Towards an Optimal Sample Delivery Method for Serial Crystallography at XFEL

Abstract
:1. Introduction
2. XFEL and Serial Crystallography
3. The Need for Sample Delivery
- Conserve maximum crystal diffraction quality by minimizing mechanical and chemical stress during sample preparation and data collection.
- Maximize the signal-to-noise ratio of Bragg peaks by minimizing background scattering from the crystal carrying medium and container.
- Minimize sample wastage with crystal delivery that synchronizes to the XFEL repetition rate.
- Allow automation and robust operation (high-throughput) to minimize latent machine time due to sample changing or clogging.
- Allow homogenous “pump” triggers including rapid mixing with ligands or light permeation for time-resolved pump-probe experiment.
- Maintain sample stability until “diffraction before destruction”.
4. Serial Crystallography Sample Preparation
5. Liquid Jet for Crystals Suspended in Solution
6. Lipidic Cubic Phase (LCP) Injector for High Viscosity Sample Injection
7. Fixed-Target Approach Offers In-Situ Serial Crystallography
8. Drop-on-Demand—Potential to Maximize Sample Efficiency
9. Other Sample Delivery Methods
10. Injector Setup for Time-Resolved Experiment
11. Sample Delivery Present and Future
12. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar]
- Wong, C.H.; Siah, K.W.; Lo, A.W. Estimation of clinical trial success rates and related parameters. Biostatistic 2019, 20, 273–286. [Google Scholar] [CrossRef]
- Scott, D.E.; Coyne, A.G.; Hudson, S.A.; Abell, C. Fragment-Based Approaches in Drug Discovery and Chemical Biology. Biochemistry 2012, 51, 4990–5003. [Google Scholar] [CrossRef] [PubMed]
- Kunig, V.; Potowski, M.; Gohla, A.; Brunschweiger, A. DNA-encoded libraries—An efficient small molecule discovery technology for the biomedical sciences. Biol. Chem. 2018, 399, 691–710. [Google Scholar] [CrossRef] [PubMed]
- Favalli, N.; Basi, G.; Scheuermann, J.; Neri, D. DNA-encoded chemical libraries - achievements and remaining challenges. FEBS Lett. 2018, 592, 2168–2180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, J.; Lee, J.T.; Wang, W.; Zhang, J.; Cho, H.; Mamo, S.; Bremer, R.; Fillette, S.; Kong, J.; Haass, N.K.; et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. USA 2008, 8, 3041–3046. [Google Scholar] [CrossRef] [Green Version]
- Harris, P.A.; Berger, S.B.; Jeong, J.U.; Nagilla, R.; Bandyopadhyay, D.; Campobasso, N.; Capriotti, C.A.; Cox, J.A.; Dare, L.; Dong, X.; et al. Discovery of a First-in-Class Receptor Interacting Protein 1 (RIP1) Kinase Specific Clinical Candidate (GSK2982772) for the Treatment of Inflammatory Diseases. J. Med. Chem. 2017, 60, 1247–1261. [Google Scholar] [CrossRef]
- Smith, G.M.; Alexander, R.S.; Christianson, D.W.; McKeever, B.M.; Ponticello, G.S.; Springer, J.P.; Randall, W.C.; Baldwin, J.J.; Habecker, C.N. Positions of His-64 and a bound water in human carbonic anhydrase II upon binding three structurally related inhibitors. Protein Sci. 1994, 3, 118–125. [Google Scholar] [CrossRef] [Green Version]
- Navia, M.A.; Fitzgerald, P.M.D.; Mckeever, B.M.; Leu, C.; Heimbach, J.C.; Herber, W.K.; Sigal, I.S.; Darke, P.L.; Springer, J.P. Three-dimensional structure of aspartyl protease from human immunodeficiency virus HIV-1. Nature 1989, 337, 615–620. [Google Scholar] [CrossRef]
- von Itzstein, M.; Wu, W.Y.; Kok, G.B.; Pegg, M.S.; Dyason, J.C.; Jin, B.; Van Phan, T.; Smythe, M.L.; White, H.F.; Oliver, S.W.; et al. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature 1993, 363, 418–423. [Google Scholar] [CrossRef]
- Batool, M.; Ahmad, B.; Choi, S. A Structure-Based Drug Discovery Paradigm. Int. J. Mol. Sci. 2019, 20, 2783. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Song, K.; Li, L.; Chen, L. Structure-Based Drug Design Strategies and Challenges. Curr. Top. Med. Chem. 2018, 18, 12. [Google Scholar] [CrossRef] [PubMed]
- Manjasetty, B.A.; Büssow, K.; Panjikar, S.; Turnbull, A.P. Current methods in structural proteomics and its applications in biological sciences. Biotech 2012, 2, 89–113. [Google Scholar] [CrossRef] [Green Version]
- Leelananda, S.P.; Lindert, S. Computational methods in drug discovery. Beilstein J. Org. Chem. 2016, 12, 2694–2718. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y. Membrane protein structural biology in the era of single particle cryo-EM. Curr. Opin. Struct. Biol. 2018, 52, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.C.; McMullan, G.; Scheres, S.H.W. How cryo-EM is revolutionizing structural biology. Trends Biochem. Sci. 2015, 40, 49–57. [Google Scholar] [CrossRef]
- Ceska, T.; Chung, C.W.; Cooke, R.; Phillips, C.; Williams, P.A. Cryo-EM in drug discovery. Biochem. Soc. Trans. 2019, 47, 281–293. [Google Scholar] [CrossRef] [Green Version]
- Renaud, J.P.; Chari, A.; Ciferri, C.; Liu, W.T.; Rémigy, H.W.; Stark, H.; Wiesmann, C. Cryo-EM in drug discovery: Achievements, limitations and prospects. Nat. Rev. Drug. Discov. 2018, 17, 471–492. [Google Scholar] [CrossRef]
- Eriksson, M.; van der Veen, J.F.; Quitmann, C. Diffraction-limited storage rings—A window to the science of tomorrow. J. Synchrotron Rad. 2014, 21, 837–842. [Google Scholar] [CrossRef]
- Meents, A.; Wiedorn, M.O.; Srajer, V.; Henning, R.; Sarrou, I.; Bergtholdt, J.; Barthelmess, M.; Reinke, P.Y.A.; Dierksmeyer, D.; Tolstikova, A.; et al. Pink-beam serial crystallography. Nat. Commun. 2017, 8, 1281. [Google Scholar] [CrossRef]
- Brown, D.G.; Boström, J. Where Do Recent Small Molecule Clinical Development Candidates Come From? J. Med. Chem. 2018, 61, 9442–9468. [Google Scholar] [CrossRef] [PubMed]
- Markus, S.A.; Nordström, K.J.V.; Fredriksson, R.; Schiöth, H.B. Mapping the human membrane proteome: A majority of the human membrane proteins can be classified according to function and evolutionary origin. BMC Biol. 2009, 7, 50. [Google Scholar]
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How many drug targets are there? Nat. Rev. Drug Discov. 2006, 5, 993–996. [Google Scholar] [CrossRef] [PubMed]
- Congreve, M.; Dias, J.M.; Marshall, F.H. Structure-based drug design for G protein-coupled receptors. Prog. Med. Chem. 2014, 53, 1–63. [Google Scholar]
- Congreve, M.; Oswald, C.; Marshall, F.H. Applying Structure-Based Drug Design Approaches to Allosteric Modulators of GPCRs. Trends Pharmacol. Sci. 2017, 38, 837–847. [Google Scholar] [CrossRef]
- Lee, Y.; Basith, S.; Choi, S. Recent Advances in Structure-Based Drug Design Targeting Class A G Protein-Coupled Receptors Utilizing Crystal Structures and Computational Simulations. J. Med. Chem. 2018, 61, 1–46. [Google Scholar] [CrossRef]
- Serrano-Vega, M.J.; Magnani, F.; Shibata, Y.; Tate, C.G. Conformational thermostabilization of the beta1-adrenergic receptor in a detergent-resistant form. Proc. Natl. Acad. Sci. USA 2008, 105, 877–882. [Google Scholar] [CrossRef] [Green Version]
- Scott, D.J.; Kummer, L.; Tremmel, D.; Plückthun, A. Stabilizing membrane proteins through protein engineering. Curr. Opin. Chem. Biol. 2013, 17, 427–435. [Google Scholar] [CrossRef]
- Chun, E.; Thompson, A.A.; Liu, W.; Roth, C.B.; Griffith, M.T.; Katritch, V.; Kunken, J.; Xu, F.; Cherezov, V.; Hanson, M.A.; et al. Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure 2012, 20, 967–976. [Google Scholar] [CrossRef] [Green Version]
- Bayburt, T.H.; Grinkova, Y.V.; Sligar, S.G. Self-assembly of discoidal phospholipid bilayer nanoparticles with membrane scaffold proteins. Nano Lett. 2002, 2, 853–856. [Google Scholar] [CrossRef]
- Knowles, T.J.; Finka, R.; Smith, C.; Lin, Y.P.; Dafforn, T.; Overduin, M. Membrane proteins solubilized intact in lipid containing nanoparticles bounded by styrene maleic acid copolymer. J. Am. Chem. Soc. 2009, 131, 7484–7485. [Google Scholar] [PubMed]
- Frauenfeld, J.; Löving, R.; Armache, J.P.; Sonnen, A.F.; Guettou, F.; Moberg, P.; Zhu, L.; Jegerschöld, C.; Flayhan, A.; Briggs, J.A.; et al. A saposin-lipoprotein nanoparticle system for membrane proteins. Nat. Methods 2016, 13, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Caffrey, M.; Porter, C. Crystallizing membrane proteins for structure determination using lipidic mesophases. J. Vis. Exp. 2010, 3, 2–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faham, S.; Boie, J.U. Bicelle crystallization: A new method for crystallizing membrane proteins yields a monomeric bacteriorhodopsin Structure. J. Mol. Biol. 2002, 316, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Gourdon, P.; Andersen, J.L.; Hein, K.L.; Bublitz, M.; Pedersen, B.P.; Liu, X.; Yatime, L.; Nyblom, M.; Nielsen, T.T.; Olesen, C.; et al. HiLiDe—Systematic Approach to Membrane Protein Crystallization in Lipid and Detergent. Cryst. Growth Des. 2011, 11, 2098–2106. [Google Scholar] [CrossRef]
- Pándy-Szekeres, G.; Munk, C.; Tsonkov, T.M.; Mordalski, S.; Harpsøe, K.; Hauser, A.S.; Bojarski, A.J.; Gloriam, D.E. GPCRdb in 2018: Adding GPCR structure models and ligands. Nucleic Acids Res. 2018, 46, D440–D446. [Google Scholar] [CrossRef] [Green Version]
- Naydenova, K.; McMullan, G.; Peet, M.J.; Lee, Y.; Edwards, P.C.; Chen, S.; Leahy, E.; Scotcher, S.; Henderson, R.; Russo, C.J. CryoEM at 100 keV: A demonstration and prospects. IUCrJ 2019, 6, 1086–1098. [Google Scholar] [CrossRef]
- Vénien-Bryan, C.; Li, Z.; Vuillard, L.; Boutin, J.A. Cryo-electron microscopy and X-ray crystallography: Complementary approaches to structural biology and drug discovery. Acta Crystallogr. F Struct. Biol. Commun. 2017, 73, 174–183. [Google Scholar] [CrossRef] [Green Version]
- Shoemaker, S.C.; Ando, N. X-rays in the Cryo-Electron Microscopy Era: Structural Biology’s Dynamic Future. Biochemistry 2018, 57, 277–285. [Google Scholar] [CrossRef]
- Pellegrini, C. The history of X-ray free-electron lasers. Eur. Phys. J. H. 2012, 37, 659–708. [Google Scholar] [CrossRef] [Green Version]
- White, W.; Robert, A.; Dunne, M. The Linac Coherent Light Source. J. Synchrotron Rad. 2015, 22, 472–476. [Google Scholar] [CrossRef]
- Yabashi, M.; Tanak, H.; Ishikawa, T. Overview of the SACLA facility. J. Synchrotron Rad. 2015, 22, 477–484. [Google Scholar] [CrossRef] [Green Version]
- Tschentscher, T.; Bressler, C.; Grünert, J.; Madsen, A.; Mancuso, A.P.; Meyer, M.; Scherz, A.; Sinn, H.; Zastrau, U. Photon beam transport and scientific instruments at the european XFEL. Appl. Sci. 2017, 7, 592. [Google Scholar] [CrossRef] [Green Version]
- Milne, C.J.; Schietinger, T.; Alba, M.; Alarcon, A.; Alex, J.; Anghei, A.; Arsov, V.; Beard, C.; Beaud, P.; Bettoni, S.; et al. SwissFEL: The Swiss X-ray free electron laser. Appl. Sci. 2017, 7, 720. [Google Scholar] [CrossRef]
- Ko, I.S.; Kang, H.S.; Heo, H.; Kim, C.; Kim, G.; Min, C.K.; Yang, H.; Baek, S.Y.; Choi, H.J.; Mun, G.; et al. Construction and commissioning of PAL-XFEL facility. Appl. Sci. 2017, 7, 479. [Google Scholar] [CrossRef]
- Neutze, R.; Wouts, R.; van der Spoel, D.; Weckert, E.; Hajdu, J. Potential for biomolecular imaging with femtosecond X-ray pulses. Nature 2002, 406, 752–757. [Google Scholar] [CrossRef]
- Martin-Gracia, J.M.; Conrad, C.E.; Coe, J.; Roy-Chowdhury, S.; Fromme, P. Serial femtosecond crystallography: A revolution in structural biology. Arch. Biochem. Biophys. 2016, 602, 32–47. [Google Scholar] [CrossRef] [Green Version]
- Neutze, R.; Huldt, G.; Hajdu, J.; Van Der Spoel, D. Potential impact of an X-ray free electron laser on structural biology. Radiat. Phys. Chem. 2004, 71, 905–916. [Google Scholar] [CrossRef]
- Spence, J.C.H. XFELs for structure and dynamics in biology. IUCrJ 2017, 4, 322–339. [Google Scholar] [CrossRef]
- Boutet, S.; Lomb, L.; Williams, G.J.; Barends, T.R.; Aquila, A.; Doak, R.B.; Weierstall, U.; DePonte, D.P.; Steinbrener, J.; Shoeman, R.L.; et al. High-resolution protein structure determination by serial femtosecond crystallography. Science 2002, 337, 362–364. [Google Scholar] [CrossRef] [Green Version]
- Johansson, L.C.; Arnlund, D.; Katona, G.; White, T.A.; Barty, A.; DePonte, D.P.; Shoeman, R.L.; Wickstrand, C.; Sharma, A.; Williams, G.J.; et al. Structure of a photosynthetic reaction centre determined by serial femtosecond crystallography. Nat. Commun. 2013, 4, 2911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishigami, I.; Zatsepin, N.A.; Hikita, M.; Conrad, C.E.; Nelson, G.; Coe, J.D.; Basu, S.; Grant, T.D.; Seaberg, M.H.; Sierra, R.G.; et al. Crystal structure of CO-bound cytochrome c oxidase determined by serial femtosecond X-ray crystallography at room temperature. Proc. Natl. Acad. Sci. USA 2017, 114, 8011–8016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Han, G.W.; Batyuk, A.; Ishchenko, A.; White, K.L.; Patel, N.; Sadybekov, A.; Zamlynny, B.; Rudd, M.T.; Hollenstein, K.; et al. Structural basis for selectivity and diversity in angiotensin II receptors. Nature 2017, 544, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Fenalti, G.; Zatsepin, N.A.; Betti, C.; Giguere, P.; Han, G.W.; Ishchenko, A.; Liu, W.; Guillemyn, K.; Zhang, H.; James, D.; et al. Structural basis for bifunctional peptide recognition at human δ-opioid receptor. Nat. Struct. Mol. Biol. 2015, 22, 265–268. [Google Scholar] [CrossRef] [Green Version]
- Ginn, H.M.; Messerschmidt, M.; Ji, X.; Zhang, H.; Axford, D.; Gildea, R.J.; Winter, G.; Brewster, A.S.; Hattne, J.; Wagner, A.; et al. Structure of CPV17 polyhedrin determined by the improved analysis of serial femtosecond crystallographic data. Nat. Commun. 2015, 6, 6435. [Google Scholar] [CrossRef] [Green Version]
- Johansson, L.C.; Stauch, B.; McCorvy, J.D.; Han, G.W.; Patel, N.; Huang, X.P.; Batyuk, A.; Gati, C.; Slocum, S.T.; Li, C.; et al. XFEL structures of the human MT2 melatonin receptor reveal the basis of subtype selectivity. Nature 2019, 569, 289–292. [Google Scholar] [CrossRef]
- Cheng, R.K.Y.; Abela, R.; Hennig, M. X-ray free electron laser: Opportunities for drug discovery. Essays Biochem. 2017, 61, 529–542. [Google Scholar] [CrossRef]
- Mishin, A.; Gusach, A.; Luginina, A.; Marin, E.; Borshchevskiy, V.; Cherezov, V. An outlook on using serial femtosecond crystallography in drug discovery. Expert Opin. Drug Discov. 2019, 14, 933–945. [Google Scholar] [CrossRef]
- Ishchenko, A.; Stauch, B.; Han, G.W.; Batyuk, A.; Shiriaeva, A.; Li, C.; Zatsepin, N.; Weierstall, U.; Liu, W.; Nango, E.; et al. Toward G protein-coupled receptor structure-based drug design using X-ray lasers. IUCrJ 2019, 6, 1106–1119. [Google Scholar] [CrossRef]
- Nango, E.; Royant, A.; Kubo, M.; Nakane, T.; Wickstrand, C.; Kimura, T.; Tanaka, T.; Tono, K.; Song, C.; Tanaka, R.; et al. A three-dimensional movie of structural changes in bacteriorhodopsin. Science 2016, 354, 1552–1557. [Google Scholar] [CrossRef]
- Shimada, A.; Kubo, M.; Baba, S.; Yamashita, K.; Hirata, K.; Ueno, G.; Nomura, T.; Kimura, T.; Shinzawa-Itoh, K.; Baba, J.; et al. A nanosecond time-resolved XFEL analysis of structural changes associated with CO release from cytochrome c oxidase. Sci. Adv. 2017, 3, e1603042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kupitz, C.; Basu, S.; Zatsepin, N.; Pande, K.; Milathianaki, D.; Frank, M.; Hunter, M.; Boutet, S.; Williams, G.J.; Koglin, J.E.; et al. Time-resolved serial crystallography captures high-resolution intermediates of photoactive yellow protein. Science 2015, 346, 1242–1246. [Google Scholar]
- Kupitz, C.; Basu, S.; Grotjohann, I.; Fromme, R.; Zatsepin, N.A.; Rendek, K.N.; Hunter, M.S.; Shoeman, R.L.; White, T.A.; Wang, D.; et al. Serial time-resolved crystallography of photosystem II using a femtosecond X-ray laser. Nature 2014, 513, 261–265. [Google Scholar] [CrossRef] [PubMed]
- White, T.A. Crystallographic data processing for free-electron laser sources. Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 1231–1240. [Google Scholar] [CrossRef]
- Martiel, I.; Muller-Werkmeister, H.M.; Cohen, A.E. Strategies for sample delivery for femtosecond crystallography. Acta Cryst. 2019, D75, 160–177. [Google Scholar] [CrossRef] [Green Version]
- Grünbein, M.L.; Kovacs, G.N. Sample delivery for serial crystallography at free-electron lasers and synchrotrons. Acta Cryst. 2019, D75, 178–191. [Google Scholar] [CrossRef]
- Sui, S.; Perry, S.L. Microfluidics: From crystallization to serial time-resolved crystallography. Struct Dyn. 2017, 4, 032202. [Google Scholar] [CrossRef]
- Southworth-Davies, R.J.; Medina, M.A.; Carmichael, I.; Garman, E.F. Observation of decreased radiation damage at higher dose rates in room temperature protein crystallography. Structure 2007, 15, 1531–1541. [Google Scholar] [CrossRef] [Green Version]
- Owen, R.L.; Rudino-Pinera, E.; Garman, E.F. Experimental determination of the radiation dose limit for cryocooled protein crystals. Proc. Natl. Acad. Sci. USA 2006, 103, 4912–4917. [Google Scholar] [CrossRef] [Green Version]
- Kupitz, C.; Grotjohann, I.; Conrad, C.; Roy-Chowdhury, S.; Fromme, R.; Fromme, P. Microcrystallization techniques for serial femtosecond crystallography using photosystem II from Thermosynechococcus elongatus as a model system. Philos. Trans. R. Soc. B 2014, 369, 20130316. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Nogly, P.; Rheinberger, J.; Kick, L.M.; Gati, C.; Nelson, G.; Deupi, X.; Standfuss, J.; Schertler, G.; Panneels, V. Batch crystallization of rhodopsin for structural dynamics using an X-ray free-electron laser. Acta Cryst. 2015, F71, 856–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beale, J.; Bolton, R.; Marshall, S.A.; Beale, E.V.; Carr, S.B.; Ebrahim, A.; Moreno-Chicano, T.; Hough, M.A.; Worrall, J.A.R.; Tews, I.; et al. Successful sample preparation for serial crystallography experiments. J. Appl. Cryst. 2019, 52, 1385–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.B.; Kim, J.M.; Seok, J.H.; Lee, J.H.; Jo, J.D.; Mun, J.Y.; Conrad, C.; Coe., J.; Nelson, G.; Hogue, B.; et al. Supersaturation-controlled microcrystallization and visualization analysis for serial femtosecond crystallography. Sci. Rep. 2018, 8, 2541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dods, R.; Båth, P.; Arnlund, D.; Beyerlein, K.R.; Nelson., G.; Liang, M.; Harimoorthy, R.; Berntsen., P.; Malmerberg., E.; Johansson, L.; et al. From Macrocrystals to Microcrystals: A Strategy for Membrane Protein Serial Crystallography. Structure 2017, 25, 1461–1468. [Google Scholar] [CrossRef] [Green Version]
- Edlund, P.; Takala, H.; Claesson, E.; Henry, L.; Dods, R.; Lehtivuori, H.; Panman, M.; Pande, K.; White, T.; Nakane, T.; et al. The room temperature crystal structure of a bacterial phytochrome determined by serial femtosecond crystallography. Sci. Rep. 2016, 6, 35279. [Google Scholar] [CrossRef] [Green Version]
- Ishchenko, A.; Cherezov, V.; Liu, W. Preparation and Delivery of Protein Microcrystals in Lipidic Cubic Phase for Serial Femtosecond Crystallography. J. Vis. Exp. 2016, Sep 02, 115. [Google Scholar] [CrossRef] [Green Version]
- Andersson, R.; Safari, C.; Båth, P.; Bosman, R.; Shilova, A.; Dahl, P.; Ghosh, S.; Dunge, A.; Kjeldsen-Jensen, R.; Nan, J.; et al. Well-based crystallization of lipidic cubic phase microcrystals for serial X-ray crystallography experiments. Acta Crystallogr. D Struct. Biol. 2019, 75, 937–946. [Google Scholar] [CrossRef] [Green Version]
- Kolek, S.A.; Bräuning, B.; Steward, P.D. A novel microseeding method for the crystallization of membrane proteins in lipidic cubic phase. Acta Crystallogr. F Struct. Biol. Commun. 2016, 72, 307–312. [Google Scholar] [CrossRef] [Green Version]
- Doye, J.P.K.; Poon, W.C.K. Protein crystallization in vivo. Curr. Opin. Colloid Interface Sci. 2006, 11, 40–46. [Google Scholar] [CrossRef] [Green Version]
- Gati, C.; Oberthuer, D.; Yefanov, O.; Bunker, R.D.; Stellato, F.; Chiu, E.; Yeh, S.M.; Aquila, A.; Basu, S.; Bean, R.; et al. Atomic structure of granulin determined from native nanocrystalline granulovirus using an X-ray free-electron laser. Proc. Natl. Acad. Sci. USA 2017, 114, 2247–2252. [Google Scholar] [CrossRef] [Green Version]
- Koopmann, R.; Cupelli, K.; Redecke, L.; Nass, K.; Deponte, D.P.; White, T.A.; Stellato, F.; Rehders, D.; Liang, M.; Andreasson, J.; et al. In vivo protein crystallization opens new routes in structural biology. Nat. Methods 2012, 9, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Gati, C.; Bourenkov, G.; Klinge, M.; Rehders, D.; Stellato, F.; Oberthür, D.; Yefanov, O.; Sommer, B.P.; Mogk, S.; Duszenko, M.; et al. Serial crystallography on in vivo grown microcrystals using synchrotron radiation. IUCrJ 2014, 1, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Gallat, F.; Matsugaki, N.; Coussens, N.P.; Yagi, K.J.; Boudes, M.; Higashi, T.; Tsuji, D.; Tatano, Y.; Suzuki, M.; Mizohata, E.; et al. In vivo crystallography at X-ray free electron lasers: The next generation of structural biology? Philos. Trans. R. Soc. B 2014, 369, 20130497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawaya, M.R.; Cascio, D.; Gingery, M.; Rodriguez, J.; Goldschmidt, L.; Colletier, J.P.; Messerschmidt, M.M.; Boutet, S.; Koglin, J.E.; Williams, G.J.; et al. Protein crystal structure obtained at 2.9 Å resolution from injecting bacterial cells into an X-ray free-electron laser beam. Proc. Natl. Acad. Sci. USA 2014, 111, 12769–12774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duyvesteyn, H.M.E.; Ginn, H.M.; Peitila, M.K.; Wagner, A.; Hattne, J.; Grimes, J.M.; Hirvonen, E.; Evans, G.; Parsy, M.; Sauter, N.K.; et al. Towards in cellulo virus crystallography. Sci. Rep. 2018, 8, 3771. [Google Scholar] [CrossRef] [PubMed]
- Jakobi, A.J.; PAsson, D.M.; Knoops, K.; Stellato, F.; Liang, M.; White, T.A.; Seine, T.; Messerschmidt, M.; Chapman, H.N.; Wilmanns, M. In cellulo serial crystallography of alcohol oxidase crystals inside yeast cells. IUCrJ 2016, 3, 88–95. [Google Scholar] [CrossRef] [Green Version]
- Darmanin, C.; Strachan, J.; Adda, C.G.; Thomas, V.; Kobe, B.; Abbey, B. Protein crystal screening and characterization for serial femtosecond nanocrystallography. Sci. Rep. 2016, 6, 25345. [Google Scholar] [CrossRef] [Green Version]
- Stevenson, H.P.; DePonte, D.P.; Makhov, A.M.; Conway, J.F.; Zeldin, O.B.; Boutet, S.; Calero, G.; Cohen, A.E. Transmission electron microscopy as a tool for nanocrystal characterization pre- and post-injector. Philos. Trans. R. Soc. B 2014, 369, 20130322. [Google Scholar] [CrossRef] [Green Version]
- Barnes, C.O.; Kovaleva, E.G.; Fu, X.; Stevenson, H.P.; Brewster, A.S.; DePonte, D.P.; Baxter, E.L.; Cohen, A.E.; Calero, G. Assessment of microcrystal quality by transmission electron microscopy for efficient serial femtosecond crystallography. Arch. Biochem. Biophys. 2016, 602, 61–68. [Google Scholar] [CrossRef] [Green Version]
- Chapman, H.N.; Fromme, P.; Barty, A.; White, T.A.; Kirian, R.A.; Aquila, A.; Hunter, M.S.; Schulz, J.; DePonte, D.P.; Weierstall, U.; et al. Femtosecond X-ray protein nanocrystallography. Nature 2011, 470, 73–78. [Google Scholar] [CrossRef]
- Gañán-Calvo, A.M. Generation of Steady Liquid Microthreads and Micron-Sized Monodisperse Sprays in Gas Streams. Phys. Rev. Lett. 1998, 80, 285–288. [Google Scholar] [CrossRef]
- DePonte, D.P.; Weierstall, U.; Schmidt, K.; Warner, J.; Starodub, D.; Spence, J.C.H.; Doak, R.B. Gas dynamic virtual nozzle for generation of microscopic droplet streams. J. Phys. D: Appl. Phys. 2008, 41, 195505. [Google Scholar] [CrossRef] [Green Version]
- Doak, R.B.; DePonte, D.P.; Nelson, G.; Camacho-Alanis, F.; Ros, A.; Spence, J.C.H.; Weierstall, U. Microscopic Linear Liquid Streams in Vacuum: Injection of Solvated Biological Samples into X-Ray Free Electron Lasers. AIP Conf. Proc. 2012, 1501, 1314–1323. [Google Scholar]
- Weierstall, U. Liquid sample delivery techniques for serial femtosecond crystallography. Philos. Trans. R. Soc. B 2014, 369, 20130337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyerlein, K.R.; Adriano, L.; Heymann, M.; Kirian, R.; Knoska, J.; Wilde, F.; Chapman, H.N.; Bajt, S. Ceramic micro-injection molded nozzles for serial femtosecond crystallography sample delivery. Rev. Sci. Instrum. 2015, 86, 125104. [Google Scholar] [CrossRef] [Green Version]
- Weierstall, U.; Spence, J.C.H.; Doak, R.B. Injector for scattering measurements on fully solvated biospecies. Rev. Sci. Instrum. 2012, 83, 035108. [Google Scholar] [CrossRef]
- Nelson, G.; Kirian, R.A.; Weierstall, U.; Zatsepin, N.A.; Faragó, T.; Baumbach, T.; Wilde, F.; Niesler, F.B.; Zimmer, B.; Ishigami, I.; et al. Three-dimensional-printed gas dynamic virtual nozzles for x-ray laser sample delivery. Optics Express. 2016, 24, 11515–11530. [Google Scholar] [CrossRef]
- Demirci, H.; Sierra, R.G.; Laksmono, H.; Shoeman, R.L.; Botha, S.; Barends, T.R.; Nass, K.; Schlichting, I.; Doak, R.B.; Gati, C.; et al. Serial femtosecond X-ray diffraction of 30S ribosomal subunit microcrystals in liquid suspension at ambient temperature using an X-ray free-electron laser. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2013, 69, 1066–1069. [Google Scholar] [CrossRef]
- Abdallah, B.G.; Zatsepin, N.A.; Roy-Chowdhury, S.; Coe, J.; Conrad, C.E.; Dörner, K.; Sierra, R.G.; Stevenson, H.P.; Camacho-Alanis, F.; Grant, T.D.; et al. Microfluidic sorting of protein nanocrystals by size for X-ray free-electron laser diffraction. Struct. Dyn. 2015, 2, 041719. [Google Scholar] [CrossRef]
- Lomb, L.; Steinbrener, J.; Bari, S.; Beisel, D.; Berndt, D.; Kieser, C.; Lukat, M.; Neef, N.; Shoeman, R.L. An anti-settling sample delivery instrument for serial femtosecond crystallography. J. Appl. Cryst. 2012, 45, 674–678. [Google Scholar] [CrossRef]
- Grunbein, M.L.; Bielecki, J.; Gorel, A.; Stricker, M.; Bean, R.; Cammarata, M.; Dörner, K.; Fröhlich, L.; Hartmann, E.; Hauf, S.; et al. Megahertz data collection from protein microcrystals at an X-ray free-electron laser. Nat. Commun. 2018, 9, 3487. [Google Scholar] [CrossRef] [PubMed]
- Wiedorn, M.O.; Oberthür, D.; Bean, R.; Schubert, R.; Werner, N.; Abbey, B.; Aepfelbacher, M.; Adriano, L.; Allahgholi, A.; Al-Qudami, N.; et al. Megahertz serial crystallography. Nat. Commun. 2018, 9, 4025. [Google Scholar] [CrossRef] [Green Version]
- Oberthuer, D.; Knoška, J.; Wiedorn, M.O.; Beyerlein, K.R.; Bushnell, D.A.; Kovaleva, E.G.; Heymann, M.; Gumprecht, L.; Kirian, R.A.; Barty, A.; et al. Double-flow focused liquid injector for efficient serial femtosecond crystallography. Sci. Rep. 2017, 7, 44628. [Google Scholar] [CrossRef] [PubMed]
- Sierra, R.G.; Laksmono, H.; Kern, J.; Tran, R.; Hattne, J.; Alonso, M.; Lassalle-Kaiser, B.; Glöckner, C.; Hellmich, J.; Schafer, D.W.; et al. Nanoflow electrospinning serial femtosecond crystallography. Acta Cryst. 2012, D68, 1584–1587. [Google Scholar] [CrossRef] [PubMed]
- Sierra, R.G.; Gati, C.; Laksmono, H.; Dao, E.H.; Gul, S.; Fuller, F.; Kern, J.; Chatterjee, R.; Ibrahim, M.; Brewster, A.S.; et al. Concentric-flow electro kinetic injector enables serial crystallography of ribosome and photosystem I. Nat. Methods 2015, 13, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Caffrey, M. A comprehensive review of the lipid cubic phase or in meso method for crystallizing membrane and soluble proteins and complexes. Acta Cryst. 2015, F71, 3–18. [Google Scholar]
- Weierstall, U.; James, D.; Wang, C.; White, T.A.; Wang, D.; Liu, W.; Spence, J.C.; Bruce Doak, R.; Nelson, G.; Fromme, P.; et al. Lipidic cubic phase injector facilitates membrane protein serial femtosecond crystallography. Nat. Commun. 2013, 5, 3309. [Google Scholar] [CrossRef]
- Shimazu, Y.; Tono, K.; Tanaka, T.; Yamanaka, Y.; Nakane, T.; Mori, C.; Terakado Kimura, K.; Fujiwara, T.; Sugahara, M.; Tanaka, R.; et al. High-viscosity sample injection device for serial femtosecond crystallography at atmospheric pressure. J. Appl. Crystallogr. 2019, 52, 1280–1288. [Google Scholar] [CrossRef] [Green Version]
- Fromme, R.; Ishchenko, A.; Metz, M.; Chowdhury, S.R.; Basu, S.; Boutet, S.; Fromme, P.; White, T.A.; Barty, A.; Spence, J.C.; et al. Serial femtosecond crystallography of soluble proteins in lipidic cubic phase. IUCrJ 2015, 2, 545–551. [Google Scholar] [CrossRef] [Green Version]
- Nam, K.H. Sample Delivery Media for Serial Crystallography. Int. J. Mol. Sci. 2019, 20, 1094. [Google Scholar] [CrossRef] [Green Version]
- Sugahara, M.; Mizohata, E.; Nango, E.; Suzuki, M.; Tanaka, T.; Masuda, T.; Tanaka, R.; Shimamura, T.; Tanaka, Y.; Suno, C.; et al. Grease matrix as a versatile carrier of proteins for serial crystallography. Nat. Methods 2015, 12, 61–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conrad, C.E.; Basu, S.; James, D.; Wang, D.; Schaffer, A.; Roy-Chowdhury, S.; Zatsepin, N.A.; Aquila, A.; Coe, J.; Gati, C.; et al. A novel inert crystal delivery medium for serial femtosecond crystallography. IUCrJ 2015, 2, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, M.; Song, C.; Suzuki, M.; Masuda, T.; Inoue, S.; Nakane, T.; Yumoto, F.; Nango, E.; Tanaka, R.; Tono, K.; et al. Oil-free hyaluronic acid matrix for serial femtosecond crystallography. Sci. Rep. 2016, 6, 24484. [Google Scholar] [CrossRef] [Green Version]
- Botha, S.; Nass, K.; Barends, T.R.; Kabsch, W.; Latz, B.; Dworkowski, F.; Foucar, L.; Panepucci, E.; Wang, M.; Shoeman, R.L.; et al. Room-temperature serial crystallography at synchrotron X-ray sources using slowly flowing free-standing high-viscosity microstreams. Acta Cryst. 2015, D71, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Sugahara, M.; Nakane, T.; Masuda, T.; Suzuki, M.; Inoue, S.; Song, C.; Tanaka, R.; Nakatsu, T.; Mizohata, E.; Yumoto, F.; et al. Hydroxyethyl cellulose matrix applied to serial crystallography. Sci. Rep. 2017, 7, 703. [Google Scholar] [CrossRef] [PubMed]
- Kovacsova, G.; Grünbein, M.L.; Kloos, M.; Barends, T.R.M.; Schlesinger, R.; Heberle, J.; Kabsch, W.; Shoeman, R.L.; Doak, R.B.; Schlichting, I. Viscous hydrophilic injection matrices for serial crystallography. IUCrJ 2017, 4, 400–410. [Google Scholar] [CrossRef] [Green Version]
- Martin-Gracia, J.M.; Conrad, C.E.; Nelson, G.; Stander, N.; Zatsepin, N.A.; Zook, J.; Zhu, L.; Geiger, J.; Chun, E.; Kissick, D.; et al. Serial millisecond crystallography of membrane and soluble protein microcrystals using synchrotron radiation. IUCrJ 2017, 4, 439–454. [Google Scholar] [CrossRef]
- Park, J.; Park, S.; Kim, J.; Park, G.; Cho, Y.; Nam, K.H. Polyacrylamide injection matrix for serial femtosecond crystallography. Sci. Rep. 2019, 9, 2525. [Google Scholar] [CrossRef] [Green Version]
- Nam, K.H. Shortening injection matrix for serial crystallography. Sci. Rep. 2020, 10, 107. [Google Scholar] [CrossRef] [Green Version]
- Sugahara, M.; Motomura, K.; Suzuki, M.; Masuda, T.; Joti, Y.; Numata, K.; Tono, K.; Yabashi, M.; Ishikawa, T. Viscosity-adjustable grease matrices for serial nanocrystallography. Sci. Rep. 2020, 10, 1371. [Google Scholar] [CrossRef]
- James, D.; Weinert, T.; Skopintsev, P.; Furrer, A.; Gashi, D.; Tanaka, T.; Nango, E.; Nogly, P.; Standfuss, J. Improving High Viscosity Extrusion of Microcrystals for Time-resolved Serial Femtosecond Crystallography at X-ray Lasers. J. Vis. Exp. 2019, 144, e59087. [Google Scholar] [CrossRef] [PubMed]
- Tosha, T.; Nomura, T.; Nishida, T.; Saeki, N.; Okubayashi, K.; Yamagiwa, R.; Sugahara, M.; Nakane, T.; Yamashita, K.; Hirata, K.; et al. Capturing an initial intermediate during the P450nor enzymatic reaction using time-resolved XFEL crystallography and caged-substrate. Nat. Commun. 2017, 8, 1585. [Google Scholar] [CrossRef] [PubMed]
- Audet, M.; White, K.L.; Breton, B.; Zarzycka, B.; Han, G.W.; Lu, Y.; Gati, C.; Batyuk, A.; Popov, P.; Velasquez, J.; et al. Crystal structure of misoprostol bound to the labor inducer prostaglandin E2 receptor. Nat. Chem. Biol. 2019, 15, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Luginina, A.; Gusach, A.; Marin, E.; Mishin, A.; Brouillette, R.; Popov, P.; Shiriaeva, A.; Besserer-Offroy, É.; Longpré, J.M.; Lyapina, E.; et al. Structure-based mechanism of cysteinyl leukotriene receptor inhibition by antiasthmatic drugs. Sci. Adv. 2019, 5, eaax2518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stauch, B.; Johansson, L.C.; McCorvy, J.D.; Patel, N.; Han, G.W.; Huang, X.P.; Gati, C.; Batyuk, A.; Slocum, S.T.; Ishchenko, A.; et al. Structural basis of ligand recognition at the human MT1 melatonin receptor. Nature 2019, 569, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Hunter, M.S.; Segelke, B.; Messerschmidt, M.; Williams, G.J.; Zatsepin, N.A.; Barty, A.; Benner, W.H.; Carlson, D.B.; Coleman, M.; Graf, A.; et al. Fixed-target protein serial microcrystallography with an X-ray free electron laser. Sci. Rep. 2014, 4, 6026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Joti, Y.; Ishikawa, T.; Song, C. Monte Carlo study for optimal conditions in single-shot imaging with femtosecond x-ray laser pulses. Appl. Phys. Lett. 2013, 103, 264101. [Google Scholar] [CrossRef] [Green Version]
- Cohen, A.E.; Soltis, S.M.; González, A.; Aguila, L.; Alonso-Mori, R.; Barnes, C.O.; Baxter, E.L.; Brehmer, W.; Brewster, A.S.; Brunger, A.T.; et al. Goniometer-based femtosecond crystallography with X-ray free electron lasers. Proc. Natl. Acad. Sci. USA 2014, 111, 17122–17127. [Google Scholar] [CrossRef] [Green Version]
- Baxter, E.; Aguila, L.; Alonso-Mori, R.; Barnes, C.O.; Bonagura, C.A.; Brehmer, W.; Brunger, A.T.; Calero, G.; Caradoc-Davies, T.T.; Chatterjee, R.; et al. High-density grids for efficient data collection from multiple crystals. Acta Crystallogr. D Struct. Biol. 2016, 72, 2–11. [Google Scholar] [CrossRef]
- Zarrine-Afsar, A.; Barends, T.R.; Müller, C.; Fuchs, M.R.; Lomb, L.; Schlichting, I.; Miller, R.J. Crystallography on a chip. Acta Cryst. 2012, D68, 321–323. [Google Scholar] [CrossRef] [Green Version]
- Roedig, P.; Vartiainen, I.; Duman, R.; Panneerselvam, S.; Stübe, N.; Lorbeer, O.; Warmer, M.; Sutton, G.; Stuart, D.I.; Weckert, E.; et al. A micro-patterned silicon chip as sample holder for macromolecular crystallography experiments with minimal background scattering. Sci. Rep. 2015, 5, 10451. [Google Scholar] [CrossRef] [PubMed]
- Roedig, P.; Duman, R.; Sanchez-Weatherby, J.; Vartiainen, I.; Burkhardt, A.; Warmer, M.; David, C.; Wagner, A.; Meents, A. Room-temperature macromolecular crystallography using a micro-patterned silicon chip with minimal background scattering. J. Appl. Cryst. 2016, 49, 968–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roedig, P.; Ginn, H.M.; Pakendorf, T.; Sutton, G.; Harlos, K.; Walter, T.S.; Meyer, J.; Fischer, P.; Duman, R.; Vartiainen, I.; et al. High-speed fixed-target serial virus crystallography. Nat. Methods 2017, 14, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Lieske, J.; Cerv, M.; Kreida, S.; Komadina, D.; Fischer, J.; Barthelmess, M.; Fischer, P.; Pakendorf, T.; Yefanov, O.; Mariani, V.; et al. On-chip crystallization for serial crystallography experiments and on-chip ligand-binding studies. IUCrJ 2019, 6, 714–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coquelle, N.; Brewster, A.S.; Kapp, U.; Shilova, A.; Weinhausen, B.; Burghammer, M.; Colletier, J. Raster scanning serial protein crystallography using micro- and nano-focused synchrotron beams. Acta Cryst. 2015, D71, 1184–1196. [Google Scholar] [CrossRef] [Green Version]
- Murray, T.D.; Lyubimov, A.Y.; Ogata, C.M.; Vo, H.; Uervirojnangkoorn, M.; Brunger, A.T.; Berger, J.M. A high-transparency, micro-patternable chip for X-ray diffraction analysis of microcrystals under native growth conditions. Acta Cryst. 2015, D71, 1987–1997. [Google Scholar] [CrossRef] [Green Version]
- Oghbaey, S.; Sarracini, A.; Ginn, H.M.; Pare-Labrosse, O.; Kuo, A.; Marx, A.; Epp, S.W.; Sherrell, D.A.; Eger, B.T.; Zhong, Y.; et al. Fixed target combined with spectral mapping: Approaching 100% hit rates for serial crystallography. Acta Crystallogr. D Struct. Biol. 2016, 72, 944–955. [Google Scholar] [CrossRef] [Green Version]
- Opara, N.; Martiel, I.; Arnold, S.A.; Braun, T.; Stahlberg, H.; Makita, M.; David, C.; Padeste, C. Direct protein crystallization on ultrathin membranes for diffraction measurements at X-ray free-electron lasers. J. Appl. Cryst. 2017, 50, 909–918. [Google Scholar] [CrossRef] [Green Version]
- Gicquel, Y.; Schubert, R.; Kapis, S.; Bourenkov, G.; Schneider, T.; Perbandt, M.; Betzel, C.; Chapman, H.N.; Heymann, M. Microfluidic Chips for In Situ Crystal X-ray Diffraction and In Situ Dynamic Light Scattering for Serial Crystallography. J. Vis. Exp. 2018, 134, e57133. [Google Scholar] [CrossRef] [Green Version]
- Doak, R.B.; Nass Kovacs, G.; Gorel, A.; Foucar, L.; Barends, T.R.M.; Grünbein, M.L.; Hilpert, M.; Kloos, M.; Roome, C.M.; Shoeman, R.L.; et al. Crystallography on a chip—without the chip: Sheet-on-sheet sandwich. Acta Crystallogr. D Struct. Biol. 2018, 74, 1000–1007. [Google Scholar] [CrossRef]
- Sui, S.; Wang, Y.; Kolewe, K.W.; Srajer, V.; Henning, R.; Schiffman, J.D.; Dimitrakopoulos, C.; Perry, S.L. Graphene-based microfluidics for serial crystallography. Lab Chip. 2016, 16, 3082–3096. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Ayhan, M.; Bandara, S.; Bowatte, K.; Kumarapperuma, I.; Gunawardana, S.; Shin, H.; Wang, C.; Zeng, X.; Yang, X. Crystal-on-crystal chips for in situ serial diffraction at room temperature. Lab Chip. 2018, 18, 2246–2256. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Baek, S.; Park, J.; Lee, K.; Kim, J.; Lee, S.J.; Chung, W.K.; Lee, J.L.; Cho, Y.; Nam, K.H. Nylon mesh-based sample holder for fixed-target serial femtosecond crystallography. Sci. Rep. 2019, 9, 6971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nass, K. Radiation damage in protein crystallography at X-ray free-electron lasers. Acta Crystallogr. D Struct. Biol. 2019, 75, 211–218. [Google Scholar] [CrossRef]
- Lyubimov, A.Y.; Murray, T.D.; Koehl, A.; Araci, I.E.; Uervirojnangkoorn, M.; Zeldin, O.B.; Cohen, A.E.; Soltis, S.M.; Baxter, E.L.; Brewster, A.S.; et al. Capture and X-ray diffraction studies of protein microcrystals in a microfluidic trap array. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 928–940. [Google Scholar] [CrossRef]
- Mueller, C.; Marx, A.; Epp, S.W.; Zhong, Y.; Kuo, A.; Balo, A.R.; Soman, J.; Schotte, F.; Lemke, H.T.; Owen, R.L.; et al. Fixed target matrix for femtosecond time-resolved and in situ serial micro-crystallography. Struct. Dyn. 2015, 2, 054302. [Google Scholar] [CrossRef] [Green Version]
- Owen, R.L.; Axford, D.; Sherrell, D.A.; Kuo, A.; Ernst, O.P.; Schulz, E.C.; Miller, R.J.; Mueller-Werkmeister, H.M. Low-dose fixed-target serial synchrotron crystallography. Acta Crystallogr. D Struct. Biol. 2017, 73, 373–378. [Google Scholar] [CrossRef]
- Sanchez-Weatherby, J.; Moraes, I. Crystal Dehydration in Membrane Protein Crystallography. In The Next Generation in Membrane Protein Structure Determination; Advances in Experimental Medicine and Biology; Moraes, I., Ed.; Springer: Cham, Switzerland, 2016; Volume 92, pp. 73–89. [Google Scholar]
- Ellson, R.; Mutz, M.; Browning, B.; Lee, L.; Miller, M.F. Roeland Papen Picoliter Inc. Transfer of Low Nanoliter Volumes between Microplates Using Focused Acoustics—Automation Considerations. JALA 2003, 8, 29–34. [Google Scholar]
- Villaseñor, A.G.; Wong, A.; Shao, A.; Garg, A.; Donohue, T.J.; Kuglstatter, A.; Harris, S.F. Nanolitre-scale crystallization using acoustic liquid-transfer technology. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 893–900. [Google Scholar] [CrossRef]
- Villaseñor, A.G.; Wong, A.; Shao, A.; Garg, A.; Kuglstatter, A.; Harris, S.F. Acoustic matrix microseeding: Improving protein crystal growth with minimal chemical bias. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 568–576. [Google Scholar] [CrossRef]
- Yin, X.; Scalia, A.; Leroy, L.; Cuttitta, C.M.; Polizzo, G.M.; Ericson, D.L.; Roessler, C.G.; Campos, O.; Ma, M.Y.; Agarwal, R.; et al. Hitting the target: Fragment screening with acoustic in situ co-crystallization of proteins plus fragment libraries on pin-mounted data-collection micromeshes. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 1177–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teplitsky, E.; Joshi, K.; Ericson, D.L.; Scalia, A.; Mullen, J.D.; Sweet, R.M.; Soares, A.S. High throughput screening using acoustic droplet ejection to combine protein crystals and chemical libraries on crystallization plates at high density. J. Struct. Biol. 2015, 191, 49–58. [Google Scholar] [CrossRef] [Green Version]
- Collins, P.M.; Ng, J.T.; Talon, R.; Nekrosiute, K.; Krojer, T.; Douangamath, A.; Brandao-Neto, J.; Wright, N.; Pearce, N.M.; von Delft, F. Gentle, fast and effective crystal soaking by acoustic dispensing. Acta Crystallogr. D Struct. Biol. 2017, 73, 246–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, A.S.; Engel, M.A.; Stearns, R.; Datwani, S.; Olechno, J.; Ellson, R.; Skinner, J.M.; Allaire, M.; Orville, A.M. Acoustically Mounted Microcrystals Yield High-Resolution X-ray Structures. Biochemistry 2011, 50, 4399–4401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samara, Y.N.; Brennan, H.M.; McCarthy, L.; Bollard, M.T.; Laspina, D.; Wlodek, J.M.; Campos, S.L.; Natarajan, R.; Gofron, K.; McSweeney, S.; et al. Using sound pulses to solve the crystal-harvesting Bottleneck. Acta Crystallogr. D Struct. Biol. 2018, 74, 986–999. [Google Scholar] [CrossRef] [PubMed]
- Roessler, C.G.; Agarwal, R.; Allaire, M.; Alonso-Mori, R.; Andi, B.; Bachega, J.F.R.; Bommer, M.; Brewster, A.S.; Browne, M.C.; Chatterjee, R.; et al. Acoustic Injectors for Drop-On-Demand Serial Femtosecond Crystallography. Structure 2016, 24, 631–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuller, F.D.; Gul, S.; Chatterjee, R.; Burgie, E.S.; Young, I.D.; Lebrette, H.; Srinivas, V.; Brewster, A.S.; Michels-Clark, T.; Clinger, J.A.; et al. Drop-on-demand sample delivery for studying biocatalysts in action at X-ray free-electron lasers. Nat. Methods. 2017, 14, 443–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsujino, S.; Tomizaki, T. Ultrasonic acoustic levitation for fast frame rate X-ray protein crystallography at room temperature. Sci. Rep. 2016, 6, 25558. [Google Scholar] [CrossRef]
- Morris, R.H.; Dye, E.R.; Axford, D.; Newton, M.I.; Beale, J.H.; Docker, P.T. Non-Contact Universal Sample Presentation for Room Temperature Macromolecular Crystallography Using Acoustic Levitation. Sci. Rep. 2019, 9, 12431. [Google Scholar] [CrossRef]
- Mafuné, F.; Miyajima, K.; Tono, K.; Takeda, Y.; Kohno, J.Y.; Miyauchi, N.; Kobayashi, J.; Joti, Y.; Nango, E.; Iwata, S.; et al. Microcrystal delivery by pulsed liquid droplet for serial femtosecond crystallography. Acta Crystallogr. D Struct. Biol. 2016, 72, 520–523. [Google Scholar] [CrossRef]
- Davy, B.; Axford, D.; Beale, J.H.; Butryn, A.; Docker, P.; Ebrahim, A.; Leen, G.; Orville, A.M.; Owen, R.L.; Aller, P. Reducing sample consumption for serial crystallography using acoustic drop ejection. J Synchrotron Radiat. 2019, 26, 1820–1825. [Google Scholar] [CrossRef] [PubMed]
- Echelmeier, A.; Kim, D.; Cruz Villarreal, J.; Coe, J.; Quintana, S.; Brehm, G.; Egatz-Gomez, A.; Nazari, R.; Sierra, R.G.; Koglin, J.E.; et al. 3D printed droplet generation devices for serial femtosecond crystallography enabled by surface coating. J. Appl. Crystallogr. 2019, 52, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Brenker, J.C.; Devendran, C.; Neild, A.; Alan, T. On-demand sample injection: Combining acoustic actuation with a tear-drop shaped nozzle to generate droplets with precise spatial and temporal control. Lab Chip. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ashtiani, D.; Venugopal, H.; Belousoff, M.; Spicer, B.; Mak, J.; Neild, A.; de Marco, A. Delivery of femtolitre droplets using surface acoustic wave based atomisation for cryo-EM grid preparation. J. Struct. Biol. 2018, 203, 94–101. [Google Scholar] [CrossRef]
- Mathews, I.I.; Allison, K.; Robbins, T.; Lyubimov, A.Y.; Uervirojnangkoorn, M.; Brunger, A.T.; Khosla, C.; DeMirci, H.; McPhillips, S.E.; Hollenbeck, M.; et al. The Conformational Flexibility of the Acyltransferase from the Disorazole Polyketide Synthase Is Revealed by an X-ray Free-Electron Laser Using a Room-Temperature Sample Delivery Method for Serial Crystallography. Biochemistry 2017, 56, 4751–4756. [Google Scholar] [CrossRef] [Green Version]
- Awel, S.; Kirian, R.A.; Wiedorn, M.O.; Beyerlein, K.R.; Roth, N.; Horke, D.A.; Oberthür, D.; Knoska, J.; Mariani, V.; Morgan, A.; et al. Femtosecond X-ray diffraction from an aerosolized beam of protein nanocrystals. J. Appl. Crystallogr. 2018, 51, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Daurer, B.J.; Okamoto, K.; Bielecki, J.; Maia, F.R.N.C.; Mühlig, K.; Seibert, M.M.; Hantke, M.F.; Nettelblad, C.; Benner, W.H.; Svenda, M.; et al. Experimental strategies for imaging bioparticles with femtosecond hard X-ray pulses. IUCrJ 2017, 4, 251–262. [Google Scholar] [CrossRef]
- Schulz, E.C.; Kaub, J.; Busse, F.; Mehrabi, P.; Muller-Werkmeister, H.M.; Pai, E.F.; Robertson, W.D.; Miller, R.J.D. Protein crystals IR laser ablated from aqueous solution at high speed retain their diffractive properties: Applications in high-speed serial crystallography. J. Appl. Cryst. 2017, 50, 1773–1781. [Google Scholar] [CrossRef]
- Monteiro, D.C.F.; Vakili, M.; Harich, J.; Sztucki, M.; Meier, S.M.; Horrell, S.; Josts, I.; Trebbin, M. A microfluidic flow-focusing device for low sample consumption serial synchrotron crystallography experiments in liquid flow. J. Synchrotron Radiat. 2019, 26, 406–412. [Google Scholar] [CrossRef]
- Stellato, F.; Oberthur, D.; Liang, M.; Bean, R.; Gati, C.; Yefanov, O.; Barty, A.; Burkhardt, A.; Fischer, P.; Galli, L.; et al. Room-temperature macromolecular serial crystallography using synchrotron radiation. IUCrJ 2014, 1, 204–212. [Google Scholar] [CrossRef]
- Nam, K.H. Stable sample delivery in viscous media via a capillary for serial crystallography. J. Appl. Cryst. 2020, 53, 45–50. [Google Scholar] [CrossRef]
- Moffat, K. Time-Resolved Crystallography. Acta Cryst. 1998, A54, 833–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, X.; He, Y.; Huang, X.; Zhao, Y. Crystallographic Snapshots of Class A β-Lactamase Catalysis Reveal Structural Changes That Facilitate -Lactam Hydrolysis. J. Biol. Chem. 2017, 292, 4022–4033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pande, K.; Hutchison, C.D.; Groenhof, G.; Aquila, A.; Robinson, J.S.; Tenboer, J.; Basu, S.; Boutet, S.; DePonte, D.P.; Liang, M.; et al. Femtosecond structural dynamics drives the trans/cis isomerization in photoactive yellow protein. Science 2016, 352, 725–729. [Google Scholar] [CrossRef] [Green Version]
- Neutze, R.; Moffat, K. Time-resolved structural studies at synchrotrons and X-ray free electron lasers: Opportunities and challenges. Curr. Opin. Struct. Biol. 2012, 22, 651–659. [Google Scholar] [CrossRef] [Green Version]
- Levantino, M.; Yorke, B.A.; Monteiro, D.C.F.; Cammarata, M.; Pearson, A.R. Using synchrotrons and XFELs for time-resolved X-ray crystallography and solution scattering experiments on biomolecules. Curr. Opin. Struct. Biol. 2015, 35, 41–48. [Google Scholar] [CrossRef]
- Nogly, P.; James, D.; Wang, D.; White, T.A.; Zatsepin, N.; Shilova, A.; Nelson, G.; Liu, H.; Johansson, L.; Heymann, M.; et al. Lipidic cubic phase serial millisecond crystallography using synchrotron radiation. IUCrJ 2015, 2, 168–176. [Google Scholar] [CrossRef]
- Weinert, T.; Olieric, N.; Cheng, R.; Brünle, S.; James, D.; Ozerov, D.; Gashi, D.; Vera, L.; Marsh, M.; Jaeger, K.; et al. Serial millisecond crystallography for routine roomtemperature structure determination at synchrotrons. Nat. Commun. 2017, 8, 542. [Google Scholar] [CrossRef]
- Aquila, A.; Hunter, M.S.; Doak, R.B.; Kirian, R.A.; Fromme, P.; White, T.A.; Andreasson, J.; Arnlund, D.; Bajt, S.; Barends, T.R.; et al. Time-resolved protein nanocrystallography using an X-ray free-electron laser. Opt. Express. 2012, 20, 2706–2716. [Google Scholar] [CrossRef]
- Suga, M.; Akita, F.; Sugahara, M.; Kubo, M.; Nakajima, Y.; Nakane, T.; Yamashita, K.; Umena, Y.; Nakabayashi, M.; Yamane, T.; et al. Light-induced structural changes and the site of O=O bond formation in PSII caught by XFEL. Nature 2017, 543, 131–135. [Google Scholar] [CrossRef]
- Tenboer, J.; Basu, S.; Zatsepin, N.; Pande, K.; Milathianaki, D.; Frank, M.; Hunter, M.; Boutet, S.; Williams, G.J.; Koglin, J.E.; et al. Time-resolved serial crystallography captures high-resolution intermediates of photoactive yellow protein. Science 2014, 346, 1242–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coquelle, N.; Sliwa, M.; Woodhouse, J.; Schirò, G.; Adam, V.; Aquila, A.; Barends, T.R.M.; Boutet, S.; Byrdin, M.; Carbajo, S.; et al. Chromophore twisting in the excited state of a photoswitchable fluorescent protein captured by time-resolved serial femtosecond crystallography. Nat. Chem. 2018, 10, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Nogly, P.; Weinert, T.; James, D.; Carbajo, S.; Ozerov, D.; Furrer, A.; Gashi, D.; Borin, V.; Skopintsev, P.; Jaeger, K.; et al. Retinal isomerization in bacteriorhodopsin captured by a femtosecond x-ray laser. Science 2018, 361, 6398. [Google Scholar]
- Barends, T.R.M.; Foucar, L.; Ardevol, A.; Nass, K.; Aquila, A.; Botha, S.; Doak, R.B.; Falahati, K.; Hartmann, E.; Hilpert, M.; et al. Direct observation of ultrafast collective motions in CO myoglobin upon ligand dissociation. Science 2015, 350, 445–450. [Google Scholar] [CrossRef]
- Cohen, A.; Doukov, T.; Soltis, M. UV-Visible Absorption Spectroscopy Enhanced X-ray Crystallography at Synchrotron and X-ray Free Electron Laser Sources. Pept. Lett. 2016, 23, 283–290. [Google Scholar] [CrossRef]
- Lincoln, C.N.; Fitzpatrick, A.E.; van Thor, J.J. Photoisomerisation quantum yield and non-linear cross-sections with femtosecond excitation of the photoactive yellow protein. Phys. Chem. Chem. Phys. 2012, 14, 15752–15764. [Google Scholar] [CrossRef]
- Hutchison, C.D.M.; Kaucikas, M.; Tenboer, J.; Kupitz, C.; Moffat, K.; Schmidt, M.; van Thor, J.J. Photocycle populations with femtosecond excitation of crystalline photoactive yellow protein. Chem. Phys. Lett. 2016, 654, 63–71. [Google Scholar] [CrossRef] [Green Version]
- Harmand, M.; Coffee, R.; Bionta, M.R.; Chollet, M.; French, D.; Zhu, D.; Fritz, D.M.; Lemke, H.T.; Medvedev, N.; Ziaja, B.; et al. Achieving few-femtosecond time-sorting at hard X-ray free-electron lasers. Nat. Photonics 2013, 7, 215–218. [Google Scholar] [CrossRef]
- Hartmann, N.; Helml, W.; Galler, A.; Bionta, M.R.; Grunert, J.; Molodtsov, S.L.; Ferguson, K.R.; Schorb, S.; Swiggers, M.L.; Carron, S.; et al. Sub-femtosecond precision measurement of relative X-ray arrival time for free-electron lasers. Nat. Photonics 2014, 8, 706–709. [Google Scholar] [CrossRef]
- Keedy, D.A.; Kenner, L.R.; Warkentin, M.; Woldeyes, R.A.; Hopkins, J.B.; Thompson, M.C.; Brewster, A.S.; Van Benschoten, A.H.; Baxter, E.L.; Uervirojnangkoorn, M.; et al. Mapping the conformational landscape of a dynamic enzyme by multitemperature and XFEL crystallography. eLife 2015, 4, e07574. [Google Scholar] [CrossRef] [Green Version]
- Causgrove, T.P.; Dyer, R.B. Nonequilibrium protein folding dynamics: Laser-induced pH-jump studies of the helix–coil transition. Chem. Phys. 2006, 323, 2–10. [Google Scholar] [CrossRef]
- Schmidt, M. Mix and Inject: Reaction Initiation by Diffusion for Time-Resolved Macromolecular Crystallography. Adv. Condensed Matt. Phys. 2013, 167276. [Google Scholar] [CrossRef] [Green Version]
- Calvey, G.D.; Katz, A.M.; Schaffer, C.B.; Pollack, L. Mixing injector enables time-resolved crystallography with high hit rate at X-ray free electron lasers. Struct. Dyn. 2016, 3, 054301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyerlein, K.R.; Dierksmeyer, D.; Mariani, V.; Kuhn, M.; Sarrou, I.; Ottaviano, A.; Awel, S.; Knoska, J.; Fuglerud, S.; Jönsson, O.; et al. Mix-and-diffuse serial synchrotron crystallography. IUCrJ 2017, 4, 769–777. [Google Scholar] [CrossRef]
- Stagno, J.R.; Bhandari, Y.R.; Conrad, C.E.; Liu, Y.; Wang, Y.X. Real-time crystallographic studies of the adenine riboswitch using an X-ray free-electron laser. FEBS J. 2017, 284, 3374–3380. [Google Scholar] [CrossRef] [Green Version]
- Olmos, J.L., Jr.; Pandey, S.; Martin-Garcia, J.M.; Calvey, G.; Katz, A.; Knoska, J.; Kupitz, C.; Hunter, M.S.; Liang, M.; Oberthuer, D.; et al. Enzyme intermediates captured "on the fly" by mix-and-inject serial crystallography. BMC Biol. 2018, 16, 59. [Google Scholar] [CrossRef] [Green Version]
- Dasgupta, M.; Budday, D.; de Oliveira, S.H.P.; Madzelan, P.; Marchany-Rivera, D.; Seravalli, J.; Hayes, B.; Sierra, R.G.; Boutet, S.; Hunter, M.S.; et al. Mix-and-inject XFEL crystallography reveals gated conformational dynamics during enzyme catalysis. Proc. Natl. Acad. Sci. USA 2019, 116, 25634–25640. [Google Scholar] [CrossRef]
- Knoška, J.; Adriano, L.; Awel, S.; Beyerlein, K.R.; Yefanov, O.; Oberthuer, D.; Peña Murillo, G.E.; Roth, N.; Sarrou, I.; Villanueva-Perez, P.; et al. Ultracompact 3D microfluidics for time-resolved structural biology. Nat Commun. 2020, 11, 657. [Google Scholar] [CrossRef] [Green Version]
- Mehrabi, P.; Schulz, E.C.; Agthe, M.; Horrell, S.; Bourenkov, G.; von Stetten, D.; Leimkohl, J.P.; Schikora, H.; Schneider, T.R.; Pearson, A.R.; et al. Liquid application method for time-resolved analyses by serial synchrotron crystallography. Nat. Methods 2019, 16, 979–982. [Google Scholar] [CrossRef]
- Opara, N.L.; Mohacsi, I.; Makita, M.; Castano-Diez, D.; Diaz, A.; Juranić, P.; Marsh, M.; Meents, A.; Milne, C.J.; Mozzanica, A.; et al. Demonstration of femtosecond X-ray pump X-ray probe diffraction on protein crystals. Struct. Dyn. 2018, 5, 054303. [Google Scholar] [CrossRef] [Green Version]
- Chrencik, J.E.; Roth, C.B.; Terakado, M.; Kurata, H.; Omi, R.; Kihara, Y.; Warshaviak, D.; Nakade, S.; Asmar-Rovira, G.; Mileni, M.; et al. Crystal Structure of Antagonist Bound Human Lysophosphatidic Acid Receptor 1. Cell. 2015, 161, 1633–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, R.; Inoue, A.; Sayama, M.; Uwamizu, A.; Yamashita, K.; Hirata, K.; Yoshida, M.; Tanaka, Y.; Kato, H.E.; Nakada-Nakura, Y.; et al. Structural nsights into ligand recognition by the lysophosphatidic acid receptor LPA6. Nature 2017, 548, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Hori, T.; Okuno, T.; Hirata, K.; Yamashita, K.; Kawano, Y.; Yamamoto, M.; Hato, M.; Nakamura, M.; Shimizu, T.; Yokomizo, T.; et al. Na+-mimicking ligands stabilize the inactive state of leukotriene B4 receptor BLT1. Nat. Chem. Biol. 2018, 14, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Asada, H.; Inoue, A.; Ngako Kadji, F.M.; Hirata, K.; Shiimura, Y.; Im, D.; Shimamura, T.; Nomura, N.; Iwanari, H.; Hamakubo, T.; et al. The Crystal Structure of Angiotensin II Type 2 Receptor with Endogenous Peptide Hormone. Structure 2019, 2126, 30442–30443. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Olieric, V.; Ma, P.; Panepucci, E.; Diederichs, K.; Wang, M.; Caffrey, M. In meso in situ serial X-ray crystallography of soluble and membrane proteins. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 1238–1256. [Google Scholar] [CrossRef] [Green Version]
- Zander, U.; Hoffmann, G.; Cornaciu, I.; Marquette, J.P.; Papp, G.; Landret, C.; Seroul, G.; Sinoir, J.; Röwer, M.; Felisaz, F.; et al. Automated harvesting and processing of protein crystals through laser photoablation. Acta Crystallogr. D Struct. Biol. 2016, 72, 454–466. [Google Scholar] [CrossRef]
- Hirata, K.; Yamashita, K.; Ueno, G.; Kawano, Y.; Hasegawa, K.; Kumasaka, T.; Yamamoto, M. ZOO: An automatic data-collection system for high-throughput structure analysis in protein microcrystallography. Acta Crystallogr. D Struct. Biol. 2019, 75, 138–150. [Google Scholar] [CrossRef] [Green Version]
- Keedy, D.A.; van den Bedem, H.; Sivak, D.A.; Petsko, G.A.; Ringe, D.; Wilson, M.A.; Fraser, J.S. Crystal cryocooling distorts conformational heterogeneity in a model Michaelis complex of DHFR. Structure 2014, 22, 899–910. [Google Scholar] [CrossRef] [Green Version]
- Fraser, J.S.; van den Bedem, H.; Samelson, A.J.; Lang, P.T.; Holton, J.M.; Echols, N.; Alber, T. Accessing protein conformational ensembles using room-temperature X-ray crystallography. Proc. Natl. Acad. Sci. USA 2011, 108, 16247–16252. [Google Scholar] [CrossRef] [Green Version]
- Naitow, H.; Matsuura, Y.; Tono, K.; Joti, Y.; Kameshima, T.; Hatsui, T.; Yabashi, M.; Tanaka, R.; Tanaka, T.; Sugahara, M.; et al. Protein-ligand complex structure from serial femtosecond crystallography using soaked thermolysin microcrystals and comparison with structures from synchrotron radiation. Acta Crystallogr. D Struct. Biol. 2017, 73, 702–709. [Google Scholar] [CrossRef] [Green Version]
- Nogly, P.; Panneels, V.; Nelson, G.; Gati, C.; Kimura, T.; Milne, C.; Milathianaki, D.; Kubo, M.; Wu, W.; Conrad, C.; et al. Lipidic cubic phase injector is a viable crystal delivery system for time-resolved serial crystallography. Nat Commun. 2016, 7, 12314. [Google Scholar] [CrossRef] [PubMed]
- Stan, C.; Milathianaki, D.; Laksmono, H.; Sierra, R.G.; McQueen, T.A.; Messerschmidt, M.; Williams, G.J.; Koglin, J.E.; Lane, T.J.; Hayes, M.J.; et al. Liquid explosions induced by X-ray laser pulses. Nature Phys. 2016, 2, 966–971. [Google Scholar] [CrossRef]
- Apel, A.K.; Cheng, R.K.Y.; Tautermann, C.S.; Brauchle, M.; Huang, C.Y.; Pautsch, A.; Hennig, M.; Nar, H.; Schnapp, G. Crystal Structure of CC Chemokine Receptor 2A in Complex with an Orthosteric Antagonist Provides Insights for the Design of Selective Antagonists. Structure 2019, 27, 427–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchison, C.D.M.; Cordon-Preciado, V.; Morgan, R.M.L.; Nakane, T.; Ferreira, J.; Dorlhiac, G.; Sanchez-Gonzalez, A.; Johnson, A.S.; Fitzpatrick, A.; Fare, C.; et al. X-ray free electron laser determination of crystal structures of dark and light states of a reversibly photoswitching fluorescent protein at room temperature. Int. J. Mol. Sci. 2017, 18, 1918. [Google Scholar] [CrossRef] [Green Version]
- Rucktooa, P.; Cheng, R.K.Y.; Segala, E.; Geng, T.; Errey, J.C.; Brown, G.A.; Cooke, R.M.; Marshall, F.H.; Doré, A.S. Towards high throughput GPCR crystallography: In Meso soaking of Adenosine A2A Receptor crystals. Sci. Rep. 2018, 8, 41. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Chicano, T.; Ebrahim, A.; Axford, D.; Appleby, M.V.; Beale, J.H.; Chaplin, A.K.; Duyvesteyn, H.M.E.; Ghiladi, R.A.; Owada, S.; Sherrell, D.A.; et al. High-throughput structures of protein ligand complexes at room temperature using serial femtosecond crystallography. IUCrJ 2019, 6, 1074–1085. [Google Scholar] [CrossRef] [Green Version]


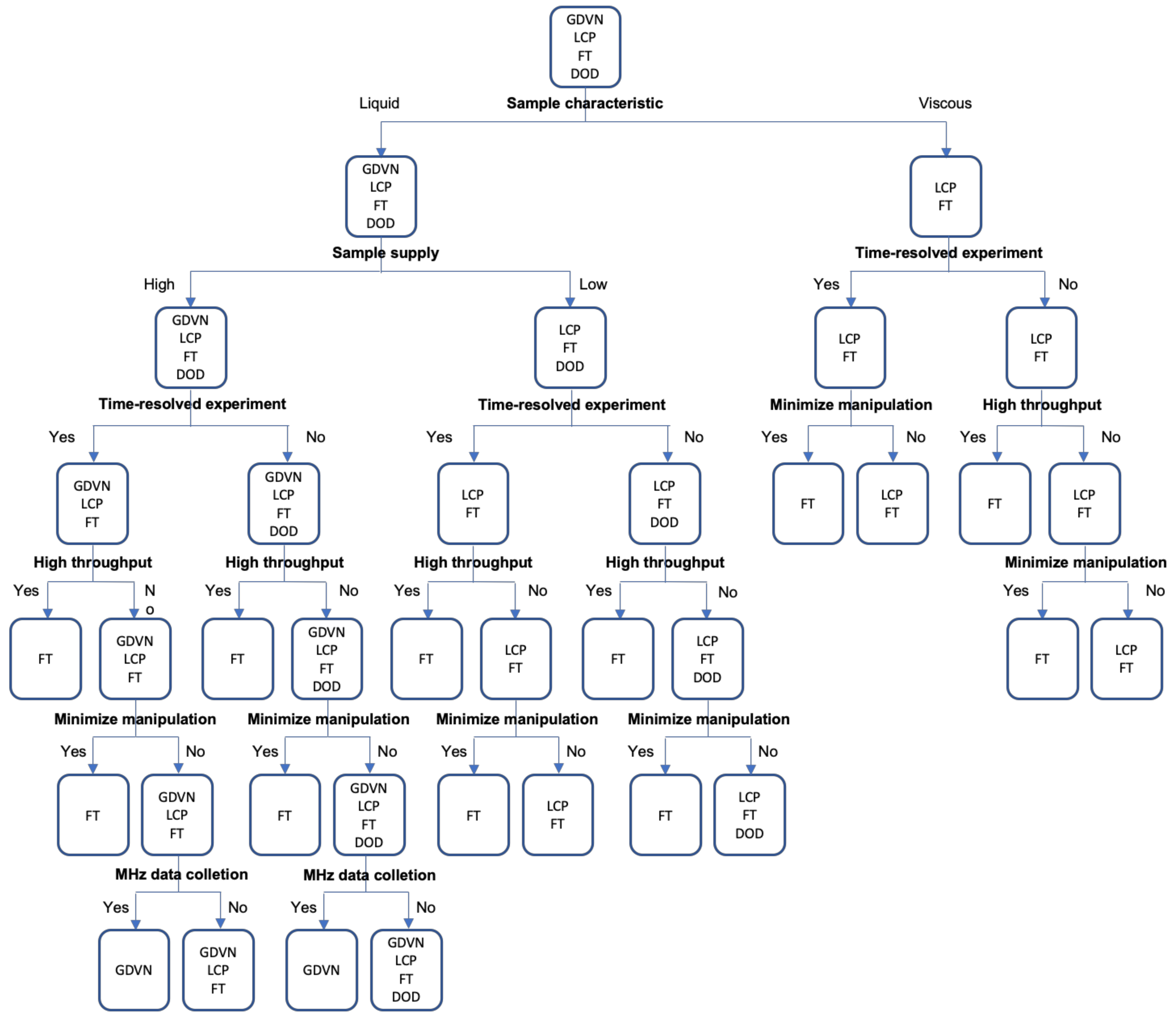{kind=link}
| Delivery Method | Pros | Cons | Key Limitation/Opportunity |
|---|---|---|---|
| Liquid jet | Low background Efficient mixing with ligand for TR-SFX | High sample consumption Prone to nozzle clogging Low throughput sample changing | High data collection efficiency at MHz XFELs |
| LCP jet | Low sample consumption Protein grown in LCP can be used directly Large variety of choice of matrixes Can also be used for crystals in suspension | Higher background (depends on matrix used) Some sample manipulation at harvest Low throughput sample changing | High throughput sample changing |
| Fixed target chip | Low sample consumption In situ crystallization High throughput sample changing Sample preparation and data collection at cryo-genic temperatures | Higher background (depends on support matrix used) Sample stability needs to be maintained before and during data collection Preferential orientation risk of certain crystal morphologies Data collection efficiency limited by robotic arms | Maximize sample and data collection efficiency by crystal pre-location (difficult to apply for viscous samples) |
| Drop-on-demand | Low sample consumption High sample efficiency | More suitable for larger crystals Higher solvent background Compatibility with TR-SFX | Cannot be applied to viscous samples |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, R.K. Towards an Optimal Sample Delivery Method for Serial Crystallography at XFEL. Crystals 2020, 10, 215. https://doi.org/10.3390/cryst10030215
Cheng RK. Towards an Optimal Sample Delivery Method for Serial Crystallography at XFEL. Crystals. 2020; 10(3):215. https://doi.org/10.3390/cryst10030215
Chicago/Turabian StyleCheng, Robert KY. 2020. "Towards an Optimal Sample Delivery Method for Serial Crystallography at XFEL" Crystals 10, no. 3: 215. https://doi.org/10.3390/cryst10030215
APA StyleCheng, R. K. (2020). Towards an Optimal Sample Delivery Method for Serial Crystallography at XFEL. Crystals, 10(3), 215. https://doi.org/10.3390/cryst10030215