3.1. Synthesis of the Target Compound and Structural Elucidation
4-Amino-5-(1
H-indol-2-yl)-1,2,4-triazol-3(2
H)-thione
1 was synthesized, according to the reported procedures in [
32], and reacted with benzyl bromide in the presence of K
2CO
3 in EtOH and stirring overnight. Coupling was explored that it proceed at sulfur to give 3-(benzylsulfanyl)-5-(1
H-indol-2-yl)-4
H-1,2,4-triazol-4-amine
2 (
Scheme 1). The compound
2 was obtained in a pure form after recrystallization. The chemical feature of the S-benzylated compound has been assigned based on NMR and X-ray diffraction analysis.
The structure of 3-(benzylsulfanyl)-5-(1
H-indol-2-yl)-4
H-1,2,4-triazol-4-amine was confirmed based upon its NMR and mass spectral data.
1H NMR displayed the methylene protons of the benzyl group at 4.45 ppm, the amino group protons appeared at 6.21 ppm, the indole and phenyl CH protons appeared between 7.03 and 7.60 ppm, and the indole NH was found at 11.74 ppm.
13C NMR demonstrated the methylene carbon of the benzyl group at 35.09 ppm, the indole and phenyl CH carbons appeared as eight signals between 102.35 and 129.05 ppm. The quaternary carbons appeared at 123.98, 127.60, 136.51, 137.46, 149.91 and 152.85 ppm. Sulfur not nitrogen alkylation was confirmed from the methylene carbon signal of the benzyl group which was found in
13C NMR at 35.09 ppm.
1H-
1H correlation spectroscopy (COSY) was used for assigning the correlation between the vicinal protons, and 2D HMQC showed the correlation between the carbons and directly attached hydrogens (all the spectrum are provided in the
Supplementary Information, Figures S1–S6)).
3.3. Hirshfeld Analysis of Molecular Packing
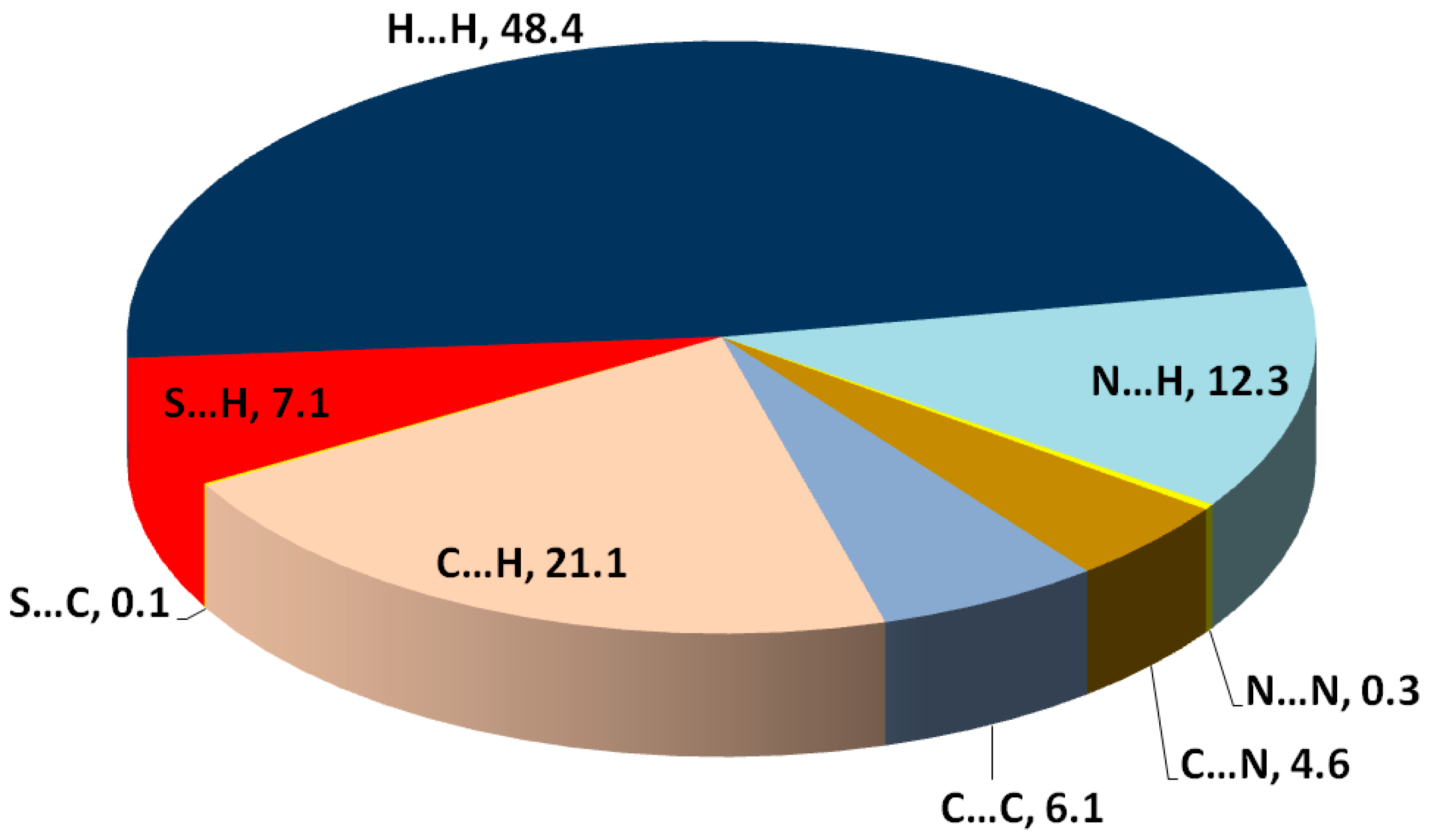
Hirshfeld topology calculations are important to analyze the different intermolecular contacts in the structure of crystalline materials. Additionally, it sheds the light on the significance, strength and percentage of each intermolecular contact. The percentages of different contacts observed in the crystal structure of
2 based on Hirshfeld calculations are shown in
Figure 3, while the complete Hirshfeld surfaces are given in
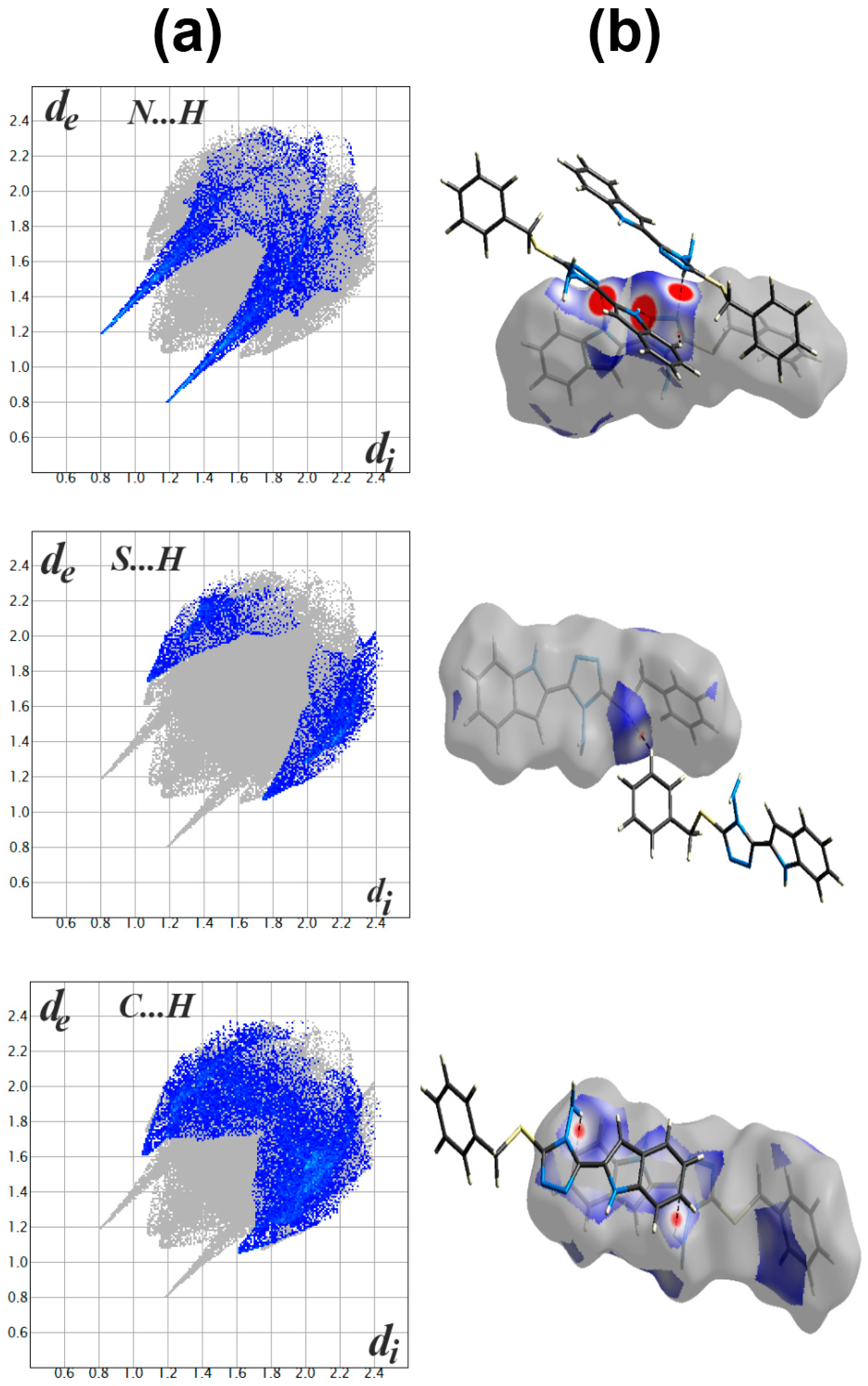
Figure S7 (Supplementary Data). The packing of molecules in the crystal is mainly dependant on significant N...H (1.994–2.595 Ǻ) and S…H (2.282 Ǻ) contacts, as well as weak C...H contact with minimum C...H distance of 2.670 Ǻ corresponding to the N-H...π interaction. Presentation of the N…H and S…H hydrogen bonds and the weak C…H interactions mapped over d
norm Hirshfeld surfaces are presented in
Figure 4. These interactions contributed by 12.3%, 7.1% and 21.1% from the whole fingerprint area, respectively. Only the N...H, C…H and S...H contacts appeared as red spots in the corresponding d
norm maps, indicating their significance (
Figure 4). The contributions of the weaker H...H, C…N and C...C interactions are 48.4%, 4.6% and 6.1%, respectively. These contacts appeared either as white or blue regions, indicating less important interactions.
The presence of some C…C (6.1%) and C…N (4.6%) contacts, as well as the blue/red triangles in the shape index Hirshfeld surface (
Figure 5), are the main characteristics for the presence of π-π stacking interactions. The shortest C…C and C…N interaction distances are presented in
Table 3. Generally, all these contacts are slightly longer than the van der Waals radii sum of the interacting elements, indicating weak π-π contacts.
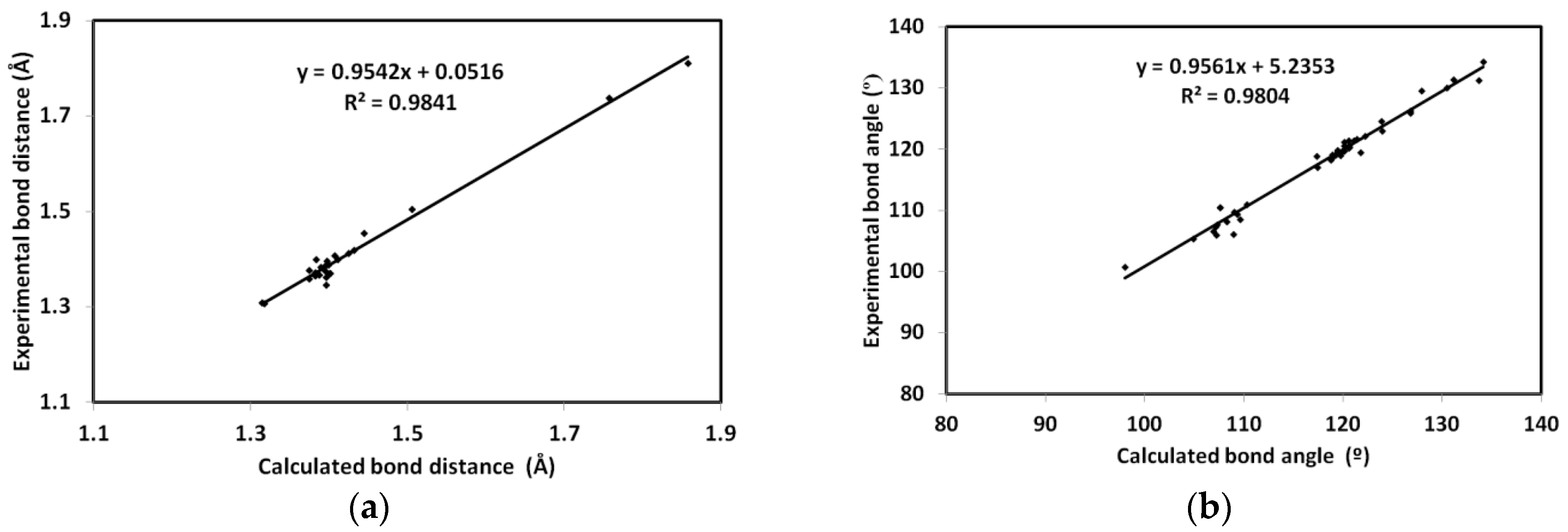
DFT studies: the optimized geometry of
2 is presented in
Figure 6. Structure matching between the computed molecular geometry with the experimental one is also presented. This structure comparison indicated very well the good agreement between the optimized and X-ray structures. In addition, the very good straight-line relations between the calculated and experimental geometric parameters further confirm this conclusion. The values of correlation coefficients are very close to 1 for both cases (
Figure 7). A complete set of bond distances and angles are given in
Table S3 (Supplementary Data).
The natural charges obtained from the NBO calculations are listed in
Table 4. All hydrogen atoms are electropositive with the highest partial charges located at the NH protons. Additionally, the sulphur atom has partial positive charge of 0.3054 e. In contrast, all nitrogen atoms have negative partial charges. The maximum negative charge is located over the amine nitrogen atom. Additionally, all carbon atoms have negative partial charges except those attached directly to the electronegative nitrogen sites. As a result of this charge distribution, the compound is predicted to be polar molecule with calculated dipole moment of 2.2557 Debye. Molecular electrostatic potential (MESP), along with the dipole vector are presented
Figure 8.
In addition, the HOMO and LUMO are important for the molecule reactivity [
33,
34,
35,
36,
37,
38,
39]. Their energies were calculated to be −5.454 and −1.070 eV, respectively. The energies of these molecular orbitals were used to calculate the reactivity descriptors using Equations (1)–(5).
Hence, the calculated ionization potential (I) and electron affinity (A) are 5.454 and 1.070 eV, respectively. Additionally, the hardness (η), electrophilicity index (ω) and chemical potential (μ) are 4.384, 1.213 and −3.262 eV, respectively.
The HOMO is located over the sulphur atom and the triazole π-system. These sites represent the ground state demand for electronic transition to the higher energy level (LUMO). The latter is distributed over the triazole and indole moieties. Hence, the HOMO to LUMO excitation represent mixed n-π* and π-π* transitions. The energy needed for this intermolecular charge transfer is 4.384 eV.
UV-Vis and NMR spectra: The experimental UV-Vis electronic spectra of 2 in ethanol showed absorption bands at 244 and 307 nm, and a shoulder at 295 nm. The assignments of these electronic transitions are presented in
Table 5 based on the TD-DFT calculations. The experimental and simulated electronic spectra are shown in
Figure 9. The experimentally observed bands were calculated at 232.3 nm (f = 0.248), 305.5 nm (f = 1.020) and 288.8 nm (f = 0.069), respectively, which correspond to HOMO→L+3 (81%), HOMO→LUMO (96%) and H-1→LUMO (92%) transitions, respectively. Presentation of the molecular orbitals involved in these electronic transitions is shown in
Figure 10.
In addition, the geometry optimization is performed in DMSO as solvent, then the NMR chemical shifts are calculated in the same solvent using TMS as internal standard. The results are summarized in
Table S4 (Supplementary Data), in comparison with the experimental data. Moreover, graphical plots of the experimental chemical shifts against the calculated data for protons and carbons are presented in
Figure 11. As can be seen from this figure, the straight lines have good correlation coefficients, indicating the good agreement between the calculated and experimental results.
NBO analysis: the electron delocalization processes from occupied orbitals to antibonding empty orbitals stabilized the system, due to the conjugation effect [
40,
41]. The stabilization energies (E
(2)) of these electron delocalization processes in 2 are listed in
Table 6. The molecule is stabilized by many σ-σ*, π→π*, n→σ* and n→π* IMCT interactions. The σ-σ* and n→σ* IMCT interactions are generally weak, with maximum stabilization of 6.79 and 9.95 kcal/mol for σ(C19-C20)→ σ*C22-C23 and n(N9)→σ*(N8-C23) IMCT interactions, respectively. In contrast, the π→π*, and n→π* are generally stronger, which stabilized the system up to 20.67 and 45.73 kcal/mol for π(C31-C33)→ π*(C28-C29) and n(N8)→ π *(N7-C24) IMCT interactions, respectively.
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}