dimensional sheets for understanding active sites in catalysis. Chem. Soc. Rev. 2015, 44, 623–636. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, N.J.; Brewer, J.R.; Wang, L.; Wu, T.S.; Wells-Kingsbury, J.; Ihrig, M.M.; Wang, G.H.; Soo, Y.L.; Mei, W.N.; Cheung, C.L. Defect engineering in cubic cerium oxide nanostructures for catalytic oxidation. Nano Lett. 2011, 11, 2666–2671. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.M.; Gilbank, A.L.; Garcia, T.; Solsona, B.; Agouram, S.; Torrente-Murciano, L. The prevalence of surface oxygen vacancies over the mobility of bulk oxygen in nanostructured ceria for the total toluene oxidation. Appl. Catal. B-Environ. 2015, 174, 403–412. [Google Scholar] [CrossRef]
- Venkataswamy, P.; Rao, K.N.; Jampaiah, D.; Reddy, B.M. Nanostructured manganese doped ceria solid solutions for CO oxidation at lower temperatures. Appl. Catal. B-Environ. 2015, 162, 122–132. [Google Scholar] [CrossRef]
- Bandara, J.; Mielczarski, J.A.; Kiwi, J.I. Adsorption mechanism of chlorophenols on iron oxides, titanium oxide and aluminum oxide as detected by infrared spectroscopy. Appl. Catal. B-Environ. 2001, 34, 307–320. [Google Scholar] [CrossRef]
- Busca, G.; Ramis, G.; Lorenzelli, V. FT-IR study of the surface-properties of polycrystalline vanadia. J. Mol. Catal. 1989, 50, 231–240. [Google Scholar] [CrossRef]
- Lichtenberger, J.; Amiridis, M.D. Catalytic oxidation of chlorinated benzenes over V2O5/TiO2 catalysts. J. Catal. 2004, 223, 296–308. [Google Scholar] [CrossRef]
- Hetrick, C.E.; Lichtenberger, J.; Amiridis, M.D. Catalytic oxidation of chlorophenol over V2O5/TiO2 catalysts. Appl. Catal. B-Environ. 2008, 77, 255–263. [Google Scholar] [CrossRef]
- Krishnamoorthy, S.; Rivas, J.A.; Amiridis, M.D. Catalytic oxidation of 1,2-dichlorobenzene over supported transition metal oxides. J. Catal. 2000, 193, 264–272. [Google Scholar] [CrossRef]
- Wang, J.; Wang, X.; Liu, X.L.; Zhu, T.Y.; Guo, Y.Y.; Qi, H. Catalytic oxidation of chlorinated benzenes over V2O5/TiO2 catalysts: The effects of chlorine substituents. Catal. Today 2015, 241, 92–99. [Google Scholar] [CrossRef]
- Li, J.W.; Zhao, P.; Liu, S.T. SnOx-MnOx-TiO2 catalysts with high resistance to chlorine poisoning for low-temperature chlorobenzene oxidation. Appl. Catal. A-Gen. 2014, 482, 363–369. [Google Scholar] [CrossRef]
- Dai, Y.; Wang, X.; Dai, Q.; Li, D. Effect of Ce and La on the structure and activity of MnOx catalyst in catalytic combustion of chlorobenzene. Appl. Catal. B-Environ. 2012, 111, 141–149. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Benzene | |
|---|---|---|
| T50 (°C) | T90 (°C) | |
| MnCO3-350 | 182 | 202 |
| MnCO3-425 | 191 | 218 |
| MnCO3-500 | 205 | 230 |
| MnCO3-575 | 213 | 241 |
| MnC2O4-350 | 195 | 223 |
| MnC2O4-425 | 202 | 229 |
| MnC2O4-500 | 213 | 240 |
| MnC2O4-575 | 223 | 252 |
| Mn(OH)2-350 | 225 | 252 |
| Mn(OH)2-425 | 225 | 252 |
| Mn(OH)2-500 | 223 | 250 |
| Mn(OH)2-575 | 227 | 255 |
| Catalyst | Specific Surface Area (m2/g) | Pore Size (nm) | Pore Volume (cm3/g) | O | |
|---|---|---|---|---|---|
| Oads c | Olatt d | ||||
| MnCO3-350 | 153.2 | 5.6 | 0.29 | 0.698 | 0.302 |
| MnCO3-425 | 105.8 | 6.5 | 0.24 | 0.587 | 0.413 |
| MnCO3-425 (B) a | 104.1 | 6.6 | 0.25 | 0.295 | 0.705 |
| MnCO3-425 (DCE) b | 37.6 | 17.5 | 0.22 | 0.271 | 0.729 |
| MnCO3-500 | 45.1 | 17.5 | 0.24 | 0.382 | 0.618 |
| MnCO3-575 | 43.9 | 17.4 | 0.29 | 0.282 | 0.718 |
| MnC2O4-350 | 58.6 | 9.6 | 0.24 | 0.285 | 0.715 |
| MnC2O4-425 | 47.7 | 9.6 | 0.24 | 0.299 | 0.701 |
| MnC2O4-500 | 33.8 | 17.7 | 0.22 | 0.375 | 0.625 |
| MnC2O4-500 (B) a | 33.3 | 17.4 | 0.20 | 0.272 | 0.728 |
| MnC2O4-500 (DCE) b | 24.2 | 31.1 | 0.32 | 0.261 | 0.739 |
| MnC2O4-575 | 25.2 | 31.5 | 0.19 | 0.274 | 0.726 |
| Mn(OH)2-350 | 30.3 | 31.0 | 0.23 | 0.433 | 0.567 |
| Mn(OH)2-425 | 30.1 | 30.5 | 0.21 | 0.304 | 0.696 |
| Mn(OH)2-425 (B) a | 37.1 | 30.9 | 0.23 | 0.212 | 0.788 |
| Mn(OH)2-425 (DCE) b | 17.2 | 46.4 | 0.24 | 0.264 | 0.736 |
| Mn(OH)2-500 | 27.0 | 31.2 | 0.36 | 0.275 | 0.725 |
| Mn(OH)2-575 | 21.6 | 46.3 | 0.85 | 0.296 | 0.704 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Zhao, H.; Song, J.; Zhu, T.; Xu, W. Structure-Activity Relationship of Manganese Oxide Catalysts for the Catalytic Oxidation of (chloro)-VOCs. Catalysts 2019, 9, 726. https://doi.org/10.3390/catal9090726
Wang J, Zhao H, Song J, Zhu T, Xu W. Structure-Activity Relationship of Manganese Oxide Catalysts for the Catalytic Oxidation of (chloro)-VOCs. Catalysts. 2019; 9(9):726. https://doi.org/10.3390/catal9090726
Chicago/Turabian StyleWang, Jian, Hainan Zhao, Jianfei Song, Tingyu Zhu, and Wenqing Xu. 2019. "Structure-Activity Relationship of Manganese Oxide Catalysts for the Catalytic Oxidation of (chloro)-VOCs" Catalysts 9, no. 9: 726. https://doi.org/10.3390/catal9090726
APA StyleWang, J., Zhao, H., Song, J., Zhu, T., & Xu, W. (2019). Structure-Activity Relationship of Manganese Oxide Catalysts for the Catalytic Oxidation of (chloro)-VOCs. Catalysts, 9(9), 726. https://doi.org/10.3390/catal9090726