Theoretical Study of a Class of Organic D-?-A Dyes for Polymer Solar Cells: Influence of Various ?-Spacers

 and
and
Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
3.1. Structural and Optoelectronic Properties
3.2. Frontier Molecular Orbitals
3.3. Electronic Properties
3.4. Absorption Spectra
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Liu, F.; Gu, Y.; Shen, X.; Ferdous, S.; Wang, H.-W.; Russell, T.P. Characterization of the Morphology of Solution-Processed Bulk Heterojunction Organic Photovoltaics. Prog. Polym. Sci. 2013, 38, 1990. [Google Scholar] [CrossRef]
- Chen, L.-M.; Hong, Z.; Li, G.; Yang, Y. Recent Progress in Polymer Solar Cells: Manipulation of Polymer: Fullerene Morphology and the Formation of Efficient Inverted Polymer Solar Cells. Adv. Mater. 2009, 21, 1434. [Google Scholar] [CrossRef]
- Ye, L.; Jing, Y.; Guo, X.; Sun, H.; Zhang, S.; Huo, L.; Hou, J. Remove the Residual Additives toward Enhanced Efficiency with Higher Reproducibility in Polymer Solar Cells. J. Phys. Chem. C 2013, 117, 14920. [Google Scholar] [CrossRef]
- Ye, L.; Zhang, S.; Ma, W.; Fan, B.; Guo, X.; Huang, Y.; Ade, H.; Hou, J. From binary to ternary solvent: Morphology fine-tuning of D/A blends in PDPP3T-based polymer solar cells. Adv. Mater. 2012, 24, 6335. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Gao, J.; Hummelen, J.C.; Wudl, F.; Heeger, A.J. Polymer Photovoltaic Cells: Enhanced Efficiencies via a Network of Internal Donor-Acceptor Heterojunctions. Science 1995, 270, 1789. [Google Scholar] [CrossRef] [Green Version]
- Heeger, A.J. 25th anniversary article: Bulk heterojunction solar cells: Understanding the mechanism of operation. Adv. Mater. 2014, 26, 10. [Google Scholar] [CrossRef]
- Cheng, Y.J.; Yang, S.H.; Hsu, C.S. Synthesis of Conjugated Polymers for Organic Solar Cell Applications. Chem. Rev. 2009, 109, 5868. [Google Scholar] [CrossRef]
- Chen, J.; Cao, Y. Development of Novel Conjugated Donor Polymers for High-Efficiency Bulk-Heterojunction Photovoltaic Devices. Acc. Chem. Res. 2009, 42, 1709. [Google Scholar] [CrossRef]
- Zhan, X.; Zhu, D. Conjugated polymers for high-efficiency organic photovoltaics. Polym. Chem. 2010, 1, 409. [Google Scholar] [CrossRef]
- Beaujuge, P.M.; Fréchet, J.M.J. Molecular Design and Ordering Effects in π-Functional Materials for Transistor and Solar Cell Applications. J. Am. Chem. Soc. 2011, 133, 20009. [Google Scholar] [CrossRef]
- He, F.; Yu, L. How Far Can Polymer Solar Cells Go? In Need of a Synergistic Approach. J. Phys. Chem. Lett. 2011, 2, 3102. [Google Scholar] [CrossRef]
- Boudreault, P.-L.T.; Najari, A.; Leclere, M. Processable Low-Bandgap Polymers for Photovoltaic Applications. Chem. Mater. 2011, 23, 456. [Google Scholar] [CrossRef]
- Spanggaard, H.; Krebs, F.C. A brief history of the development of organic and polymeric photovoltaics. Sol. Energy Mater. Sol. Cells 2004, 83, 125. [Google Scholar] [CrossRef]
- Li, Y.; Zou, Y. Conjugated Polymer Photovoltaic Materials with Broad Absorption Band and High Charge Carrier Mobility. Adv. Mater. 2008, 20, 2952. [Google Scholar] [CrossRef]
- Shaheen, S.E.; Brabec, C.J.; Sariciftci, N.S.; Padinger, F.; Fromherz, T.; Hummelen, J.C. 2.5% efficient organic plastic solar cells. Appl. Phys. Lett. 2001, 78, 841. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Zheng, L.; Sun, D.; Deng, X.; Yu, G.; Cao, Y. Efficient polymer photovoltaic devices based on blend of MEH-PPV and C60 derivatives. Synth. Met. 2003, 135–136, 825–826. [Google Scholar] [CrossRef]
- Dang, M.T.; Hirsch, L.; Wantz, G. P3HT:PCBM, Best Seller in Polymer Photovoltaic Research. Adv. Mater. 2011, 23, 3597. [Google Scholar] [CrossRef]
- Li, G.; Shrotriya, V.; Huang, J.; Yao, Y.; Moriarty, T.; Emery, K.; Yang, Y. High-efficiency solution processable polymer photovoltaic cells by self-organization of polymer blends. Nat. Mater. 2005, 4, 864–868. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Goto, T.; Tamao, K. Silole-Thiophene Alternating Copolymers with Narrow Band Gaps. Angew. Chem. Int. Ed. 2000, 39, 1695–1697. [Google Scholar] [CrossRef]
- Murata, H.; Kafafi, Z.H.; Uchida, M. Efficient organic light-emitting diodes with undoped active layers based on silole derivative. Appl. Phys. Lett. 2002, 80, 189–191. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.Y.; Lam, W.Y.; Luo, J.D.; Ho, Y.L.; Tang, B.Z.; Zhu, D.B.; Wong, M.; Kwok, H.S. Highly efficient organic light-emitting diodes with a silole-based compound. Appl. Phys. Lett. 2002, 81, 574–575. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wang, K.; Xu, F.; Tang, Z.; Zheng, W.; Zhang, J.; Li, C.; Yu, T.; You, X. Synthesis and photovoltaic performances of donor– p–acceptor dyes utilizing 1,3,5-triazine as π-spacers. Tetrahedron Lett. 2011, 52, 6492–6496. [Google Scholar] [CrossRef]
- Liu, T.; Meng, D.; Cai, Y.; Sun, X.; Li, Y.; Huo, L.; Liu, F.; Wang, Z.; Russell, T.P.; Sun, Y. High-Performance Non-Fullerene Organic Solar Cells Based on a Selenium-Containing Polymer Donor and a Twisted Perylene Bisimide Acceptor. Adv. Sci. 2016, 3, 1600117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandberg, H.G.O.; Frey, G.L.; Shkunov, M.N.; Sirringhaus, H.; Friend, R.H.; Nielsen, M.M.; Kumpf, C. Ultrathin Regioregular Poly(3-hexyl thiophene) Field-Effect Transistors. Langmuir 2002, 18, 10176–10182. [Google Scholar] [CrossRef]
- Shuto, A.; Kushida, T.; Fukushima, T.; Kaji, H.; Yamaguchi, S. π Extended Planarized Triphenylboranes with Thiophene Spacers. Org. Lett. 2013, 15, 6234–6237. [Google Scholar] [CrossRef]
- Yang, H.; LeFevre, S.W.; Ryu, C.Y.; Bao, Z. Solubility-driven thin film structures of regioregular poly (3-hexyl thiophene) using volatile solvents. Appl. Phys. Lett. 2007, 90, 172116. [Google Scholar] [CrossRef]
- Yao, H.; Yu, R.; Shin, T.J.; Zhang, H.; Zhang, S.; Jang, B.; Uddin, M.A.; Woo, H.Y.; Hou, J. A Wide Bandgap Polymer with Strong π–π Interaction for Efficient Fullerene-Free Polymer Solar Cells. Adv. Energy Mater. 2016, 6, 1600742. [Google Scholar] [CrossRef]
- Li, Y. Molecular Design of Photovoltaic Materials for Polymer Solar Cells: Toward Suitable Electronic Energy Levels and Broad Absorption. Acc. Chem. Res. 2012, 45, 723. [Google Scholar] [CrossRef]
- Zhou, H.; Yang, L.; You, W. Rational Design of High Performance Conjugated Polymers for Organic Solar Cells. Macromolecules 2012, 45, 607. [Google Scholar] [CrossRef] [Green Version]
- Henson, Z.B.; Mullen, K.; Bazan, G.C. Design strategies for organic semiconductors beyond the molecular formula. Nat. Chem. 2012, 4, 699. [Google Scholar] [CrossRef]
- Yu, R.L.; Price, S.C.; You, W. Structure-property optimizations in donor polymers via electronics, substituents, and side chains toward high effiency solar cells. Macromol. Rapid Commun. 2012, 33, 1162. [Google Scholar]
- Dang, M.T.; Hirsch, L.; Wantz, G.; Wuest, J.D. Controlling the morphology and performance of bulk heterojunctions in solar cells. Lessons learned from the benchmark poly(3-hexylthiophene):[6,6]-phenyl-C61-butyric acid methyl ester system. Chem. Rev. 2013, 113, 3734–3765. [Google Scholar] [CrossRef] [PubMed]
- Ohshita, J.; Kai, H.; Takata, A.; Iida, T.; Kunai, A.; Ohta, N.; Komaguchi, K.; Shiotani, M.; Adachi, A.; Sakamaki, K.; et al. Effects of Conjugated Substituents on the Optical, Electrochemical, and Electron-Transporting Properties of Dithienosiloles. Organometallics 2001, 20, 4800−4805. [Google Scholar] [CrossRef]
- Bonnier, C.; Machin, D.D.; Abdi, O.; Koivisto, B.D. Manipulating non-innocent π -spacers: The challenges of using 2,6-disubstituted BODIPY cores within donor– acceptor light-harvesting motifs. Org. Biomol. Chem. 2013, 11, 3756. [Google Scholar] [CrossRef] [Green Version]
- Goh, C.; Kline, R.J.; McGehee, M.D.; Kadnikova, E.N.; Fréchet, J.M. Molecular-weight-dependent mobilities in regioregular poly(3-hexyl-thiophene) diodes. J. Appl. Phys. Lett. 2005, 86, 122110. [Google Scholar] [CrossRef] [Green Version]
- Hua, Y.; He, J.; Zhang, C.; Qin, C.; Han, L.; Zhao, J.; Chen, T.; Wong, W.-Y.; Wong, W.-K.; Zhu, X. Effects of Various π-Conjugated Spacers in Thiadiazole[3,4-c]pyridine-Cored Panchromatic Organic Dyes for Dye-Sensitized Solar Cells. J. Mater. Chem. A 2015, 3, 3103–3112. [Google Scholar] [CrossRef]
- Tan, D.; Wan, J.; Xu, X.; Lee, Y.W.; Woo, H.Y.; Fenga, K.; Peng, Q. Naphthobistriazole-Based Wide Bandgap Donor Polymers for Efficient Non-Fullerene Organic Solar Cells: Significant Fine-Tuning Absorption and Energy Level by Backbone Fluorination. Nano Energy 2018, 53, 258–269. [Google Scholar]
- Xu, X.; Zhang, G.; Li, Y.; Peng, Q. The recent progress of wide bandgap donor polymers towards non-fullerene organic solar cells. Chin. Chem. Lett. 2019, 30, 809–825. [Google Scholar] [CrossRef]
- Huang, F.; Yip, H.-L.; Cao, Y. Polymer Photovoltaics: Materials, Physics, and Device Engineering, RSC Polymer Chemistry Series No. 17; Royal Society of Chemistry: London, UK, 2016. [Google Scholar]
- Bin, H.J.; Gao, L.; Zhang, Z.G.; Yang, Y.K.; Zhang, Y.D.; Zhang, C.F.; Chen, S.S.; Xue, L.W.; Yang, C.; Xiao, M.; et al. 11.4% Efficiency non-fullerene polymer solar cells with trialkylsilyl substituted 2D-conjugated polymer as donor. Nat. Commun. 2016, 7, 13651. [Google Scholar] [CrossRef] [Green Version]
- Bin, H.J.; Zhang, Z.G.; Gao, L.; Chen, S.S.; Zhong, L.; Xue, L.W.; Yang, C.; Li, Y.F. Nonfullerene polymer solar cells based on alkylthio and fluorine substituted 2D-conjugated polymers reach 9.5% efficiency. J. Am. Chem. Soc. 2016, 138, 4657–4664. [Google Scholar] [CrossRef]
- Fan, Q.P.; Su, W.Y.; Meng, X.Y.; Guo, X.; Li, G.D.; Ma, W.; Zhang, M.J.; Li, Y.F. High performance non-fullerene polymer solar cells based on fluorine substituted wide bandgap copolymers without extra treatments. Solar RRL 2017, 1, 1700020. [Google Scholar] [CrossRef]
- Fan, Q.P.; Su, W.Y.; Wang, Y.; Guo, B.; Jiang, Y.F.; Guo, X.; Liu, F.; Russell, T.P.; Zhang, M.J.; Li, Y.F. Synergistic effect of fluorination on both donor and acceptor materials for high performance non-fullerene polymer solar cells with 13.5% efficiency. Sci. Chin. Chem. 2018, 61, 531–537. [Google Scholar] [CrossRef]
- Fei, Z.P.; Eisner, F.D.; Jiao, X.C.; Azzouzi, M.; Rohr, J.A.; Han, Y.; Shahid, M.; Chesman, A.S.R.; Easton, C.D.; McNeill, C.R.; et al. An alkylated indacenodithieno[3,2-b]thiophene based nonfullerene acceptor with high crystallinity exhibiting single junction solar cell efficiencies greater than 13% with low voltage losses. Adv. Mater. 2018, 30, 1800728. [Google Scholar] [CrossRef] [PubMed]
- Tang, A.L.; Xiao, B.; Chen, F.; Zhang, J.Q.; Wei, Z.X.; Zhou, E.J. The introduction of fluorine and sulfur atoms into benzotriazole based p-type polymers to match with a benzotriazole-containing n-type small molecule: ‘‘The same-acceptor-strategy’’ to realize high open-circuit voltage. Adv. Energy Mater. 2018, 8, 1801582. [Google Scholar] [CrossRef]
- Xu, X.P.; Bi, Z.Z.; Ma, W.; Wang, Z.S.; Choy, W.C.H.; Wu, W.L.; Zhang, G.J.; Li, Y.; Peng, Q. Highly efficient ternary-blend polymer solar cells enabled by a nonfullerene acceptor and two polymer donors with a broad composition tolerance. Adv. Mater. 2017, 29, 1704271. [Google Scholar] [CrossRef]
- Xu, X.P.; Li, Z.J.; Bi, Z.Z.; Yu, T.; Ma, W.; Feng, K.; Li, Y.; Peng, Q. Highly efficient nonfullerene polymer solar cells enabled by a copper(I) coordination strategy employing a 1,3,4-oxadiazole-containing wide bandgap copolymer donor. Adv. Mater. 2018, 30, 1800737. [Google Scholar] [CrossRef]
- Ye, L.; Zhang, S.Q.; Huo, L.J.; Zhang, M.J.; Hou, J.H. Realizing over 10% efficiency in polymer solar cell by device optimization. Acc. Chem. Res. 2014, 47, 1595–1603. [Google Scholar] [CrossRef]
- Zhang, G.J.; Xu, X.P.; Bi, Z.Z.; Ma, W.; Tang, D.S.; Li, Y.; Peng, Q. Fluorinated and Alkylthiolated Polymeric Donors Enable both Efficient Fullerene and Nonfullerene Polymer Solar Cells. Adv. Funct. Mater. 2018, 28, 1706404. [Google Scholar] [CrossRef]
- Zhang, H.; Yao, H.F.; Hou, J.X.; Zhu, J.; Zhang, J.Q.; Li, W.N.; Yu, R.N.; Gao, B.W.; Zhang, S.Q.; Hou, J.H. Over 14% Efficiency in Organic Solar Cells Enabled by Chlorinated Nonfullerene Small-Molecule Acceptors. Adv. Mater. 2018, 30, 1800613. [Google Scholar] [CrossRef]
- Zhao, W.C.; Li, S.S.; Yao, H.F.; Zhang, S.Q.; Zhang, Y.; Yang, B.; Hou, J.H. Molecular optimization enables over 13% efficiency in organic solar cells. J. Am. Chem. Soc. 2017, 139, 7148–7151. [Google Scholar] [CrossRef]
- Hou, J.; Park, M.-H.; Zhang, S.; Yao, Y.; Chen, L.-M.; Li, J.-H.; Yang, Y. Bandgap and Molecular Energy Level Control of Conjugated Polymer Photovoltaic Materials Based on Benzo[1,2-b:4,5-b′]dithiophene. Macromoleculaes 2008, 41, 6012–6018. [Google Scholar] [CrossRef] [Green Version]
- Hou, L.; Hou, J. Benzo[1,2-b:4,5-b′]dithiophene-based conjugated polymers: Band gap and energy level control and their application in polymer solar cells. Polym. Chem. 2011, 2, 2453. [Google Scholar]
- Feng, K.; Yuan, J.; Bi, Z.; Ma, W.; Xu, X.; Zhang, G.; Peng, Q. Low-Energy-Loss Polymer Solar Cells with 14.52% Efficiency Enabled by Wide-Band-Gap Copolymers. iScience 2019, 12, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef] [Green Version]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A.02; Gaussian Inc.: Wallingford, UK, 2016. [Google Scholar]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular Orbital Methods. 9. Extended Gaussian-type Basis for Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–730. [Google Scholar] [CrossRef]
- Tang, C.W.; VanSlyke, S.A. Organic electroluminescent diodes. Appl. Phys. Lett. 1987, 51, 913–920. [Google Scholar] [CrossRef]
- Tang, C.W.; VanSlyke, S.A.; Chen, C.H. Electroluminescence of doped organic thin films. J. Appl. Phys. 1989, 65, 3610–3620. [Google Scholar] [CrossRef]
- Tamao, K.; Yamaguchi, S.; Shiozaki, M.; Nakagawa, Y.; Ito, Y. Thiophene-silole cooligomers and copolimers. J. Am. Chem. Soc. 1992, 114, 5867–5869. [Google Scholar] [CrossRef]
- Tamao, K.; Yamaguchi, S.; Ito, Y.; Matsuzaki, Y.; Yamabe, T.; Fukushima, M.; Mori, S. Silole-Containing π-Conjugated Systems. 3.1 A Series of Silole-Thiophene Cooligomers and Copolymers: Synthesis, Properties, and Electronic Structures. Macromolecules 1995, 28, 8668–8675. [Google Scholar] [CrossRef]
- Tamao, K.; Ohno, S.; Yamaguchi, S. Silole–pyrrole co-oligomers: Their synthesis, structure and UV-VIS absorption spectra. Chem. Commun. 1996, 1873–1874. [Google Scholar] [CrossRef]
- Tamao, K.; Yamaguchi, S. Regio-controlled intramolecular reductive cyclization of diynes. Pure Appl. Chem. 1996, 8, 139–144. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Tamao, K. Theoretical Study of the Electronic Structure of 2,2′-Bisilole in Comparison with 1,1′-Bi-1,3-cyclopentadiene: σ*–π* Conjugation and a Low-Lying LUMO as the Origin of the Unusual Optical Properties of 3,3′,4,4′-Tetraphenyl-2,2′-bisilole. Bull. Chem. Soc. Jpn. 1996, 69, 2327–2336. [Google Scholar] [CrossRef]
- Kim, D.-H.; Ohshita, J.; Lee, K.-H.; Kunugi, Y.; Kunai, A. Synthesis of p-conjugated oligomers containing dithienosilole units. Organometallics 2006, 25, 1511–1516. [Google Scholar] [CrossRef]
- Ohshita, J. Conjugated Oligomers and Polymers Containing Dithienosilole Units. J. Macromol. Chem. Phys. 2009, 210, 1360–1370. [Google Scholar] [CrossRef]
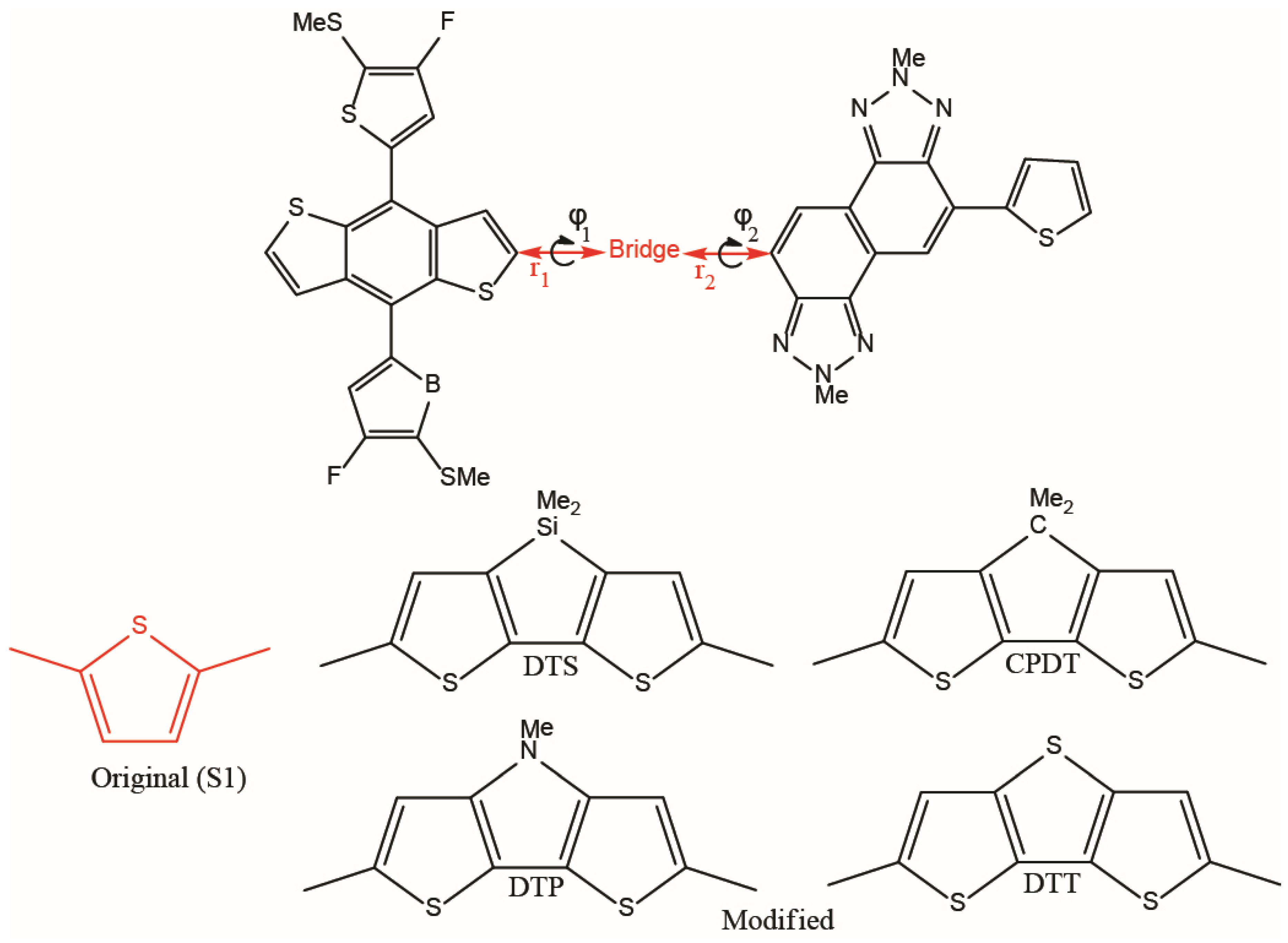

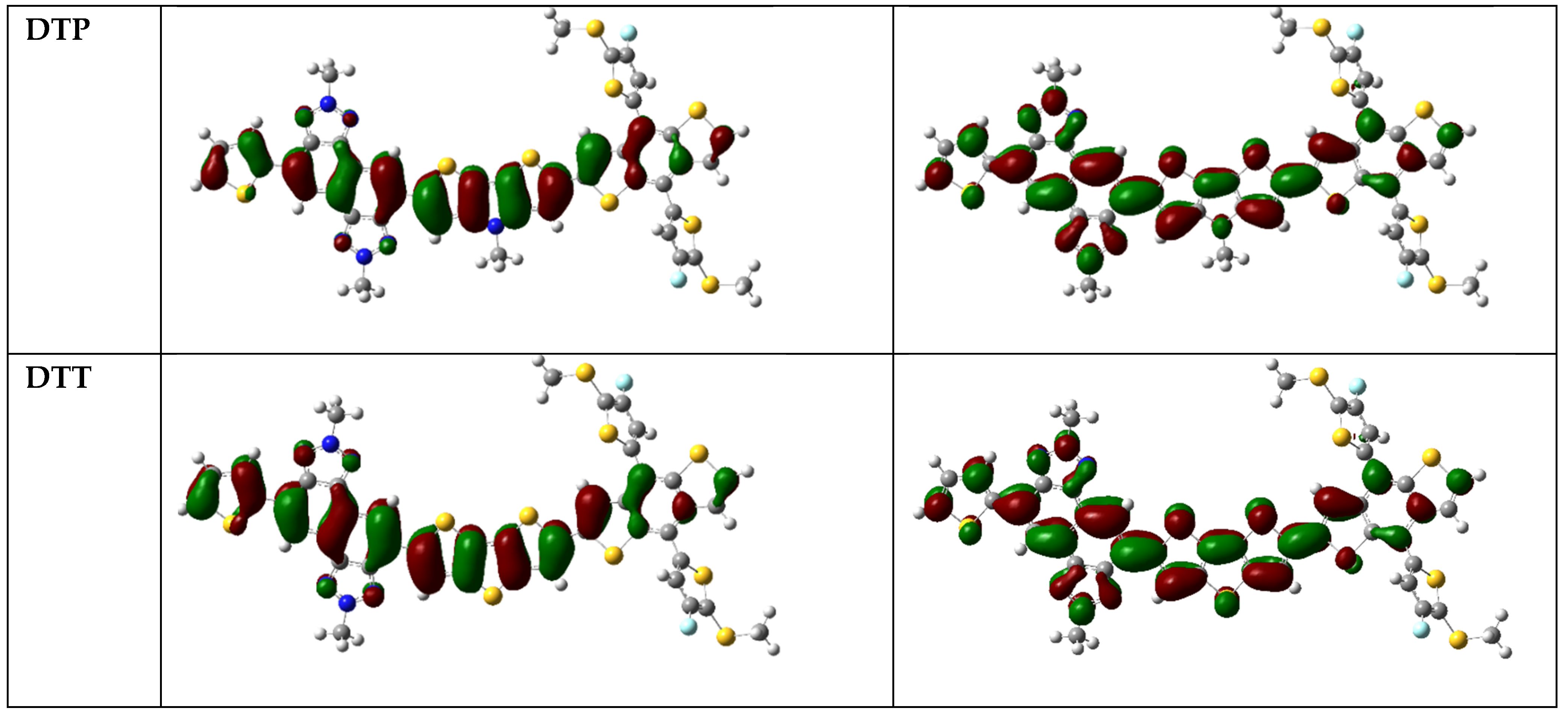
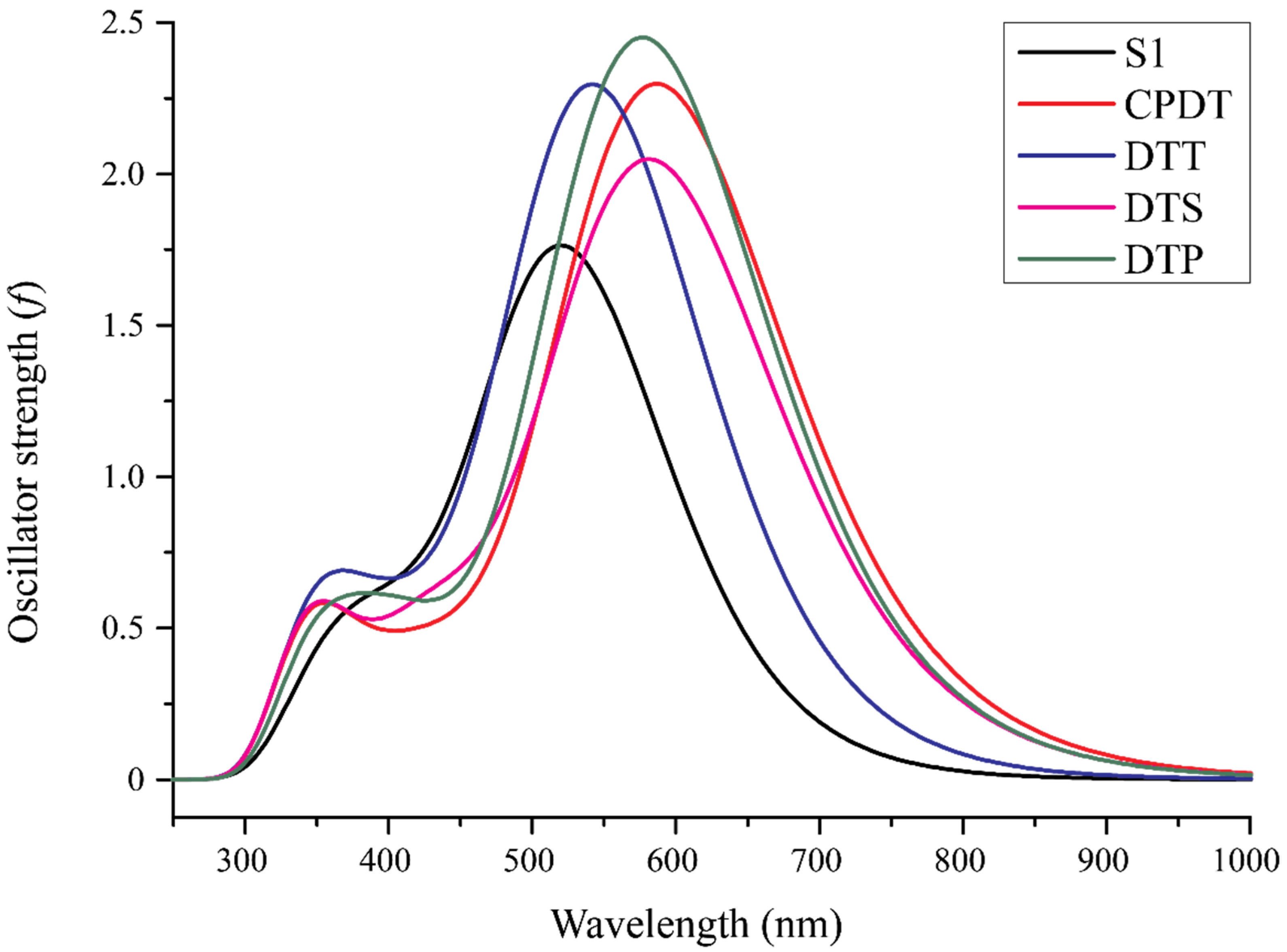

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| r1 | r2 | φ1 | φ2 | raver | φaver | ||
|---|---|---|---|---|---|---|---|
| 0S1 (1) | Gas | 1.443 | 1.454 | 11 | 3 | 1.449 | 7 |
| Solvent | 1.444 | 1.455 | 12 | 2 | 1.450 | 7 | |
| CPDT (2) | Gas | 1.440 | 1.451 | 16 | 4 | 1.446 | 10 |
| Solvent | 1.441 | 1.452 | 14 | 5 | 1.447 | 10 | |
| DTS (3) | Gas | 1.441 | 1.452 | 3 | 2 | 1.447 | 2 |
| Solvent | 1.442 | 1.453 | 0.8 | 2 | 1.448 | 2 | |
| DTP (4) | Gas | 1.440 | 1.451 | 11 | 0.5 | 1.446 | 6 |
| Solvent | 1.441 | 1.452 | 11 | 0.4 | 1.447 | 6 | |
| DTT (5) | Gas | 1.443 | 1.453 | 20 | 2 | 1.448 | 11 |
| Solvent | 1.444 | 1.454 | 18 | 3 | 1.449 | 11 |
| Anion | Cation | |||
|---|---|---|---|---|
| Δr (Å) | Δφ (°) | Δr (Å) | Δφ (°) | |
| S1 | 0.052 | 10 | 0.044 | 13 |
| CPDT | 0.050 | 16 | 0.038 | 17 |
| DTS | 0.048 | 2 | 0.038 | 3 |
| DTP | 0.045 | 8 | 0.037 | 8 |
| DTT | 0.048 | 18 | 0.040 | 18 |
| HOMO | LUMO | Egap | ||
|---|---|---|---|---|
| S1 | Gas Phase | −5.28 | −2.56 | 2.7 |
| Solvent | −5.40 | −2.67 | 2.74 | |
| CPDT | Gas Phase | −5.03 | −2.56 | 2.47 |
| Solvent | −5.11 | −2.64 | 2.46 | |
| DTS | Gas Phase | −5.08 | −2.59 | 2.50 |
| Solvent | −5.17 | −2.67 | 2.50 | |
| DTP | Gas Phase | −4.98 | −2.49 | 2.49 |
| Solvent | −5.08 | −2.59 | 2.49 | |
| DTT | Gas Phase | −5.23 | −2.60 | 2.63 |
| Solvent | −5.31 | −2.67 | 2.64 | |
| Dye | IEv | IEa | EAv | EAa | λh | λe |
|---|---|---|---|---|---|---|
| S1 | 6.21 | 6.11 | 1.73 | 1.85 | 0.23 | 0.22 |
| CPDT | 5.93 | 5.82 | 1.80 | 1.92 | 0.22 | 0.21 |
| DTS | 5.97 | 5.86 | 1.82 | 1.94 | 0.23 | 0.21 |
| DTP | 5.89 | 5.79 | 1.75 | 1.85 | 0.20 | 0.19 |
| DTT | 6.12 | 6.01 | 1.84 | 1.96 | 0.22 | 0.20 |
| Dye | λ (nm) | F | Orbital Transition | Singlet Electronic State Transition |
|---|---|---|---|---|
| S1 | 525 53754 | 1.7 | H → L (99%) | So → S1 |
| CPDT | 588 | 2.2 | H → L (99%) | |
| DTS | 584 | 2.0 | H → L (99%) | |
| DTP | 578 | 2.4 | H → L (99%) | |
| DTT | 545 | 2.2 | H → L (98%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trang, N.V.; Dung, T.N.; Cuong, N.T.; Hai, L.T.H.; Escudero, D.; Nguyen, M.T.; Nguyen, H.M.T. Theoretical Study of a Class of Organic D-?-A Dyes for Polymer Solar Cells: Influence of Various ?-Spacers. Crystals 2020, 10, 163. https://doi.org/10.3390/cryst10030163
Trang NV, Dung TN, Cuong NT, Hai LTH, Escudero D, Nguyen MT, Nguyen HMT. Theoretical Study of a Class of Organic D-?-A Dyes for Polymer Solar Cells: Influence of Various ?-Spacers. Crystals. 2020; 10(3):163. https://doi.org/10.3390/cryst10030163
Chicago/Turabian StyleTrang, Nguyen Van, Tran Ngoc Dung, Ngo Tuan Cuong, Le Thi Hong Hai, Daniel Escudero, Minh Tho Nguyen, and Hue Minh Thi Nguyen. 2020. "Theoretical Study of a Class of Organic D-?-A Dyes for Polymer Solar Cells: Influence of Various ?-Spacers" Crystals 10, no. 3: 163. https://doi.org/10.3390/cryst10030163
APA StyleTrang, N. V., Dung, T. N., Cuong, N. T., Hai, L. T. H., Escudero, D., Nguyen, M. T., & Nguyen, H. M. T. (2020). Theoretical Study of a Class of Organic D-?-A Dyes for Polymer Solar Cells: Influence of Various ?-Spacers. Crystals, 10(3), 163. https://doi.org/10.3390/cryst10030163