1. Introduction
The development of innovative and efficient energy storage technologies, able to support a wide penetration of renewables at different scales, is an urgent matter [
1,
2]. It mainly comprises electric (EES), mechanical (MES) and thermal energy storage (TES) technologies. In particular, the relevant role of TES in the whole energy system is confirmed by the most recent Eurostat analysis, reporting energy consumption at households dominated by space heating (SH) (63.6%), followed by domestic hot water (DHW) (14.8%) [
3]. These values gain even more relevance considering that, again according to Eurostat, the residential sector accounts for 26.1% of the final energy consumption in Europe. This means that, overall, SH and DHW causes roughly 20% of the final energy consumption in EU, corresponding to about 3900 TWh/y [
4]. In particular, a market analysis related to currently employed SH and DHW equipment installed in EU, demonstrated that the market is still dominated by gas boilers, both non-condensing, up to 67%, and condensing ones, about 9% of total energy consumption. The other technologies comprise district heating networks (13%) and electric radiators (6%). The current diffusion of solar thermal collectors, representing the direct renewable sources, is limited to 2% [
4]. This study confirms the reduced market penetration of renewables in the domestic heating sector, thus calling for innovative TES solutions acting as enabling technologies to unlock the potential of the renewable heating systems.
The TES technologies are commonly classified depending on the physical process underpinning the TES operation, namely, sensible, latent, and thermochemical. The sensible TES [
5,
6] exploits the increasing temperature level of the storage media, to store thermal energy to be used subsequently. Accordingly, the overall storage capacity is directly dependent on the specific heat and temperature difference under which it is operated. This represents, especially for operation temperature below 100 °C, the most common technology, since it exploits water as storage media. Nevertheless, it is characterized by low TES density and long-term storage limitation due to the unavoidable heat losses towards the surrounding environment, which reduce the amount of stored energy over time.
The latent TES [
7] exploits the phase change (e.g., solid/liquid) occurring in the storage media when thermal energy is provided. The storage materials are often referred as phase change materials (PCMs). In this case, the most relevant thermophysical parameter useful to evaluate the TES density is the latent heat of phase transition. Of course, a certain portion of energy is also stored sensibly, due to the occurring temperature variation in the storage media. This technology is currently mainly commercialized for applications where the system needs to be operated in a narrow temperature range, exploiting the ability of latent TES to work almost isothermally at the phase change temperature. Examples are the preservation of vaccines and perishable food [
8]. Even if characterized by higher energy storage density, thanks to the latent heat which is usually one order of magnitude higher than the specific heat, latent TESs are still suffering from heat dissipation, preventing their long-term reliability, as well as poor heat transfer efficiency due to the low thermal conductivity of the most common PCMs [
9], which can be improved by using innovative nanofluids as heat transfer fluid [
10].
The thermochemical TES [
11,
12] is based on the reversible reaction occurring between two components, to which a reaction heat is associated. Accordingly, during the charging phase, the thermal energy is supplied to the storage media to separate the two components through the endothermic reaction. While during the discharging phase, the reverse exothermic reaction of re-combination is performed, thus releasing the stored heat. This technology can be based either on chemical reactions, usually suitable for high temperature storage [
13] or on physical interactions, characterized by lower heat and operating temperature [
14]. This technology has gained a lot of attention thanks to the higher storage density achievable through the occurring reaction, as well as the possibility of keeping the energy stored almost indefinitely, as long as the two components are kept separated and no re-combination reaction is allowed (i.e., seasonal storage [
15]). On the contrary, some of the main barriers towards its diffusion are represented by the highest system complexity, slow kinetic reaction, and issues in the long-term stability of the materials.
The thermochemical TES process commonly proposed for low-temperature storage (i.e., below 100 °C) is usually referred as “sorption” [
12] TES. Indeed, in this operating temperature range, it is possible to exploit physical or weak chemical reactions between a sorbate (often referred as working fluid) and a sorbent. Unlike the other technologies, the sorption TES needs to be operated at least between two different temperature levels, like reverse heat pumping cycles [
16]. Indeed, during charging phase, it exploits the high-temperature source to be stored and an ambient sink where the reaction heat is dumped; while, during discharging phase, it exploits ambient heat as a source to promote the re-combination process and it discharges the recovered heat at the end-user temperature level. This technology can be applied both as open and closed cycle. In the former one, the TES exchanges both mass and heat with the ambient conditions, accordingly it usually needs to exploit ambient humidity as sorbate [
17,
18]. In the latter one, only heat is exchanged with the ambient conditions, and the sorbate is continuously circulated inside the system, following the subsequent charging and discharging phases. The most common sorbate in closed systems is again water, even if other fluids were proposed that can be operated even at temperature below 0 °C, such as ammonia [
19], ethanol [
20], and methanol [
21]. While, regarding the sorbent, the main classification is between solid and liquid sorbents. In the solid sorbents, the reaction with the sorbate occurs on the available surface, mainly by means of weak physical interactions. The most common solid sorbents are zeolites [
22], silica gel [
23], zeotypes [
24], and metal–organic frameworks [
25]. In the liquid sorbents, the reaction occurs between a sorbate and a liquid solution. In this case, the most common proposed solutions are LiBr/water [
26], NaOH/water [
27] and, more recently, ionic liquids/water [
28]. Both approaches show some limitations, such as the limited storage capacity achievable by pure physisorption processes, which is directly linked to the available specific pore volume of the solid sorbents [
29], and the mass transfer limitation occurring in the reaction between a sorbate and a liquid solution [
27]. A class of materials that can exploit the solid and liquid sorption storage features, also helping in overcoming the above listed limitations, is the composite sorbents class [
14]. These materials are based on a solid porous matrix which is embedded with a salt. Accordingly, the interaction between the composites and sorbates combine several mechanisms: (i) the physisorption occurring on the matrix surface; (ii) the reaction with the embedded salt; and (iii) absorption by the salt solution forming inside the pores. This can substantially increase the achievable sorption capacity, which is reflected in higher storage density and, at the same time, it limits the mass transfer resistance in the reaction between salt and sorbate, since the grain size of the salt confined inside the pores is in the nano-scale.
In the literature, different matrixes and salts were proposed to synthesize the composites for TES applications. Usually, the main requests for the matrix is to have a large pore volume and an average pore size in the range 2–50 nm (i.e., mesoporous structure), thus allowing embedding of enough salt with reasonable crystal size. The selection of salt is strongly related to the boundary operating conditions.
The most common application for sorption TES is focused on SH provision, since, when low-temperature emission systems (i.e., radiant floors) are employed, the delivering temperature is low enough to achieve high storage density. For instance, in [
30] an optimization of composite sorbents based on commercial mesoporous silica gels and LiCl was presented, aiming at maximizing the storage density for SH applications. The different samples were characterized from the morphological point of view as well as by detailed sorption equilibrium measurements. Starting from these characterizations, the theoretical storage density was estimated, showing promising storage densities up to 1200 J/g
ads. A different matrix was recently proposed for the same application, the multi-wall carbon nanotube (MWCNT) [
21,
31]. Composites based on LiCl and LiBr were synthesized in [
21], showing an extremely high sorption capacity, both using water and methanol as sorbate. Specifically, the investigation performed for SH applications under seasonal operating conditions provided a theoretical storage capacity up to 1.70 kJ/g
ads, for the composite using LiCl and water. Under the same conditions, the LiBr one looks more attractive when methanol is used as sorbate, achieving up to 0.46 kJ/g
ads. Another possible matrix proposed for the application is the vermiculite [
32]. Differently from other matrixes, vermiculite is usually macro-porous, thus having higher pore volume but also wider pore size, causing a bigger crystal size of the embedded salt. This reflects on the higher sorption TES density achievable, which, as reported in [
32], can be as high as 2.3 kJ/g
ads for seasonal operating conditions. On the other hand, this can pose limitations on the sorption kinetic due to a larger salt grain size in the matrix. Concerning DHW provision, only a few examples were reported in the literature. For instance, a 4-kW prototype based on zeolite 13X and water with an open sorption TES was realized and tested in [
33,
34]. Of course, in this case the charging temperature is much higher than other cases, due to the high hydrophilicity of zeolite 13X. After a wide testing campaign, a storage density of about 0.29 GJ/m
3 was obtained at prototype level. A composite based on LiOH and expanded graphite was presented in [
35]. It was tested under conditions suitable for DHW provision. The employed charging temperature was 110 °C and the achieved theoretical storage density was about 1 kJ/g
ads for the composite that showed a stable behavior after repeated cycles. Nevertheless, the kinetic performance demonstrated the need to further investigate the applicability on a large scale.
Starting from the literature analysis reported before, this paper presents, for the first time, a detailed comparison of different porous matrixes embedding two salts, optimized for SH and DHW storage applications. Water was selected as the sorbate, because it is environmentally benign and have a large evaporation heat. The comparative analysis starts from the structural and morphological characterization of the synthesized samples to end up with equilibrium and sorption storage capacity analysis under realistic operating conditions, to identify the most promising solutions for a large-scale sorption TES, able to supply both SH and DHW, thus increasing the share of renewables for heating purposes in domestic applications.
3. Results
3.1. X-ray Diffraction
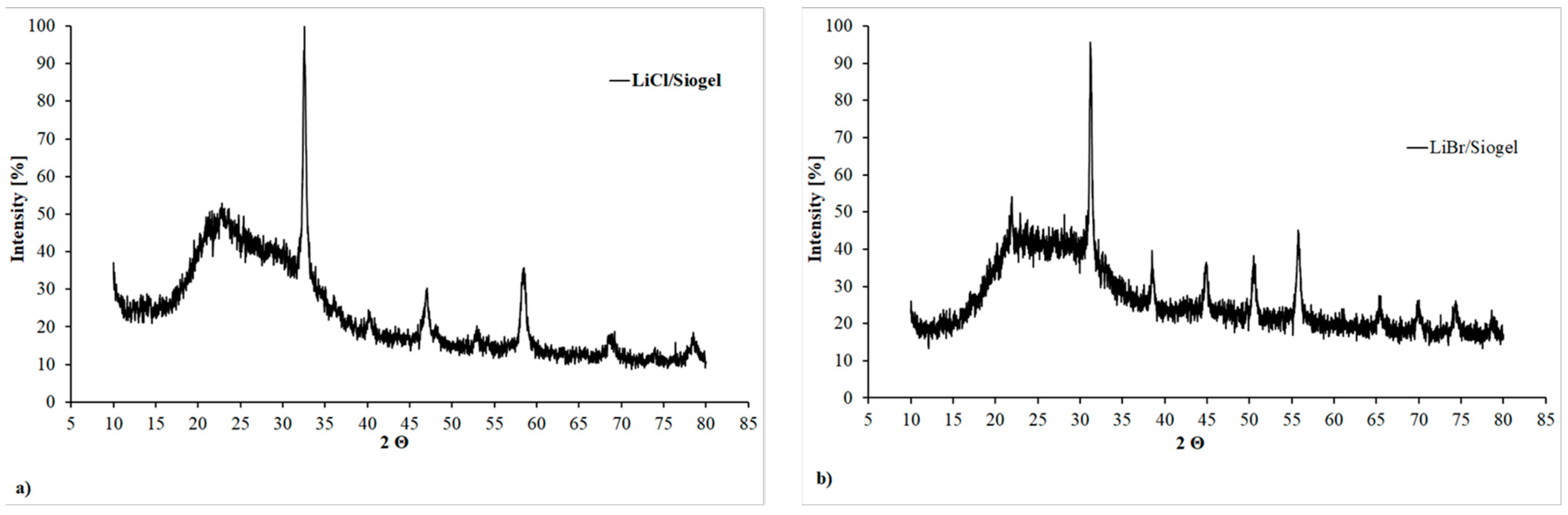
For each studied composite sorbent sample two different XRD patterns was recorded, the first one taken at RT (i.e., 25 °C), the second one after that the sample was kept for 30 min at 90 °C. The 2Θ range was always between 10° and 80°.
As reference case,
Figure 6a,b reports the XRD patters obtained at 25 °C of two samples based on Siogel and LiCl (
Figure 6a) and LiBr (
Figure 6b).
As expected, both XRD patterns reveal that composite samples present a relevant amorphous phase constituted by the silica gel matrix and also by the water molecules adsorbed in composite samples at 25 °C. Nevertheless, some peaks indicating the presence of crystalline structure are still present (
Figure 4). These peaks can be assigned to the crystalline hydrates LiCl·H
2O and LiBr·H
2O formed through the reaction of the salts with ambient humidity [
46].
Generally, an increasing crystallinity grade is common to all the studied composites passing from 25 °C to 90 °C and this confirms that the material is properly and quickly dehydrated without any kinetic hindrance. This is a feature related to the nano-confinement of small salt grains inside the porous structure.
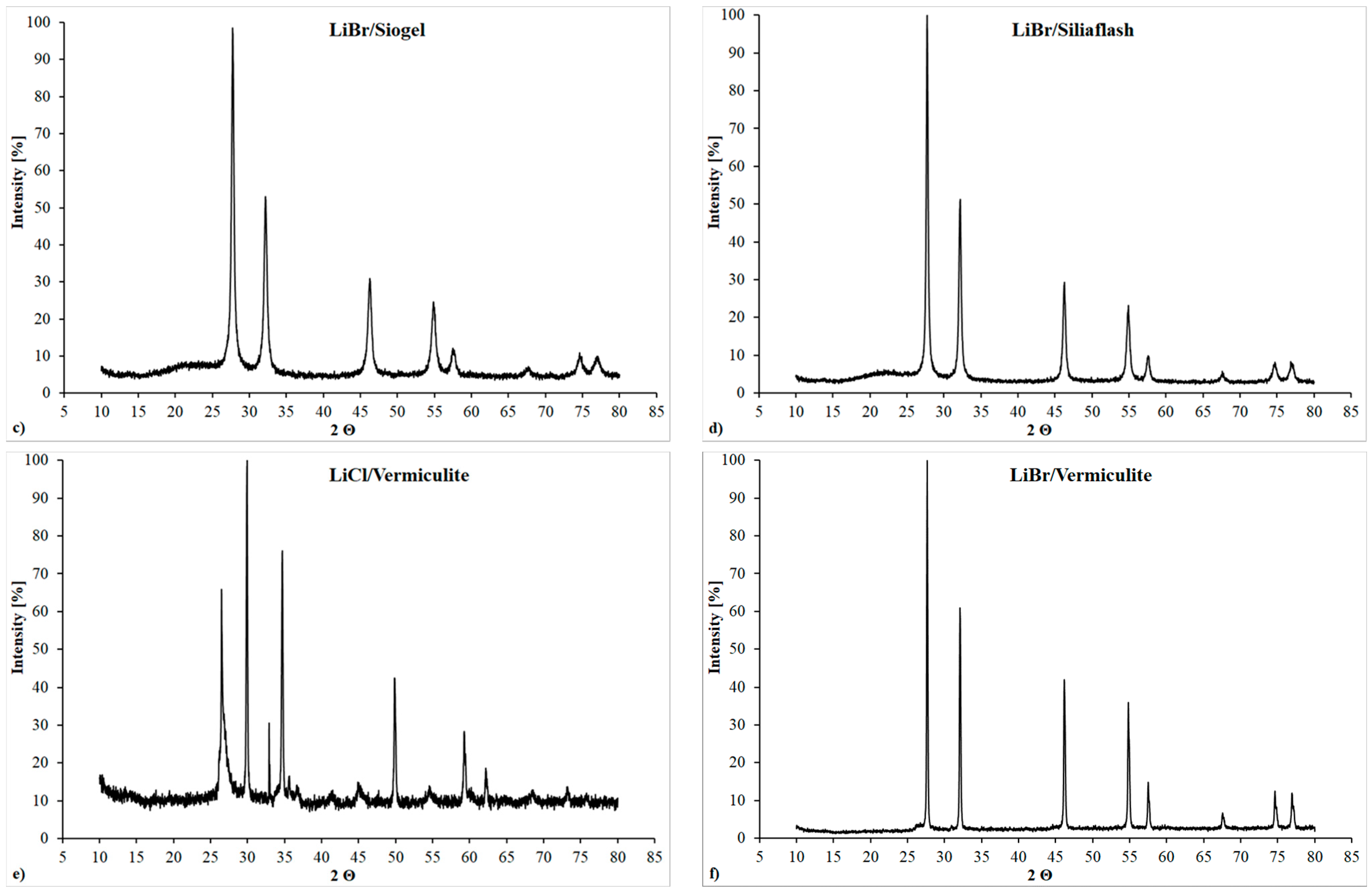
The XRD patterns of LiCl based samples that employ silica gel as matrix (
Figure 7a,b), at 90 °C, showed a highest and clearer diffraction peaks. This can be justified by the releasing of the adsorbed water present at low temperature, which implies presence of amorphous phase. This reported behavior is even more evident for the LiBr-based samples that employ silica gel as matrix (
Figure 7c,d), probably because at 25 °C, the salt is mainly dissolved in the adsorbed water and forms the aqueous LiBr solution inside pores. After having heated up the material, the crystalline pattern of the LiBr salt is more evident.
For the composite sorbents that employ LiCl and vermiculite as host matrix (
Figure 7e,f), several peaks at 2Θ = 26, 33, 42, 45, 54 in the XRD patterns at 90 °C may be attributed to the crystalline vermiculite structure. For LiBr/vermiculite the intensity of these peaks is negligible compared to the pure LiBr due to its larger content.
The size of coherently scattering domains is around to 14 nm and 20 nm for siogel-based composites and SiliaFlash based composites, respectively. These values agree with the pore size found for the pristine matrices, even though there might be a slight effect of the salt deposited outside the pores, causing the dimensions slightly larger than the pore size. For vermiculite-based composites the size of coherently scattering domains is about 33 nm, which is somewhat larger that for silica-based composites due to larger pore size of the vermiculite.
3.2. Nitrogen Physisorption
Figure 8a–d report the N
2 ad/desorption isotherms obtained for silica gel based composite sorbents. The results demonstrate that all the N
2 isotherms at 77 K of the composite sorbents belong to Type IV(a), according to the IUPAC classification [
37].
Table 5 summarizes the results in terms of BET surface area and pore volume. The physisorption tests allow to estimate the real amount of salt deposited as well as to analyze the space left for the water vapor flux inside the material, knowing the pore volume before and after impregnation process.
Table 5 also shows the features of the porous structure of the prepared composites based on silica gel matrix, as well as the theoretical pore volume V
p.th (cm
3/g), calculated considering the salt is completely deposited inside silica pores and does not block them, according to the following equation
where V
p.m (cm
3/g) is the matrix pore volume, C
s (wt %) is the salt content, and
ρs (g/cm
3) is the salt density.
As expected, these parameters follow the same trend of the pure porous matrixes. The pore volume reduction seems to be in line with amount of embedded salt, thus confirming that the salt can be considered deposited inside the pores.
The samples based on LiCl present a theoretical pore volume slightly smaller than the experimental one, which implies that salt is partially located out of pores. While, composite sorbents based on LiBr present a theoretical pore volume larger than experimental one. This could indicate that the pores are partially blocked by either LiBr particles deposited on the external surface of the composite’s grains, or by LiBr crystals placed inside pores necks. In any case, it is evident that most of the salt is actually embedded inside the pores, since a maximum deviation of about 10% was encountered.
3.3. Equilibrium Curves of Water Adsorption
The equilibrium curves of water vapor adsorption, reported in
Figure 9a–f, were collected by measuring the ad/sorption isotherms at 35°C. The choice of this temperature was established considering the discharge temperature of a storage system under SH operating conditions and were used for an initial screening of adsorbent materials performance.
At low relative humidity < 3 and 5% for LiCl/silica and LiCl/vermiculite-based composites a small mass of water is adsorbed on the matrix surface (
Figure 9d–f). At increasing humidity, the LiCl reacts with water resulting in formation of the hydrate LiCl·H
2O that reveals as the step on adsorption isotherms. At increasing humidity, the hydrate deliquesces and the forming solution absorbs water vapor that corresponds to the gradual increase in the uptake.
The isotherms of the composites employing LiBr are shifted toward a lower humidity that indicates its stronger affinity to water. Again, the step attributed to the formation of LiBr·H
2O is observed, which is followed by the smooth uptake growth corresponding to the water absorption by LiBr aqueous solution inside pores. From the obtained isotherms it is easy to observe that the behavior of composite sorbents depends on the matrix and on the embedded salt. Generally, the sorbents made by silica gel matrices show smoother de/adsorption de/increment of the uptake (
Figure 9a,b,d,e), while the sorbents based on vermiculite present distinct steps of de/adsorption (
Figure 9c–f). This difference is probably due to a larger size of the salt crystals inside pores.
The maximum uptake is higher for composite sorbents based on the vermiculite and this result is in line with the salt content.
3.4. Coupled TG/DSC
In order to evaluate the achievable energy storage density, the sorption heat was measured for the prepared composites under pure water vapor atmosphere, to simulate the typical operation of STES working with a closed cycle. Specifically, according to the boundaries reported in
Table 2, an evaporation pressure p
ev of 8.7 mbar, corresponding to 5 °C of evaporation temperature was considered for all the tests. The initial temperature, T
in_ads, of the isobaric adsorption stage was selected in such a way, that the value ΔF
in_ads = RT
in_adsln(p
s(T
in_ads)/p
ev) equals the value of ΔF
char of the appropriate STES cycle (
Table 2). In accordance with the Polanyi principle of temperature invariance, the uptake w(ΔF
in_ads) = w(ΔF
char). Thus, the TG/DSC tests imitate the selected STES cycles. Accordingly, starting temperature T
in_ads = 66 and 80°C, were considered for SH and DHW storage applications, respectively. For comparison purpose, the tests were performed also at two higher temperatures T
in_ads = 80 and 9 °C for SH and DHW storage applications, respectively. The adsorption temperature, T
ads, was selected according to
Table 2, namely, 35 °C for SH and 55 °C for DHW.
Figure 10 reports, for comparison purposes, some reference dynamic evolutions obtained during the TG/DSC characterization. Specifically, the tests over Siogel-based and vermiculite-based composites employing LiCl and LiBr, under the same operating conditions are described. Each evolution reports the measured heat flow, along with the peak integration needed to evaluate the sorption heat, the mass variation and the first derivative of the mass variation, dm/dt. Since all the measurements refer to an adsorption stage, a mass increasing is always obtained. As can be highlighted by
Figure 10a,b, the obtained evolution for composites based on LiCl, employing Siogel and vermiculite are quite different. Indeed, while the sample based on silica gel has a single non-symmetrical adsorption peak, which also corresponds to a single peak of the dm/dt signal, in the vermiculite sample the heat flow signal is characterized by two consecutive peaks that are also associated with two peaks observed in the dm/dt signal. This difference can be justified with the larger size of the salt grains embedded in the vermiculite pores, which slow down the hydration reaction with the water vapor, especially at such a low absolute pressure. This effect is even more pronounced for the LiBr-based composites. Also in this case, Siogel and vermiculite are compared. A narrow reaction peak is obtained for the silica gel-based sample, while a much broader peak is obtained for the vermiculite-based one. Indeed, in the latter case, after an initial heat flux peak, corresponding to a dm/dt peak, a plateau is observed, until the reaction process is over, and the heat flux as well as the dm/dt suddenly drop. This behavior can be ascribed to the large amount of LiBr embedded in the vermiculite pores, as represented in
Table 5, and a larger crystal size, which is reflected in a behavior of the salt closer to the bulk rather than to the nano-confined one. Of course, from the practical operation point of view, this behavior implies that the vermiculite-based samples are characterized by a slower reaction kinetics, which can limit the discharging power during operation.
The synthesized samples were tested under the above conditions. Each test consisted of four consecutive adsorption drops inside the TG/DSC apparatus, in order to guarantee the replicability of the measurement. All the samples showed a good reproducibility of the results, with a deviation of 5% maximum. The heat flux peaks were integrated to obtain the sorption heat and the corresponding water uptake variation was calculated.
Figure 11 summarizes the achieve results. For each sample and each temperature drop tested, the orange bar represents the uptake variation in (g/g), while the green bar represents the sorption heat in (GJ/m
3), evaluated considering the sample densities reported in
Table 3.
As expected, both water uptake variation and sorption heat obtained for DHW operation are much smaller than the ones measured for SH applications. Interestingly, the obtained results are almost unaffected by the initial adsorption temperatures applied. This means that, at the investigated water vapor pressure, the composite sorbents already reached their minimum adsorption capacity at the lowest starting adsorption temperature, namely 66 °C for SH and 80 °C for DHW. Looking at the achieved results, composite sorbents based on vermiculite achieve the highest water uptake variation, namely about 0.18 g/g for DHW and 0.52 g/g for SH. The silica gel-based ones show, on average, a lower water exchange capacity ranging between 15% (LiBr composites) and 25% (LiCl composites) of the ones obtained with the vermiculite-based composites. This is due to the higher amount of salt confined in the pores of the vermiculite. Specifically, these values are in line with the salt content of the synthesized samples. Indeed, the LiCl embedded in the silica gel samples is roughly 30% lower than content embedded in the vermiculite, while for the LiBr, this reduction is of about 20%. This confirms that the salt represents the active part of the composite, which mainly sorbs water vapor.
On the other hand, looking at the corresponding volumetric sorption heat, the composites employing silica gel as matrix achieve the highest values, since the density is about 45% higher than the one of vermiculite samples. This represents a crucial parameter in order to obtain compact TES. Interestingly, the highest sorption heat for SH applications is obtained with the Siliaflash sample, which reaches up to 0.7 GJ/m3. This value is reduced to 0.6 GJ/m3 when Siogel is employed. This difference is due to the higher pore volume of the Siliaflash, which allows embedding a higher quantity of salt in the matrix. As highlighted, the LiCl/vermiculite achieves the lowest volumetric sorption heat, down to 0.4 GJ/m3. Considering that water based sensible heat storage, operating with a temperature gradient of 40 K, can achieve about 0.17 GJ/m3, it is clear that these composites can help in achieving higher TES density. Furthermore, the absence of heat losses over long stand-by periods makes this approach even more profitable. Similarly, for DHW applications, silica gel-based composites achieve the highest volumetric sorption heat, about 0.4 GJ/m3, while the vermiculite-based one reaches up to 0.3 GJ/m3. Also, in this case, the advantage over sensible water storage is relevant, especially since, due to the higher delivering temperature, the temperature gradient under which the sensible storage can operate is lower than the one that exploitable for DHW applications, thus further limiting the overall volumetric storage density.


 ,
, 















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}