Crystal Structure and Supramolecular Architecture of Inorganic Ligand-Coordinated Salen-Type Schiff Base Complex: Insights into Halogen Bond from Theoretical Analysis and 3D Energy Framework Calculations

 ,
,
Abstract
:
1. Introduction
2. Experimental
2.1. Material and Physical Measurements
2.2. Synthesis of the Complex
Synthesis of [MnIII(3-Br-5Cl-salen)(Cl)(H2O)] (1)
2.3. X-Ray Crystal Structure Analysis
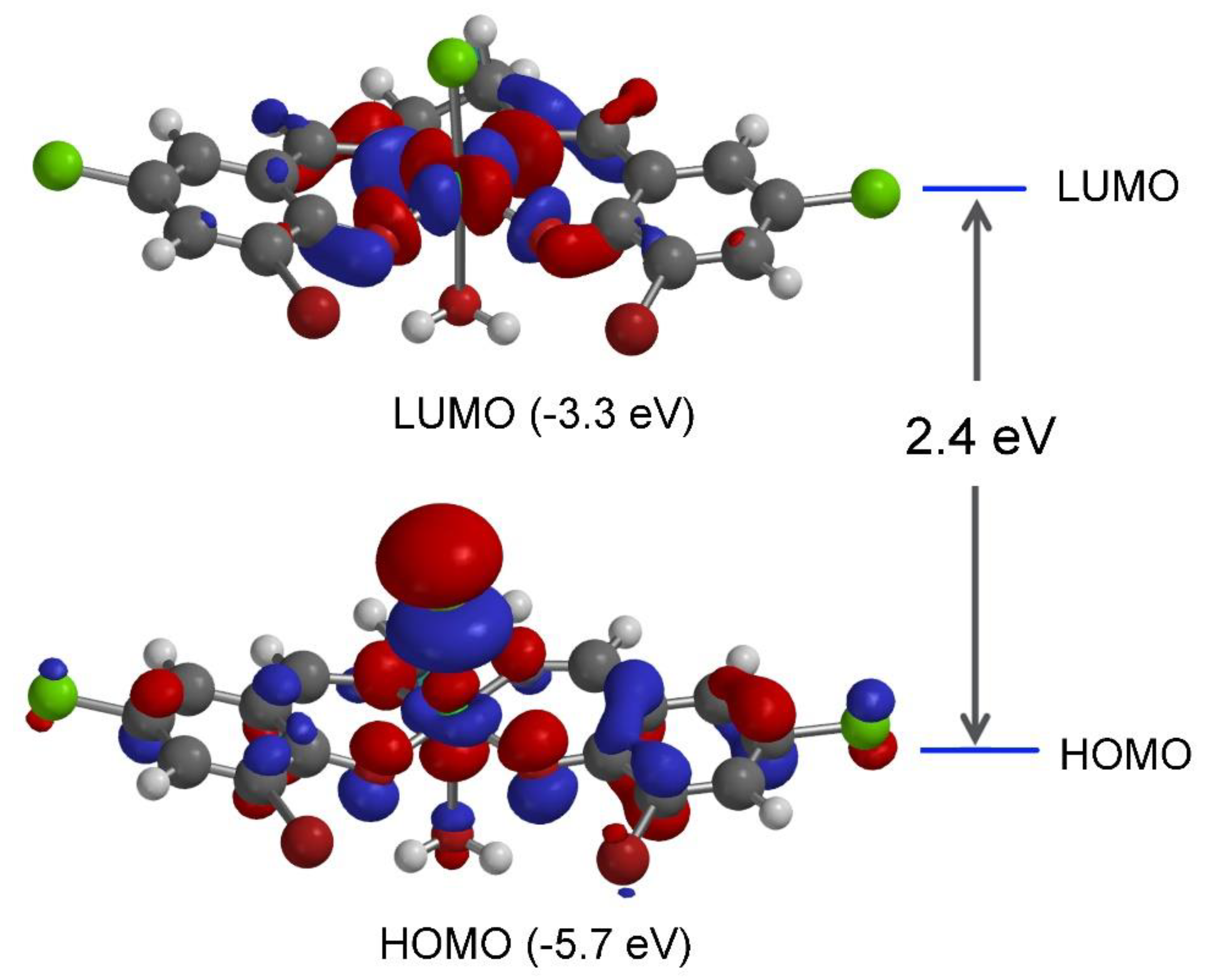
2.4. Optimized Structure and HOMO-LUMO
2.5. Hirshfeld Surface Analysis
2.6. Intermolecular Interaction Energy
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Voorhaar, L.; Hoogenboom, R. Supramolecular polymer networks: Hydrogels and bulk materials. Chem. Soc. Rev. 2016, 45, 4013–4031. [Google Scholar] [CrossRef]
- Okesola, B.O.; Smith, D.K. Applying low-molecular weight supramolecular gelators in an environmental setting-self-assembled gels as smart materials for pollutant removal. Chem. Soc. Rev. 2016, 45, 4226–4251. [Google Scholar] [CrossRef] [Green Version]
- Pang, H.; Peng, J.; Sha, J.; Tian, A.; Zhang, P.; Chen, Y.; Zhu, M. A molecular crown analogue templated by Keggin polyanions: Synthesis, structure, and electrochemical and luminescent properties. Z. Nat. B 2015, 70, 547–553. [Google Scholar]
- Xia, Q.; Yuan, C.; Li, Y.; Cui, Y. Design and assembly of a chiral composite metal-organic framework for efficient asymmertric sequential transformation of alkenes to amino alcohols. Chem. Commun. 2019, 55, 9136–9139. [Google Scholar] [CrossRef]
- Kirillov, A.M. Hexamethylenetetramine: An old new building block for design of coordination polymers. Coord. Chem. Rev. 2011, 255, 1603–1622. [Google Scholar] [CrossRef]
- Zhang, D.S.; Zhang, Y.Z.; Gao, J.; Liu, H.L.; Hu, H.; Geng, L.L.; Zhang, X.; Li, Y.W. Structure modulation from unstable to stable MOFs by regulating secondary N-donor ligands. Dalton Trans. 2018, 47, 14025–14032. [Google Scholar] [CrossRef] [PubMed]
- Martins, G.A.V.; Gieck, C.; Coluccia, S.; Marchese, L.; Pastore, H.O. One Step Further in Understanding the Role of Hydrogen Bonds in Directing the Synthesis of CHA Analogue Molecular Sieves. J. Phys. Chem. C 2009, 113, 10675–10680. [Google Scholar] [CrossRef]
- Ong, W.-J.; Tan, L.-L.; Chai, S.-P.; Yong, S.-T. Graphene oxide as a structure-directing agent for the two-dimensional interface engineering of sandwich-like graphene–g-C3N4 hybrid nanostructures with enhanced visible-light photoreduction of CO2 to methane. Chem. Commun. 2015, 51, 858–861. [Google Scholar] [CrossRef] [PubMed]
- Thota, B.N.S.; Urner, L.H.; Haag, R. Supramolecular Architectures of Dendritic Amphiphiles in Water. Chem. Rev. 2016, 116, 2079–2102. [Google Scholar] [CrossRef]
- Gilday, L.C.; Robinson, S.W.; Barendt, T.A.; Langton, M.J.; Mullaney, B.R.; Beer, P.D. Halogen Bonding in Supramolecular Chemistry. Chem. Rev. 2015, 115, 7118–7195. [Google Scholar] [CrossRef]
- Metrangolo, P.; Resnati, G. Halogen Bonding: Where We Are and Where We Are Going. Cryst. Growth Des. 2012, 12, 5835–5838. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, S.M.; Scanlon, J.D.; Jimenez-Izal, E.; Ugalde, J.M.; Infante, I. On the directionality of halogen bonding. Phys. Chem. Chem. Phys. 2013, 15, 10350–10357. [Google Scholar] [CrossRef] [PubMed]
- Gaspard, D.; Seddon, K.R.; Robertson, P.K.J.; Gunaratne, H.Q.N. Halogen-bond mediated efficient storage of extremely volatile perfluoroiodides in ionic liquids. Chem. Commun. 2019, 55, 9088–9091. [Google Scholar] [CrossRef] [PubMed]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M. Halogen Bonding: A Halogen-Centered Noncovalent Interaction Yet to Be Understood. Inorganics 2019, 7, 40. [Google Scholar] [CrossRef] [Green Version]
- Buntara Sanjeeva, K.; Pigliacelli, C.; Gazzera, L.; Dichiarante, V.; Baldelli Bombelli, F.; Metrangolo, P. Halogen bond-assisted self-assembly of gold nanoparticles in solution and on a planar surface. Nanoscale 2019, 11, 18407–18415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheiner, S. On the capability of metal-halogen groups to participate in halogen bonds. CrystEngComm 2019, 21, 2875–2883. [Google Scholar] [CrossRef]
- Mirocki, A.; Sikorski, A. Influence of Halogen Substituent on the Self-Assembly and Crystal Packing of Multicomponent Crystals Formed from Ethacridine and Meta-Halobenzoic Acids. Crystals. 2020, 10, 79. [Google Scholar] [CrossRef] [Green Version]
- Priimagi, A.; Cavallo, G.; Metrangolo, P.; Resnati, G. The Halogen Bond in the Design of Functional Supramolecular Materials: Recent Advances. Acc. Chem. Res. 2013, 46, 2686–2695. [Google Scholar] [CrossRef] [Green Version]
- Pfrunder, M.C.; Micallef, A.S.; Rintoul, L.; Arnold, D.P.; McMurtrie, J. Interplay between the Supramolecular Motifs of Polypyridyl Metal Complexes and Halogen Bond Networks in Cocrystals. Cryst. Growth Des. 2016, 16, 681–695. [Google Scholar] [CrossRef] [Green Version]
- Aakeröy, C.B.; Wijethunga, T.K.; Desper, J.; Đaković, M. Crystal Engineering with Iodoethynylnitrobenzenes: A Group of Highly Effective Halogen-Bond Donors. Cryst. Growth Des. 2015, 15, 3853–3861. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Wijethunga, T.K.; Desper, J.; Đaković, M. Electrostatic Potential Differences and Halogen-Bond Selectivity. Cryst. Growth Des. 2016, 16, 2662–2670. [Google Scholar] [CrossRef]
- Liu, R.; Wang, H.; Jin, W.J. Soft-Cavity-type Host–Guest Structure of Cocrystals with Good Luminescence Behavior Assembled by Halogen Bond and Other Weak Interactions. Cryst. Growth Des. 2017, 17, 3331–3337. [Google Scholar] [CrossRef]
- Dumele, O.; Wu, D.; Trapp, N.; Goroff, N.; Diederich, F. Halogen Bonding of (Iodoethynyl)benzene Derivatives in Solution. Org. Lett. 2014, 16, 4722–4725. [Google Scholar] [CrossRef]
- Awwadi, F.F.; Willett, R.D.; Twamley, B.; Turnbull, M.M.; Landee, C.P. Dual Behavior of Bromine Atoms in Supramolecular Chemistry: The Crystal Structure and Magnetic Properties of Two Copper(II) Coordination Polymers. Cryst. Growth Des. 2015, 15, 3746–3754. [Google Scholar] [CrossRef]
- Lisac, K.; Cinčić, D. Simple design for metal-based halogen-bonded cocrystals utilizing the M–Cl⋯I motif. CrystEngComm 2018, 20, 5955–5963. [Google Scholar] [CrossRef]
- Ding, X.; Tuikka, M.; Rissanen, K.; Haukka, M. Extended Assemblies of Ru(bpy)(CO)2X2 (X = Cl, Br, I) Molecules Linked by 1,4-Diiodotetrafluoro-Benzene (DITFB) Halogen Bond Donors. Crystals 2019, 9, 319. [Google Scholar] [CrossRef] [Green Version]
- Awwadi, F.F.; Turnbull, M.M.; Alwahsh, M.I.; Haddad, S.F. May halogen bonding interactions compete with Cu⋯Cl semi-coordinate bonds? Structural, magnetic and theoretical studies of two polymorphs of trans-bis(5-bromo-2-chloro pyridine)dichlorocopper(ii) and trans-bis(2,5-dichloropyridine)dichlorocopper(ii). New J. Chem. 2018, 42, 10642–10650. [Google Scholar] [CrossRef]
- Stilinović, V.; Grgurić, T.; Piteša, T.; Nemec, V.; Cinčić, D. Bifurcated and Monocentric Halogen Bonds in Cocrystals of Metal(II) Acetylacetonates with p-Dihalotetrafluorobenzenes. Cryst. Growth Des. 2019, 19, 1245–1256. [Google Scholar] [CrossRef]
- Li, Y.; Wu, Q.; Lecren, L.; Clérac, R. Synthesis, structure and magnetism of new polynuclear transition metal aggregates assembled with Schiff-base ligand and anionic N-donor ligands. J. Mol. Struct. 2008, 890, 339–345. [Google Scholar] [CrossRef]
- Senapati, T.; Pichon, C.; Ababei, R.; Mathonière, C.; Clérac, R. Cyanido-bridged Fe(III)-Mn(III) heterobimetallic materials built from Mn(III) Schiff base complexes and di- or tri-cyanido Fe(III) precursors. Inorg. Chem. 2012, 51, 3796–3812. [Google Scholar] [CrossRef] [PubMed]
- Agilent Technologies: CrysAlisPRO Software system, v.; Agilent Technologies UK Ltd.: Oxford, UK, 2015.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2010, 42, 339–341. [Google Scholar] [CrossRef]
- Spartan 14; Wavefunction Inc.: Irvine, CA, USA, 2014.
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm. 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Jayatilaka, D.; Grimwood, D.J. Tonto: A Fortran Based Object-Oriented System for Quantum Chemistry and Crystallography. In Proceedings of the Computational Science—ICCS 2003, Berlin/Heidelberg, Germany, 2–4 June 2003; Sloot, P.M.A., Abramson, D., Bogdanov, A.V., Gorbachev, Y.E., Dongarra, J.J., Zomaya, A.Y., Eds.; Springer: Berlin/Heidelberg, Germany, 2003; pp. 142–151. [Google Scholar]
- Available online: www.ccdc.cam.ac.uk.data_request/cif (accessed on 8 October 2019).
- Martinez, D.; Motevalli, M.; Watkinson, M. Aquachloro[N,N’-ethylenebis(salicylideneiminato)]manganese(III). Acta Crystallogr. Sect. C 2002, 58, m258–m260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Shi, Q.; Li, Y.-G.; Wang, E.B. Synthesis, crystal structure and magnetic properties of new MnIII–CuII heterometallic aggregates based on multidentate Schiff-base ligands. J. Coord. Chem. 2008, 61, 3080–3091. [Google Scholar] [CrossRef]
- Spek, A.L.J.; Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Zhan, C.G.; Nichols, J.A.; Dixon, D.A. Ionization potential, electron affinity, electronegativity, hardness, and electron excitation energy: Molecular properties from density functional theory orbital energies. J. Phys. Chem. A 2003, 107, 4184–4195. [Google Scholar] [CrossRef] [Green Version]
- Tepper, R.; Schubert, U.S. Halogen Bonding in Solution: Anion Recognition, Templated Self-Assembly, and Organocatalysis. Angew. Chem. Int. Ed. 2018, 57, 6004–6016. [Google Scholar] [CrossRef]
- Andersson, M.P.; Uvdal, P. New Scale Factors for Harmonic Vibrational Frequencies Using the B3LYP Density Functional Method with the Triple-ζ Basis Set 6–311+G(d,p). J. Phys. Chem. A 2005, 109, 2937–2941. [Google Scholar] [CrossRef]
- Demircioğlu, Z.; Özdemir, F.A.; Dayan, O.; Şerbetçi, Z.; Özdemir, N. Synthesis, X-ray diffraction method, spectroscopic characterization (FT-IR, 1H and 13C NMR), antimicrobial activity, Hirshfeld surface analysis and DFT computations of novel sulfonamide derivatives. J. Mol. Struct. 2018, 1161, 122–137. [Google Scholar] [CrossRef]
- Pople, J.A.; Schleyer, P.V.; Hehre, W.J.; Radom, L. AB INITIO Molecular Orbital Theory; John Wiley & Sons: Hoboken, NJ, USA, 1986. [Google Scholar]
- DiCésare, N.; Belletête, M.; Marrano, C.; Leclerc, M.; Durocher, G. Conformational Analysis (ab Initio HF/3–21G*) and Optical Properties of Symmetrically Disubstituted Terthiophenes. J. Phys. Chem. A 1998, 102, 5142–5149. [Google Scholar] [CrossRef]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer model energies and energy frameworks: Extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ 2017, 4, 575–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Empirical Formula | C16H12Br3Cl3Mn1N2O3 |
|---|---|
| Mr | 601.37 |
| Temperature/K | 293(2) |
| Wavelength/Å | 0.71073 |
| Crystal system | Monoclinic |
| space group | P 21/c |
| a/Å | 12.4164(12) |
| b/Å | 13.5291(18) |
| c/Å | 13.2867(9) |
| β/deg | 122.347(6) |
| Volume/Å3 | 1142.6(3) |
| Z | 4 |
| Calculated density | 1885.6(3) Mg/m3 |
| Absorption coefficient | 5.384 mm^-1 |
| F(000) | 1168 |
| Theta range for data collection | 3.52 to 25.00 deg |
| Limiting indices | −13 ≤ h ≤ 14, −15 ≤ k ≤ 16, −15 ≤l ≤ 10 |
| Reflections collected/unique | 4827/2812 [R(int) = 0.0513] |
| Completeness to theta = 25.00 | 99.7% |
| Refinement method | Full-matrix least-squares on F^2 |
| Goodness-of-fit on F^2 | 0.962 |
| Final R indices [I>2sigma(I)] | R1 = 0.0619, wR2 = 0.0978 |
| R indices (all data) | R1 = 0.0986, wR2 = 0.1098 |
| D-H···A | D-H, Å | H···A, Å | D···A, Å | D-H···A, deg |
| O(3)-H(3A)···Cl(3)i | 0.85 | 2.52 | 3.303(8) | 154 |
| O(3)-H(3B)···Br(2)ii | 0.85 | 2.84 | 3.381(9) | 123 |
| O(3)-H(3B)···O(2)ii | 0.85 | 2.4 | 3.216(9) | 161 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Q.; Xiao, J.-C.; Zhou, C.; Sun, J.-R.; Huang, M.-F.; Xu, X.; Li, T.; Tian, H. Crystal Structure and Supramolecular Architecture of Inorganic Ligand-Coordinated Salen-Type Schiff Base Complex: Insights into Halogen Bond from Theoretical Analysis and 3D Energy Framework Calculations. Crystals 2020, 10, 334. https://doi.org/10.3390/cryst10040334
Wu Q, Xiao J-C, Zhou C, Sun J-R, Huang M-F, Xu X, Li T, Tian H. Crystal Structure and Supramolecular Architecture of Inorganic Ligand-Coordinated Salen-Type Schiff Base Complex: Insights into Halogen Bond from Theoretical Analysis and 3D Energy Framework Calculations. Crystals. 2020; 10(4):334. https://doi.org/10.3390/cryst10040334
Chicago/Turabian StyleWu, Qiong, Jian-Chang Xiao, Cun Zhou, Jin-Rong Sun, Mei-Fen Huang, Xindi Xu, Tianyu Li, and Hui Tian. 2020. "Crystal Structure and Supramolecular Architecture of Inorganic Ligand-Coordinated Salen-Type Schiff Base Complex: Insights into Halogen Bond from Theoretical Analysis and 3D Energy Framework Calculations" Crystals 10, no. 4: 334. https://doi.org/10.3390/cryst10040334