
Reactive Crystallization Kinetics of K2SO4 from Picromerite-Based MgSO4 and KCl



Abstract
:1. Introduction
1.1. Reaction, Thermodynamic and Crystallization Kinetics Framework
1.1.1. Dissolution and Reaction of Picromerite
1.1.2. Thermodynamic Supersaturation
1.1.3. Crystallization Kinetics
2. Materials and Methods
2.1. Reagents
2.2. Experimental Setup and Process Analytical Technology (PAT)
2.2.1. Focused Beam Reflectance Measurement (FBRM)
2.2.2. Attenuated Total Reflectance-Fourier Transform Infrared (ATR-FTIR) Spectroscopy
2.2.3. Temperature Measurement
2.2.4. Technobis Crystalline PV
2.2.5. X-ray Powder Diffraction Phase (XRPD) Analysis
2.2.6. Chemical Analysis by Ion Chromatography (IC)
2.3. Experimental Procedure
2.3.1. Preparation of the Saturated Solution of Magnesium Sulfate from Picromerite
2.3.2. Preparation of Saturated Solution of KCl
2.3.3. Operating Conditions of the Reactive Crystallization Process
2.3.4. Mixing of Reagents and Generation of Initial Supersaturation (S0)
3. Results and Discussion
3.1. Absorbance and Temperature in the Reactive Crystallization Process
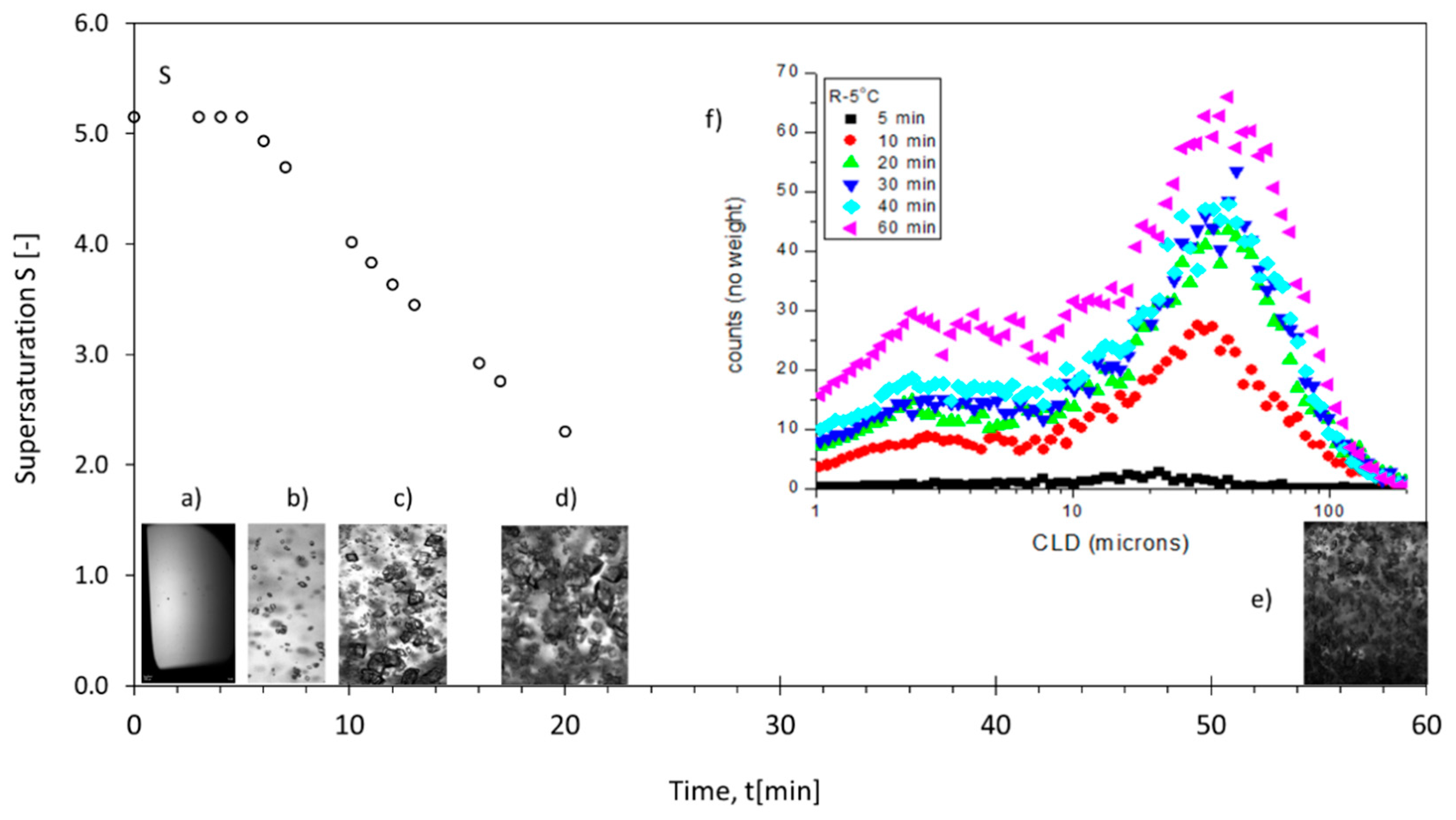
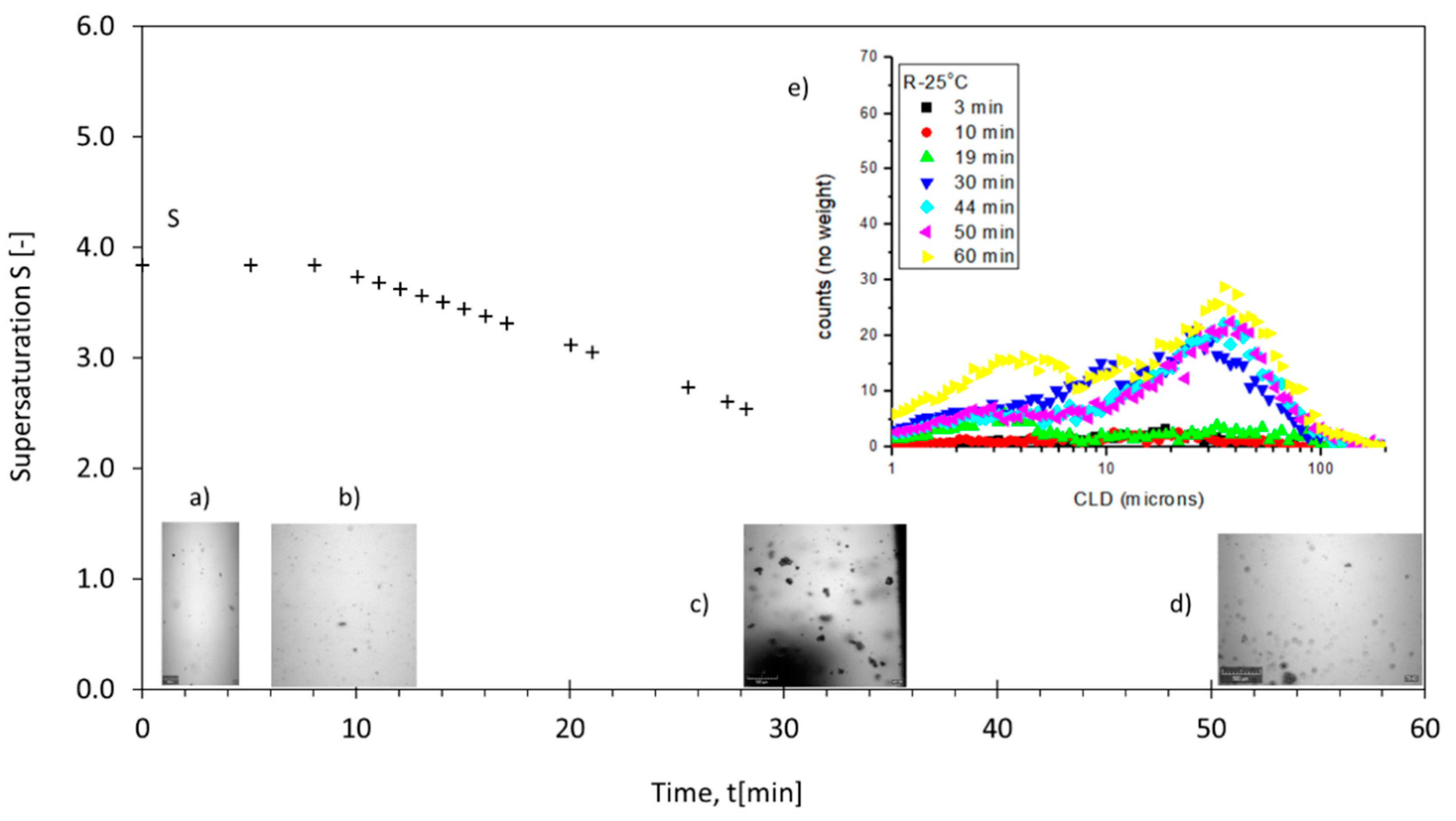
3.2. Supersaturation Profiles, Reactive Crystallization Images and CLD
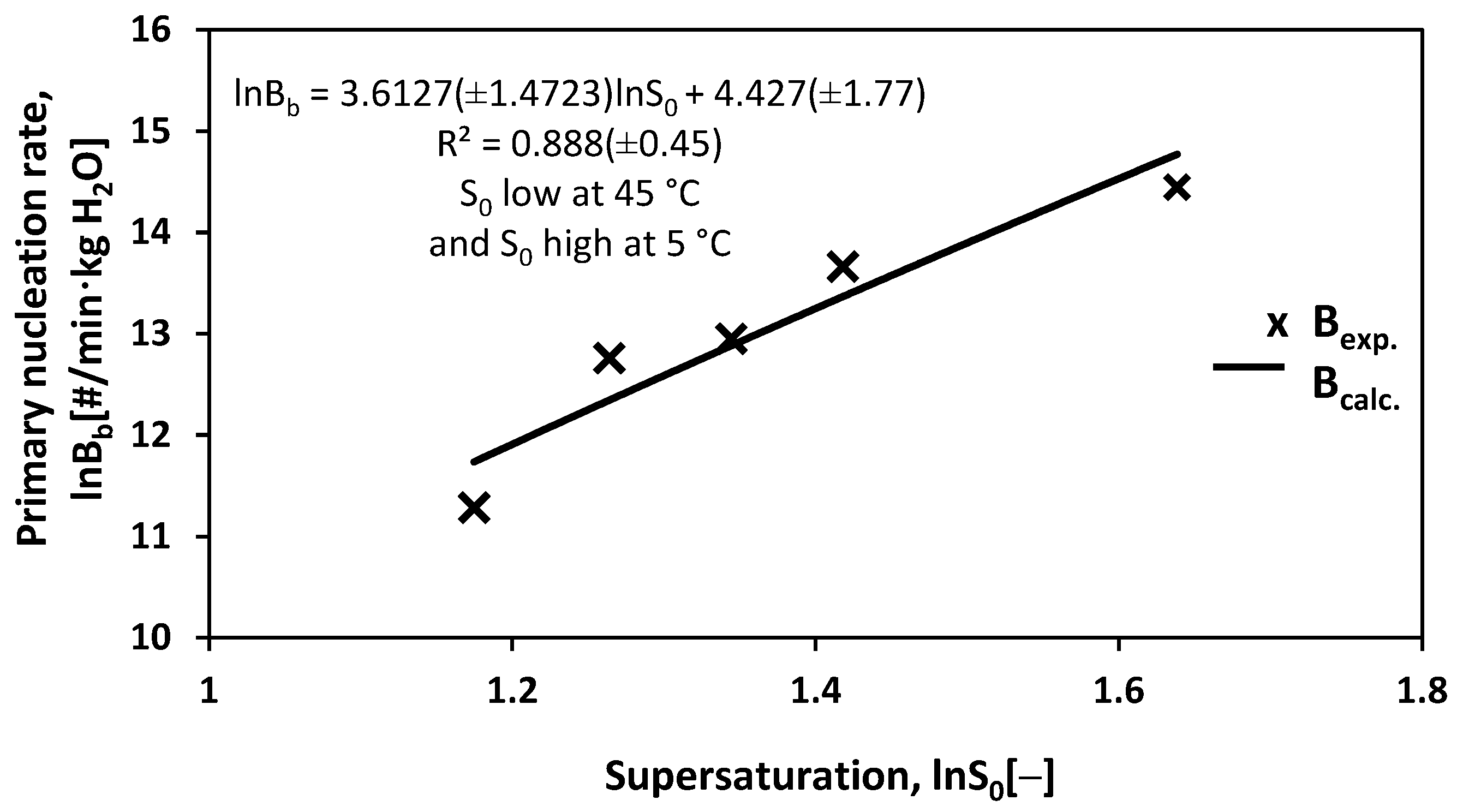
3.3. Primary Nucleation Rate Bb
3.4. Secondary Nucleation Rate B
3.5. Crystal Growth Rate of K2SO4
3.6. Crystal Suspension Density MT
3.7. Activation Energy E
3.8. K2SO4 Product Quality
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sanchez, J. Fertirrigación, Pricipios, Factores y Aplicaciones. In Seminario de Fertirrigación Apukai-Comex; Apukai-Comex: Lima, Perú, 2000; pp. 1–26. [Google Scholar]
- Vergara-Edwards, L.; Parada-Frederick, N. Study of the Phase Chemistry of the Salar de Atacama Brines. In Sixth International Symposium on Salt; Salt Institute-Documentary: Santiago, Chile, 1983; Volume II, pp. 345–366. [Google Scholar]
- Alonso, H. Origen De Los Componentes Y Balance Salino. Rev. Geol. Chile. 1996, 23, 113–122. [Google Scholar]
- Risacher, F. Estudio Econòmico Del Salar de Uyuni; ORSTOM en Bolivie Mission de la Paz: La Paz, Bolvia, 1989. [Google Scholar]
- Hidemaro, I.; Kenichi, H. Process of Producing a Potassium Sulfate Salt. U.S. Patent US4342737A, 3 August 1982. [Google Scholar]
- Abu-eishah, S.I.; Bani-kananeh, A.A.; Allawzi, M.A. K2SO4 Production via the Double Decomposition Reaction of KCl and Phosphogypsum. Chem. Eng. J. 2000, 76, 197–207. [Google Scholar] [CrossRef]
- Holdengraber, C.; Lampert, S. Process for Producing Potassium Sulfate from Potash and Sodium Sulfate. U.S. Patent US6143271A, 7 November 2000. [Google Scholar]
- Grzmil, B.U.; Kic, B. Single-Stage Process for Manufacturing of Potassium Sulphate from Sodium Sulphate. Chem. Pap. 2005, 59, 476–480. [Google Scholar]
- Mubarak, Y.A. Integrated Process for Potassium Sulfate and a Mixture of Ammonium Chloride/Potassium Sulfate Salts Production. Int. J. Eng. Technol. 2018, 7, 185–197. [Google Scholar] [CrossRef] [Green Version]
- Marx, H.; Kaps, S.; Schultheis, B.; Pfânder, M. Potassium Sulfate—A Precious by-Product for Solar Salt Works. In Proceedings of the World Salt Symposium 2018, Park City, UT, USA, 11 June 2018. [Google Scholar]
- Ryan, J.H.; Lukes, J.A.; Neitzel, U.E.; Duyster, H.P.; Great Salt Lake Minerals and Chemicals Corp. Method for the Production of Potassium Sulfate from Potassium-Containing Double Salts of Magnesium Sulfate. U.S. Patent US3634041A, 12 January 1972. [Google Scholar]
- Jannet, D.B.; M’nif, A.; Rokbani, R. Natural Brine Valorisation: Application of the System K+, Mg2+/Cl−, SO42−/H2O at 25 °C. Desalination 2004, 167, 319–326. [Google Scholar] [CrossRef]
- Voigt, W. Solubility of Inorganic Salts and Their Industrial Importance. In Developments and Applications in Solubility; RSC Publishing: Freiberg, Germany, 2007; Chapter 24; pp. 390–406. [Google Scholar] [CrossRef]
- Fezei, R.; Hammi, H.; Adel, M. Study of the Sylvite Transformation into Arcanite At 25 °C. World J. Agric. Sci. 2008, 4, 390–397. [Google Scholar]
- Goncharik, I.I.; Shevchuk, V.V.; Krut, N.P.; Smychnik, A.D.; Kudina, O.A. Synthesis of Potassium Sulfate by Conversion of Potassium Chloride and Magnesium Sulfate. Inorg. Synth. Ind. Inorg. Chem. 2014, 87, 1804–1809. [Google Scholar] [CrossRef]
- Jha, S.K.; Karthika, S.; Radhakrishnan, T.K. Modelling and Control of Crystallization Process. Resour. Technol. 2017, 3, 94–100. [Google Scholar] [CrossRef]
- Togkalidou, T.; Tung, H.H.; Sun, Y.; Andrews, A.T.; Braatz, R.D. Parameter Estimation and Optimization of a Loosely Bound Aggregating Pharmaceutical Crystallization Using in Situ Infrared and Laser Backscattering measurements. Ind. Eng. Chem. Res. 2004, 43, 6168–6181. [Google Scholar] [CrossRef]
- Shoji, M. Kinetic Studies for Industrial Crystallization to Improve Crystal Size Distribution and Crystal Shape. Available online: https://tuat.repo.nii.ac.jp/index.php?action=pages_view_main&active_action=repository_action_common_download&attribute_id=16&block_id=39&file_no=1&item_id=908&item_no=1&page_id=13 (accessed on 1 December 2012).
- Nagy, Z.K.; Chew, J.W.; Fujiwara, M.; Braatz, R.D. Advances in the Modeling and Control of Batch Crystallizers. IFAC Proc. Vol. 2004, 37, 83–90. [Google Scholar] [CrossRef]
- Tavare, N.S. Characterization of Crystallization Kinetics from Batch Experiments. Sep. Purif. Rev. 1993, 22, 93–210. [Google Scholar] [CrossRef]
- Nemdili, L.; Koutchoukali, O.; Mameri, F.; Gouaou, I.; Koutchoukali, M.S.; Ulrich, J. Crystallization Study of Potassium Sulfate-Water System, Metastable Zone Width and Induction Time Measurements Using Ultrasonic, Turbidity and 3D-ORM Techniques. J. Cryst. Growth 2018, 500, 44–51. [Google Scholar] [CrossRef]
- Bari, A.H.; Chawla, A.; Pandit, A.B. Ultrasonics Sonochemistry Sono-crystallization Kinetics of K2SO4: Estimation of Nucleation, Growth, Breakage and Agglomeration Kinetics. Ultrason. Sonochem. 2017, 35, 196–203. [Google Scholar] [CrossRef]
- Luo, M.; Liu, C.; Xue, J.; Li, P.; Yu, J. Determination of Metastable Zone Width of Potassium Sulfate in Aqueous Solution by Ultrasonic Sensor and FBRM. J. Cryst. Growth 2017, 469, 144–153. [Google Scholar] [CrossRef]
- Bari, A.H.; Pandit, A.B. Sequential Crystallization Parameter Estimation Method for Determination of Nucleation, Growth, Breakage, and Agglomeration Kinetics. Ind. Eng. Chem. Res. 2018, 57, 1370–1379. [Google Scholar] [CrossRef]
- Gougazeh, M.; Omar, W.; Ulrich, J. Growth and Dissolution kKinetics of Potassium Sulfate in Pure Solutions and in the Presence of Cr3+ Ions. Cryst. Res. Technol. 2009, 1210, 1205–1210. [Google Scholar] [CrossRef]
- Kubota, N.; Fukazawa, J.; Yashiro, H.; Mullin, J.W. Impurity Effect of Chromium (III) on the Growth and Dissolution Rates of Potassium Sulfate Crystals. J. Cryst. Growth 1995, 149, 113–119. [Google Scholar] [CrossRef]
- Mullin, J.W.; Gaska, C. Potassium Sulfate Crystal Growth Rates in Aqueous Solution. J. Chem. Eng. Data 1973, 18, 217–220. [Google Scholar] [CrossRef]
- Avila, S.T. Control del Proceso de Cristalización Continuo No-isotérmico Empleando Lógica Difusa; Celaya: Guanajuato, México, 2010. [Google Scholar]
- Chianese, A.; Di Berardino, F.; Jones, A.G. On the Effect of Secondary Nucleation on the Crystal Size Distribution from a Seeded Batch Crystallizer. Chem. Eng. Sci. 1993, 48, 551–560. [Google Scholar] [CrossRef]
- Mohamed-Kheir, A.K.M.; Tavare, N.S.; Garside, .J. Crystallization Kinetics of Potassium Sulphate in a 1 M3 Batch Cooling Crystallizer; Pergamon Press: Oxford, UK, 1987. [Google Scholar]
- Taguchi, K.; Garside, J.; Tavare, N.S. Nucleation and Growth Kinetics of Barium Sulphate in Batch Precipitation. J. Cryst. Growth 1996, 163, 318–328. [Google Scholar] [CrossRef]
- Lu, L.; Hua, Q.; Tang, J.; Liu, Y.; Liu, L.; Wang, B. Reactive Crystallization Kinetics of Magnesium Hydroxide in the Mg(NO3)2–NaOH System. Cryst. Res. Technol. 2018, 53, 1–10. [Google Scholar] [CrossRef]
- Mignon, D.; Manth, T.; Offermann, H. Kinetic Modelling of Batch Precipitation Reactions. Chem. Eng. Sci. 1996, 51, 2565–2570. [Google Scholar] [CrossRef]
- Pandit, A.V.; Vivek, V.R. Chord Length Distribution to Particle Sized Distribution. AIChE J. 2016, 62, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Heath, A.R.; Fawell, P.D.; Bahri, P.A.; Swift, J.D. Estimating Average Particle Size by Focused Beam Reflectance Measurement (FBRM). Part. Part. Syst. Charact. 2002, 19, 84–95. [Google Scholar] [CrossRef]
- Trifkovic, M.; Sheikhzadeh, M.; Rohani, S. Kinetics Estimation and Single and Multi-Objective Optimization of a Seeded, Anti-solvent, Isothermal Batch Crystallizer. Ind. Eng. Chem. Res. 2008, 47, 1586–1595. [Google Scholar] [CrossRef]
- Óciardhá, C.T.; Hutton, K.W.; Mitchell, N.A.; Frawley, P.J. Simultaneous Parameter Estimation and Optimization of a Seeded Antisolvent Crystallization. Cryst. Growth Des. 2012, 12, 5247–5261. [Google Scholar] [CrossRef]
- Pitzer, K.S. Activity Coefficients in Electrolyte Solutions, 2nd ed.; Pitzer, K.S., Ed.; CRC Press Taylor & Francis Group: Berkeley, CA, USA, 2018. [Google Scholar] [CrossRef]
- Harvie, C.E.; Møller, N.; Weare, J.H. The Prediction of Mineral Solubilities in Natural Waters: The Na-K-Mg-Ca-H-Cl-SO4-OH-HCO3-CO3-CO2-H2O system to High Ionic Strengths at 25 °C. Geochim. Cosmochim. Acta 1984, 48, 723–751. [Google Scholar] [CrossRef]
- Møller, N. The Prediction of Mineral Solubilities in Natural Waters: A Chemical Equilibrium Model for the Na-Ca-Cl-SO4-H2O System, to High Temperature and Concentration. Geochim. Cosmochim. Acta 1988, 52, 821–837. [Google Scholar] [CrossRef]
- Marion, G.M.; Farren, R.E. Mineral Solubilities in the Na-K-Mg-Ca-Cl-SO4-H2O system: A re-evaluation of the Sulfate Chemistry in the Spencer-Moller-Weare Model. Geochim. Cosmochim. Acta 1999, 63, 1305–1318. [Google Scholar] [CrossRef]
- Holmes, H.F.; Baes, C.F.; Mesmer, R.E. Isopiestic Studies of Aqueous Solutions at Elevated Temperatures. J. Chem. Thermodyn. 1978, 10, 983–996. [Google Scholar] [CrossRef]
- De Lima, M.C.P.; Pitzer, K.S. Thermodynamics of Saturated Electrolyte Mixtures of NaCl with Na2SO4 and with MgCl2. J. Solut. Chem. 1983, 12, 187–199. [Google Scholar] [CrossRef] [Green Version]
- Phutela, R.C.; Pitzer, K.S. Heat Capacity and other Thermodynamic Properties of Aqueous Magnesium Sulfate to 473 K. J. Phys. Chem. 1986, 90, 895–901. [Google Scholar] [CrossRef]
- Linke, W.F.; Seidell, A. Solubilities of Inorganic and Metal Organic Compounds, 4th ed.; Van Nostrand: Washington, DC, USA, 1958. [Google Scholar]
- Jimenez, Y.P.; Taboada, M.E.; Galleguillos, H.R. Solid-Liquid Equilibrium of K2SO4 in Solvent Mixtures at Different Temperatures. Fluid Phase Equilib. 2009, 284, 114–117. [Google Scholar] [CrossRef]
- Tavare, N.S.; Gaikar, G.V. Precipitation of Salicylic Acid: Hydrotropy and Reaction. Ind. Eng. Chemisry Res. 1991, 30, 722–728. [Google Scholar] [CrossRef]
- Vollmer, U.; Raisch, J. Control of Batch Crystallization-A System Inversion Approach. Chem. Eng. Process. Process. Intensif. 2006, 45, 874–885. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, X.; Ching, C.B. Application of Preferential Crystallization for Different Types of Racemic Compounds. Ind. Eng. Chem. Res. 2009, 48, 7266–7275. [Google Scholar] [CrossRef]
- Li, H.; Kawajiri, Y.; Grover, M.A.; Rousseau, R.W. Modeling of Nucleation and Growth Kinetics for Unseeded Batch Cooling Crystallization. Ind. Eng. Chem. Res. 2017, 56, 4060–4073. [Google Scholar] [CrossRef]
- Lyczko, N.; Espitalier, F.; Louisnard, O.; Schwartzentruber, J. Effect of Ultrasound on the Induction Time and the Metastable Zone Widths of Potassium Sulphate. Chem. Eng. J. 2002, 86, 233–241. [Google Scholar] [CrossRef] [Green Version]
- Garside, J.; Gibilaro, L.G.; Tavare, N.S. Evaluation of Crystal Growth Kinetics From a Desupersaturation Curve Using Initial Derivatives. Chem. Eng. Sci. 1982, 37, 1625–1628. [Google Scholar] [CrossRef]
- Garside, J.; Gaska, C.; Mullin, J.W. Crystal Growth Rate Studies with Potassium Sulphate in a Fluidized Bed Crystallizer. J. Cryst. Growth 1972, 13–14, 510–516. [Google Scholar] [CrossRef]
- Steyer, C.; Promotionskommission, M.; Lorenz, H.; Sundmacher, K.; Mangold, M. Precipitation of Barium Sulfate in a Semi-Batch Stirred Tank Reactor: Influence of Feeding Policy on Particle Size and Morphology; Universitätsbibl Otto von Guericke University Library: Magdeburg, Germany, 2012. [Google Scholar]
- Jones, A.G.; Budz, J.; Mullin, J.W. Batch Crystallization and Solid-Liquid Separation of Potassium Sulphate. Chem. Eng. Sci. 1987, 42, 619–629. [Google Scholar] [CrossRef]
- Jones, A.G.; Mullin, J.W. Programmed Cooling Crystallization of Potassium Sulphate Solutions. Chem. Eng. Sci. 1974, 29, 105–118. [Google Scholar] [CrossRef]
- Jones, A.G.; Chianese, A. Fines Destruction During Batch Crystallization. Chem. Eng. Commun. 1987, 62, 5–16. [Google Scholar] [CrossRef]
- Senaputra, A.; Jones, F.; Fawell, P.D.; Smith, P.G. Focused beam reflectance Measurement for Monitoring the Extent and Efficiency of Flocculation in Mineral Systems. AIChE J. 2014, 60, 251–265. [Google Scholar] [CrossRef]
- Song, X.; Zhang, L.; Zhao, J.; Xu, Y.; Sun, Z.; Li, P.; Yu, J. Preparation of Calcium Sulfate Whiskers Using Waste Calcium Chloride by Reactive Crystallization. Cryst. Res. Technol. 2011, 46, 166–172. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temperature [°C] | Ionic Pair | ||||
|---|---|---|---|---|---|
| 5 | K+-Cl− | 3.38 | 18.31 | 8 | |
| Mg2+-Cl− | 36.29 | 157.02 | 87 | ||
| Mg2+-SO42− | 19.49 | 311.56 | −19.88 | 23 | |
| 15 | K+-Cl− | 4.16 | 20.35 | −1 | |
| Mg2+-Cl− | 35.69 | 160.81 | 76 | ||
| Mg2+-SO42− | 20.69 | 324.94 | −28.60 | 20 | |
| 25 | K+-Cl− | 4.80 | 21.88 | -8 | |
| Mg2+-Cl− | 35.11 | 165.12 | 65 | ||
| Mg2+-SO42− | 21.51 | 336.63 | −32.77 | 17 | |
| 35 | K+-Cl− | 5.33 | 23.18 | −14 | |
| Mg2+-Cl− | 34.54 | 165.12 | 55 | ||
| Mg2+-SO42− | 22.09 | 347.10 | −32.97 | 15 | |
| 45 | K+-Cl− | 5.75 | 24.37 | −18 | |
| Mg2+-Cl− | 33.98 | 175.30 | 45 | ||
| Mg2+-SO42− | 22.55 | 356.78 | −29.71 | 13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Albis, A.; Jiménez, Y.P.; Graber, T.A.; Lorenz, H. Reactive Crystallization Kinetics of K2SO4 from Picromerite-Based MgSO4 and KCl. Crystals 2021, 11, 1558. https://doi.org/10.3390/cryst11121558
Albis A, Jiménez YP, Graber TA, Lorenz H. Reactive Crystallization Kinetics of K2SO4 from Picromerite-Based MgSO4 and KCl. Crystals. 2021; 11(12):1558. https://doi.org/10.3390/cryst11121558
Chicago/Turabian StyleAlbis, Abad, Yecid P. Jiménez, Teófilo A. Graber, and Heike Lorenz. 2021. "Reactive Crystallization Kinetics of K2SO4 from Picromerite-Based MgSO4 and KCl" Crystals 11, no. 12: 1558. https://doi.org/10.3390/cryst11121558
APA StyleAlbis, A., Jiménez, Y. P., Graber, T. A., & Lorenz, H. (2021). Reactive Crystallization Kinetics of K2SO4 from Picromerite-Based MgSO4 and KCl. Crystals, 11(12), 1558. https://doi.org/10.3390/cryst11121558