
Mechanical and Electronic Properties of DNTF Crystals under Different Pressure

Abstract
:1. Introduction
2. Materials and Methods
3. Results
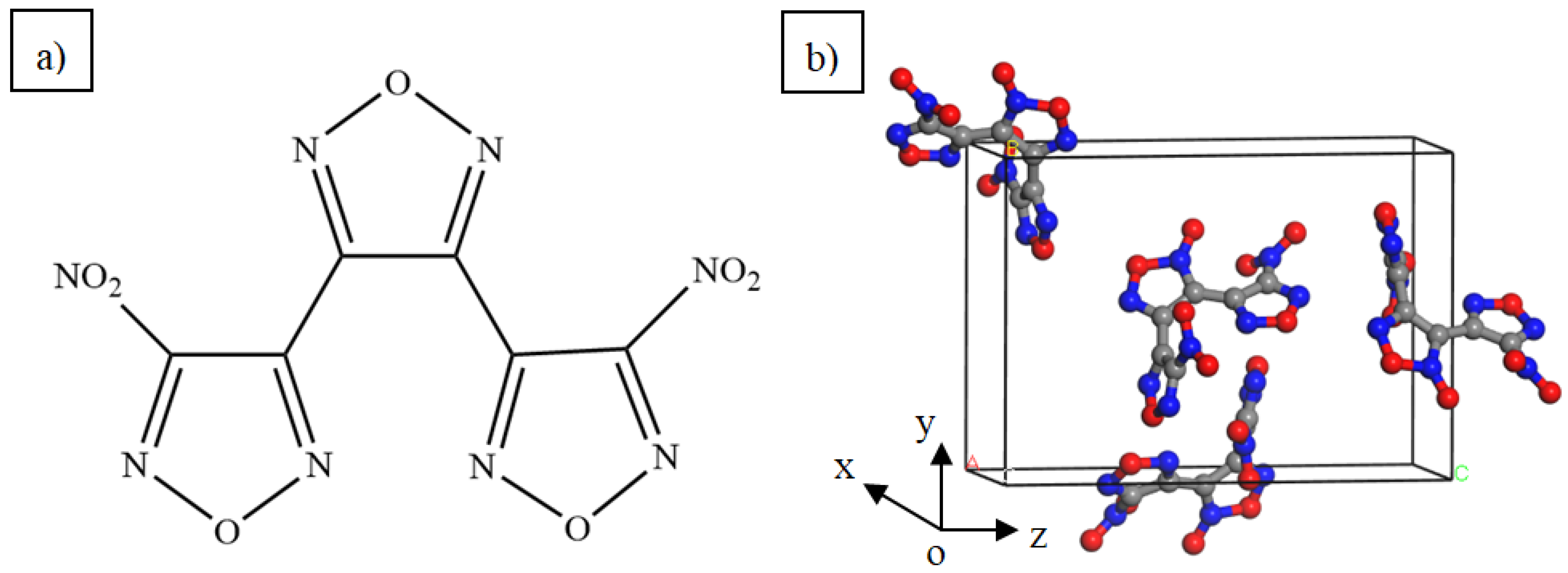
3.1. Lattice Constants and Stability
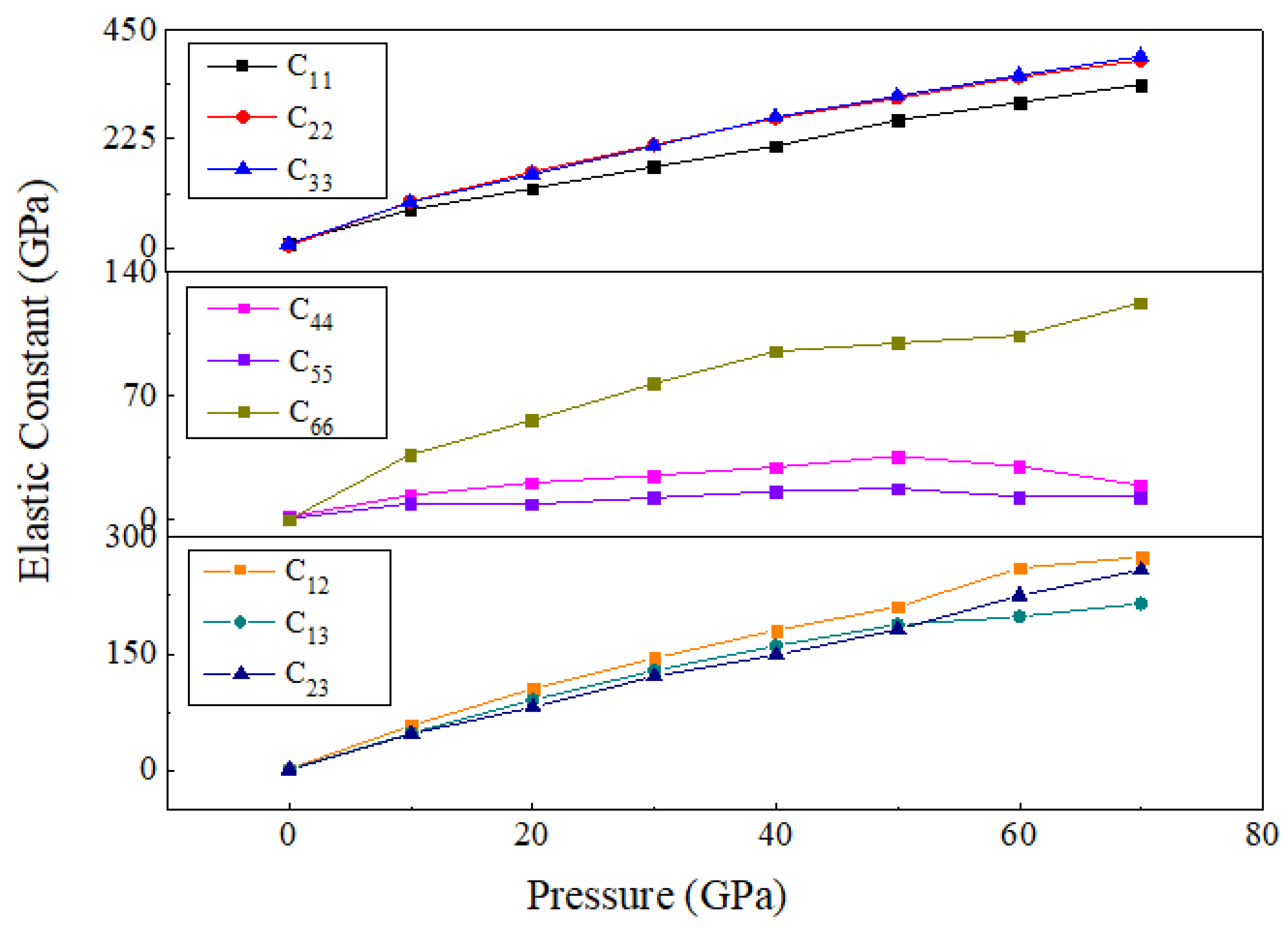
3.2. Mechanical Properties
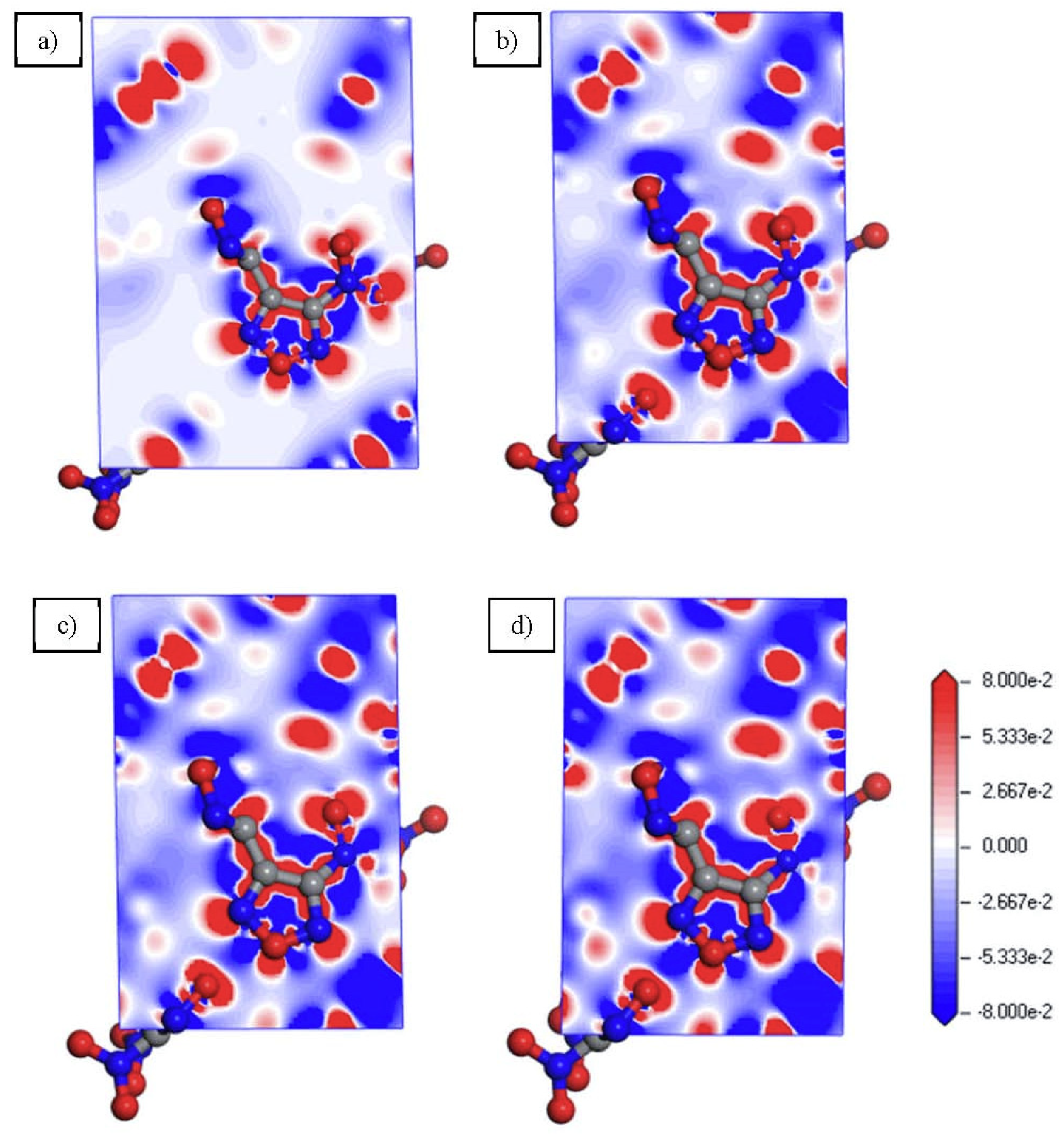
3.3. Electronic Properties
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zhang, J.; Zhou, J.; Bi, F.; Wang, B. Energetic materials based on poly furazan and furoxan structures. Chin. Chem. Lett. 2020, 31, 2375–2394. [Google Scholar] [CrossRef]
- Ji, J.; Zhu, W. Thermal decomposition mechanisms of benzotrifuroxan: 2, 4, 6-trinitrotoluene cocrystal using quantum molecular dynamics simulations. Chem. Phys. Lett. 2021, 778, 138820. [Google Scholar] [CrossRef]
- Wu, C.-L.; Zhang, S.-H.; Gou, R.-J.; Ren, F.-D.; Han, G.; Zhu, S.-F. Theoretical insight into the effect of solvent polarity on the formation and morphology of 2, 4, 6, 8, 10, 12-hexanitrohexaazaisowurtzitane (CL-20)/2, 4, 6-trinitro-toluene (TNT) cocrystal explosive. Comput. Theor. Chem. 2018, 1127, 22–30. [Google Scholar] [CrossRef]
- Larin, A.A.; Shaferov, A.V.; Epishina, M.A.; Melnikov, I.N.; Muravyev, N.V.; Ananyev, I.V.; Fershtat, L.L.; Makhova, N.N. Pushing the energy-sensitivity balance with high-performance bifuroxans. ACS Appl. Energy Mater. 2020, 3, 7764–7771. [Google Scholar] [CrossRef]
- Xiong, H.; Cheng, G.; Zhang, Z.; Yang, H. C 8 n 12 o 10: A promising energetic compound with excellent detonation performance and desirable sensitivity. New J. Chem. 2019, 43, 7784–7789. [Google Scholar] [CrossRef]
- He, C.; Gao, H.; Imler, G.H.; Parrish, D.A.; Shreeve, J.M. Boosting energetic performance by trimerizing furoxan. J. Mater. Chem. A 2018, 6, 9391–9396. [Google Scholar] [CrossRef]
- Zhao, F.; Chen, P.; Hu, R.; Yang, L.; Zhang, Z.; Zhou, Y.; Yang, X.; Gao, Y.; Gao, S.; Shi, Q. Thermochemical properties and non-isothermal decomposition reaction kinetics of 3, 4-dinitrofurazanfuroxan (DNTF). J. Hazard. Mater. 2004, 113, 67–71. [Google Scholar]
- Xu, C.; An, C.; He, Y.; Zhang, Y.; Li, Q.; Wang, J. Direct ink writing of DNTF based composite with high performance. Propellants Explos. Pyrot. 2018, 43, 754–758. [Google Scholar] [CrossRef]
- Liu, N.; Zeman, S.; Shu, Y.; Wu, Z.; Wang, B.; Yin, S. Comparative study of melting points of 3, 4-bis (3-nitrofurazan-4-yl) furoxan (DNTF)/1, 3, 3-trinitroazetidine (TNAZ) eutectic compositions using molecular dynamic simulations. RSC Adv. 2016, 6, 59141–59149. [Google Scholar] [CrossRef]
- He, Y.; Guo, X.; Long, Y.; Huang, G.; Ren, X.; Xu, C.; An, C. Inkjet Printing of GAP/NC/DNTF Based Microscale Booster with High Strength for PyroMEMS. Micromachines 2020, 11, 415. [Google Scholar] [CrossRef]
- Song, L.; Zhao, F.; Xu, S.; Ju, X. Uncovering the action of ethanol controlled crystallization of 3, 4-bis (3-nitrofurazan-4-yl) furoxan crystal: A molecular dynamics study. J. Mol. Graph. Model. 2019, 92, 303–312. [Google Scholar] [CrossRef]
- Sinditskii, V.P.; Burzhava, A.V.; Sheremetev, A.B.; Aleksandrova, N.S. Thermal and Combustion Properties of 3, 4-Bis (3-nitrofurazan-4-yl) furoxan (DNTF). Propellants Explos. Pyrot. 2012, 37, 575–580. [Google Scholar] [CrossRef]
- Song, L.; Zhao, F.; Xu, S.; Ju, X.; Ye, C. Crystal Morphology of 3, 4-Bis (3-nitrofurazan-4-yl) furoxan in Methanol and Acetic Acid/Water Solutions by Spiral Growth Mechanism. Propellants Explos. Pyrot. 2020, 45, 1125–1136. [Google Scholar] [CrossRef]
- Kazakov, A.I.; Dashko, D.V.; Nabatova, A.V.; Stepanov, A.; Lempert, D.B. Thermochemical and Energy Characteristics of DNTF and DNFF. Combust. Explos. Shock 2018, 54, 147–157. [Google Scholar] [CrossRef]
- Gu, J.; Liu, L.; Chen, H.; Chen, W.; Guo, Z.; Chen, L. Study on the thermal decomposition kinetics of DNTF. E3S Web of Conferences. EDP Sci. 2021, 245, 01027. [Google Scholar]
- Wu, Q.; Yang, C.; Pan, Y.; Xiang, F.; Liu, Z.; Zhu, W.; Xiao, H. First-principles study of the structural transformation, electronic structure, and optical properties of crystalline 2,6-diamino-3,5-dinitropyrazine-1-oxide under high pressure. J. Mol. Model. 2013, 19, 5159–5170. [Google Scholar] [CrossRef]
- Wang, F.; Du, H.; Zhang, J.; Gong, X. First-Principle Study on High-Pressure Behavior of Crystalline Polyazido-1,3,5-triazine. J. Phys. Chem. C 2012, 116, 6745–6753. [Google Scholar] [CrossRef]
- Wu, Q.; Zhu, W.; Xiao, H. Pressure effects on structural, electronic, absorption, and thermodynamic properties of crystalline 2,4,6-triamino-3,5-dinitropyridine-1-oxide: A DFT study. J. Phys. Org. Chem. 2013, 26, 589–595. [Google Scholar] [CrossRef]
- Sun, F.; Zhang, G.; Ren, X.; Wang, M.; Xu, H.; Fu, Y.; Tang, Y.; Li, D. First-principles studies on phase stability, anisotropic elastic and electronic properties of Al-La binary system intermetallic compounds. Mater. Today Commun. 2020, 24, 101101. [Google Scholar] [CrossRef]
- Wu, Q.; Zhu, W.; Xiao, H. First-principles study of the high-pressure behavior of solid 1, 7-diamino-1, 7-dinitrimino-2, 4, 6-trinitro-2, 4, 6-triazaheptane. Comput. Chem. 2014, 1030, 38–43. [Google Scholar] [CrossRef]
- Wu, Q.; Zhu, W.; Xiao, H. Dispersion-corrected DFT study on the structure and absorption properties of crystalline 5-nitro-2,4-dihydro-1,2,4-triazole-3-one under compression. Struct. Chem. 2015, 26, 477–484. [Google Scholar] [CrossRef]
- Sheremetev, A.B.; Ivanova, E.A.; Spiridonova, N.P.; Melnikova, S.F.; Tselinsky, I.V.; Suponitsky, K.Y.; Antipin, M.Y. Desilylative nitration of C, N-disilylated 3-amino-4-methylfurazan. J. Heterocycl. Chem. 2005, 42, 1237–1242. [Google Scholar] [CrossRef]
- Mouhat, F.; Coudert, F.-X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B Condens. Matter Mater. Phys. 2014, 90, 224104. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Liu, Q.; Liu, F.; Liu, Z. High-pressure structural, mechanical, and vibrational properties of NAPTO: The first fused-ring energetic material with a 2D layered structure. Solid State Commun. 2021, 331, 114293. [Google Scholar] [CrossRef]
- Sun, F.; Zhang, G.; Liu, H.; Xu, H.; Fu, Y.; Li, D. Effect of transition-elements substitution on mechanical properties and electronic structures of B2-AlCu compounds. Results Phys. 2020, 21, 103765. [Google Scholar] [CrossRef]
- Huang, B.; Duan, Y.; Hu, W.; Sun, Y.; Chen, S. Structural, anisotropic elastic and thermal properties of MB (M = Ti, Zr and Hf) monoborides. Ceram. Int. 2015, 41, 6831–6843. [Google Scholar] [CrossRef]
- Liu, W.; Niu, Y.; Li, W. Theoretical prediction of the physical characteristic of Na3MO4 (M = Np and Pu): The first-principles calculations. Ceram. Int. 2020, 46, 25359–25365. [Google Scholar] [CrossRef]
- Tian, J.; Zhao, Y.; Wen, Z.; Hou, H.; Han, P. Physical properties and Debye temperature of Al7Cu2Fe alloy under various pressures analyzed by first-principles. Solid State Commun. 2017, 257, 6–10. [Google Scholar] [CrossRef]
- Zhu, W.H. First-principles band gap criterion for impact sensitivity of energetic crystals: A review. Struct. Chem. 2010, 21, 657–665. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pressure | a(Å) | b(Å) | c(Å) |
|---|---|---|---|
| 0 GPa | 6.821 | 10.983 | 15.410 |
| Exp. [22] | 6.662 | 10.740 | 15.093 |
| 10 GPa | 5.847 | 10.381 | 14.132 |
| 20 GPa | 5.519 | 10.154 | 13.618 |
| 30 GPa | 5.311 | 9.994 | 13.315 |
| 40 GPa | 5.164 | 9.872 | 13.074 |
| 50 GPa | 5.039 | 9.775 | 12.900 |
| 60 GPa | 4.922 | 9.714 | 12.748 |
| 70 GPa | 4.820 | 9.703 | 12.548 |
| 80 GPa | 4.760 | 9.874 | 12.090 |
| Phase | Species | C11 | C12 | C13 | C22 | C23 | C33 | C44 | C55 | C66 |
|---|---|---|---|---|---|---|---|---|---|---|
| DNTF | Present | 8.70 | 1.75 | 1.09 | 3.39 | 0.70 | 7.40 | 1.72 | 0.63 | 0.07 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nan, H.; Jia, X.; Wang, X.; Liu, H.; Jiang, F.; Zhang, P. Mechanical and Electronic Properties of DNTF Crystals under Different Pressure. Crystals 2021, 11, 1180. https://doi.org/10.3390/cryst11101180
Nan H, Jia X, Wang X, Liu H, Jiang F, Zhang P. Mechanical and Electronic Properties of DNTF Crystals under Different Pressure. Crystals. 2021; 11(10):1180. https://doi.org/10.3390/cryst11101180
Chicago/Turabian StyleNan, Hai, Xianzhen Jia, Xuanjun Wang, Heping Liu, Fan Jiang, and Peng Zhang. 2021. "Mechanical and Electronic Properties of DNTF Crystals under Different Pressure" Crystals 11, no. 10: 1180. https://doi.org/10.3390/cryst11101180