1. Introduction
K-ion batteries (KIBs) attract considerable scientific and industrial attention as a cheap and efficient large-scale energy storage technology for balancing the intermittent nature of renewable electricity generation. The perspective of using KIBs is favored by the high abundance of potassium in the Earth’s crust, availability of its deposits, and low price, as well as faster K
+ diffusion in aprotic electrolytes compared to that of Li [
1]. An interest in KIBs has been further ignited by the possibility to use a broad class of cheap, easily scalable and high-energy positive electrode materials—Prussian Blue analogs (PBAs). These materials, particularly iron and manganese-based PBAs, K
2−δMn[Fe(CN)
6]
1−ɣ∙nH
2O, demonstrate exceptional energy density and capacity retention of up to 80% after 7000 cycles [
2,
3]. However, various structural defects and non-stoichiometry related to water incorporation in the crystal lattice can drastically affect the electrochemical performance of PBAs as a possible cathode material in a potassium electrochemical system [
3,
4,
5]. For instance, the presence of water can lower the coulombic efficiency, slow down the kinetics, and result in a poor cycling stability [
6]. It is assumed that in K
2−δMn[Fe(CN)
6]
1−ɣ∙nH
2O, water can reside at three different positions: (i) at the K sites, completely replacing K
+; (ii) near the K sites, coordinating to a potassium ion in spacious voids; or (iii) at the [Fe(CN)
6]
4− anion vacancy, via the formation of six water units surrounding manganese ions [
5], in the form of H
2O molecules or OH-groups [
7]. In the first two cases, the water is called interstitial or zeolite water because it is only weakly bonded to the crystal lattice. This water type can be eliminated by sufficient drying at high enough temperatures. However, in the third case, the water molecules coordinate manganese, forming much stronger bonds which are unlikely to be removed without a significant distortion of the lattice.
Tuning the amount of water in PBAs can be achieved by several approaches: (i) the use of alternative solvents during synthesis, such as ethanol [
8,
9], methanol [
10], cyclohexane [
11,
12], or their mixtures with water; (ii) increasing the concentration of K
+ ions during synthesis [
13,
14,
15]; (iii) adding chelating agents to maintain a constantly low concentration of transition metal ions [
3,
4,
16]; or (iv) additional vacuum drying of the synthesized samples at elevated temperatures. Some efforts to understand the influence of water on the performance and stability were conducted only for Na-based PBAs, showing that vacuum drying at 100 °C drastically affects the interstitial water content and triggers considerable changes in the crystal structure and electrochemical properties [
17]. Some authors mentioned a K-based PBA drying procedure, but its impact on the structure and electrochemical performance was not examined [
3,
15,
18].
The objective of this work was to study the influence of the water content in K2−δMn[Fe(CN)6]1−ɣ∙nH2O on the crystal structure, electrochemical properties and K+ diffusion coefficients.
2. Materials and Methods
K2−δMn[Fe(CN)6]1−ɣ∙nH2O (KMFCN) was synthesized by two different co-precipitation methods. To synthesize the material with large crystallites, two solutions were prepared: (i) 150 g of KCl (SigmaAldrich, Madrid, Spain, 99%) and 8.448 g of K4Fe(CN)6∙3H2O (RusKhim, Moscow, Russia, 98%) were dissolved in 800 mL of H2O with the addition of 8 mL of HCl (RusKhim, Moscow, Russia, conc. 36 wt.%), and (ii) 7.916 g of MnCl2∙4H2O (SigmaAlrdich, ACS reagent, Bangalore, India, ≥98%) was dissolved in 400 mL of H2O. The solutions were mixed dropwise, using peristatic pumps at 20 mL/min and 10 mL/min rates, respectively, in air under vigorous stirring. The resulting solution was aged for 2 h, centrifuged and washed with deionized water 3 times. The obtained powder was divided into two equal parts. The first part was dried in air at 75 °C (ad-KMFCN) for 12 h, while the other part was dried under vacuum at 100 °C (vd-KMFCN) for 12 h. To synthesize the sample with small particles the following procedure was applied. In 10 mL of deionized water, 0.988 g of K3Fe(CN)6 (RusKhim, Moscow, Russia, 99%) were dissolved and added into a mixture of 0.735 g of Mn(CH3COO)2∙4H2O (Sigma-Alrdich, Saint Louis, MO, USA, >99%), 0.100 g of polyvinylpyrrolidone (PVP K-30, Mw 40,000, Sigma-Aldrich, Beijing, China) and 0.244 g of sodium citrate (Na3Cit∙5.5H2O, RusHim, Moscow, Russia, 99.5%) dissolved in 10 mL of ethanol and 10 mL of deionized water. The light grey precipitate was centrifugated and washed 3 times with deionized water. The obtained powder was dried according to the same procedures for the first method.
The obtained powders were analyzed by powder X-ray diffraction (XRD), with data collected in Bragg–Brentano reflection geometry on a Bruker D8 Advance diffractometer (λ
CuKα1 = 1.54051 Å, λ
CuKα2 = 1.54433 Å) equipped with the energy dispersive detector LYNXEYE XE. Time-of-flight neutron powder diffraction (TOF-NPD) data were collected on the POWGEN diffractometer (BL-11A beamline) at the Oak Ridge National Laboratory pulsed spallation source (Oak Ridge, TN, USA). The data were collected at 10 K and 300 K temperatures. Crystal structures were refined by the Rietveld method using the TOPAS software [
19], and the symmetry changes were tested using ISODISTORT [
20]. The occupancies of each site (except oxygen and hydrogen for differently dried samples) were constrained between samples considering that the composition must remain constant over different temperatures (in all four refinements, the chemical compositions were kept the same except the water and hydroxyl content). The occupancies of the site constituting the [Fe(CN)
6]
4− group were also constrained as for a single entity.
Thermogravimetric analysis with mass-spectroscopy of emitted products (TGA-MS) was performed with a TG-DSC STA-449 apparatus (Netzsch, Weimar, Germany) under Ar flow (50 mL/min) combined with a mass spectrometer, QMS 403 D Aëolos (Netzsch, Weimar, Germany). The powders were heated at a 5 K/min rate in the 50–600 °C range. The water content was additionally investigated by attenuated total reflectance Fourier-transform infrared spectroscopy (ATR-FTIR) with a stand-alone FTIR microscope LUMOS (Bruker, Ettlingen, Germany) equipped with a germanium ATR crystal and liquid-N2-cooled MCT detector. Spectra were recorded in the 4000–600 cm−1 range.
Transmission electron microscopy (TEM) studies were done as follows: a tiny amount of powder sample was ground with an agate mortar and pestle under ethyl alcohol. Then, one drop of the resulted suspension was deposited onto holey-carbon-coated copper grids. HAADF-STEM images and EDX compositional maps in the STEM mode (STEM-EDX) were acquired on a probe aberration-corrected FEI Titan electron microscope at 200 kV equipped with a Super-X system for energy-dispersive X-ray (EDX) analysis.
Galvanostatic cycling profiles were registered using two electrode stainless steel cells with aluminum current collectors with a metallic potassium counter electrode. The electrolyte was 0.5 M KPF
6 (AcrosOrganics, Carlsbad, CA, USA, extra pure, 99%) in EC:PC, 1:1 by vol. (EC: SigmaAldrich, product of USA, anhydrous, 99%, PC: SigmaAldrich, product of USA, anhydrous, 99.7%), with the addition of 2 mass. % of fluoroethylene carbonate (FEC, SigmaAldrich, Santiago, Chile, ≥99%). The electrode slurry contained the active material, PVdF (GelonLib, Linyi, China, HSV900) and carbon SuperP (TimCAL, Changzhou, China) in a 65:10:25 mass ratio. For galvanostatic measurements, the typical mass loading was ~1 mg cm
−2. For the electrochemical measurements in three-electrode cells, the loading was ~0.4 mg cm
−2 to minimize the thickness and inhomogeneity of the electrodes. Cycling voltammograms (CVs; scan rate, 50 µVs
−1), potentiostatic current transients and impedance spectra were registered in three-electrode stainless-steel cells with the same electrolyte using a Biologic VMP3 potentiostat. The reference electrode was a K
2−δFe[Fe(CN)
6]
1−ɣ-based electrode, which was charged to reach the potential of the first two-phase plateau prior to the measurements. The potential of this reference electrode was 3.329 V (vs. K
+/K). Using a stable reference electrode allows for the avoidance of the influence of the large polarization of the metallic potassium electrode, which appears to be due to the high reactivity of K with the electrolyte components [
21]. The working electrode (WE) was a thin PBA-based electrode, and the counter electrode (CE) was metallic potassium. The potentiostatic intermittent titration technique (PITT) was performed in 10 mV steps in the 3.85–3.96 V and 4.0–4.15 V (vs. K
+/K) potential ranges (in the single-phase regions of the intercalation isotherm). The electrochemical impedance spectroscopy (EIS) measurements were performed in the 10 mHz–100 kHz frequency range with a 5 mV alternating potential amplitude. Apparent diffusion coefficients (D) were estimated based on PITT data using the model detailed in [
22] for spherical finite diffusion.
3. Results and Discussion
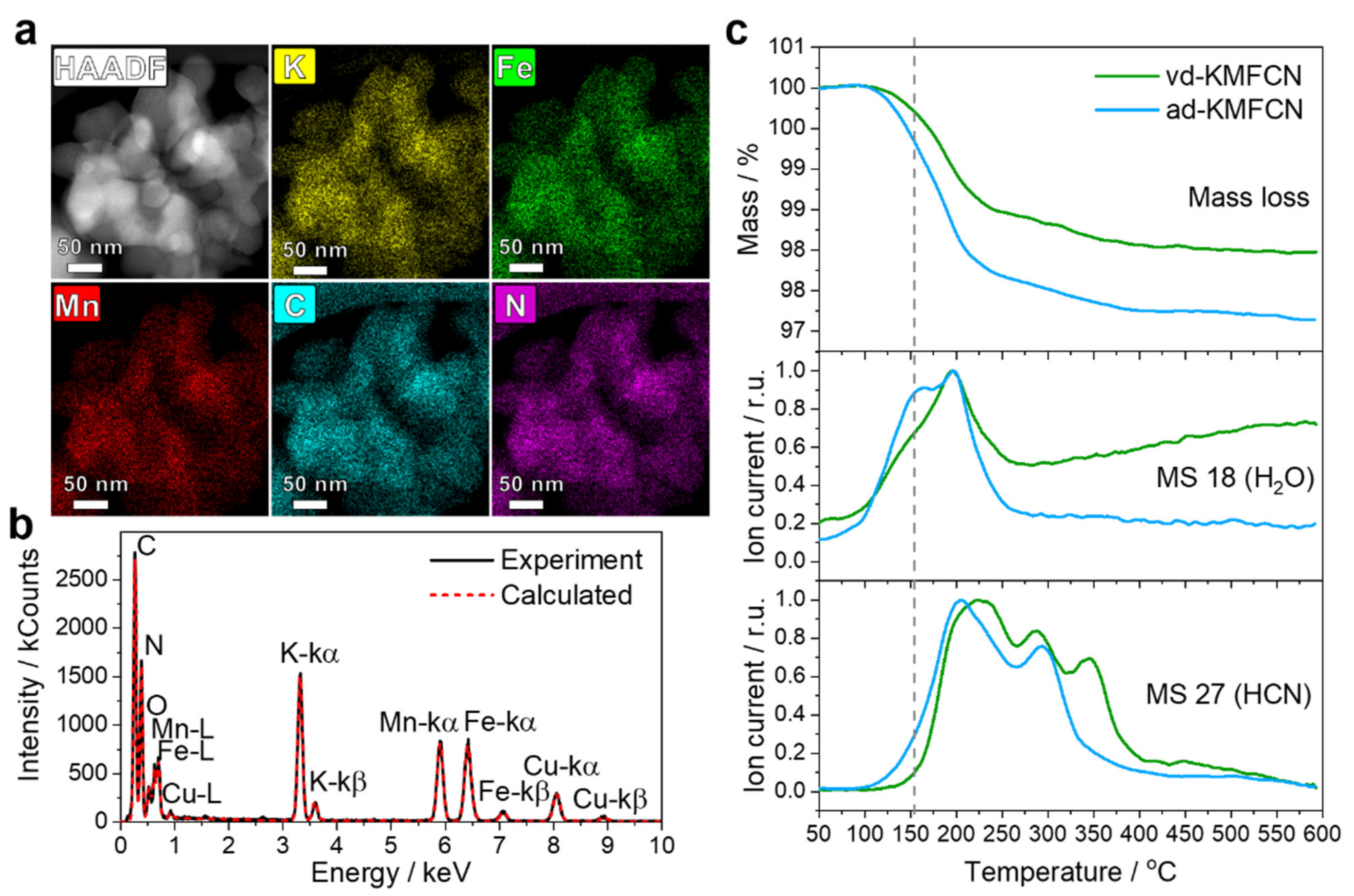
Single-phase KMFCN samples with large particles about 50–70 nm in size were synthesized for the structural analysis. According to STEM-EDX, the elemental ratio of K, Mn and Fe in both ad-KMFCN and vd-KMFCN was the same, such that their composition could be described as K
1.78 ± 0.08Mn[Fe(CN)
6]
0.94 ± 0.04 (
Figure 1a,b).
The comparative TGA-MS study of air and vacuum dried samples predictably demonstrated a smaller mass loss for vd-KMFCN, suggesting a lower water content. Additionally, the air-dried sample showed stronger water emission at around 150 °C, presumably related to interstitial water (
Figure 1c). Interestingly, for the air-dried sample, the onset and offset temperatures for releasing HCN were lower, suggesting a hydrolytic effect of interstitial water on CN
- groups. Another feature of these materials is the multistep elimination of HCN molecules as can be seen from the
m/
z 27 graph data (
Figure 1c). Since water plays a significant role in this process, we may conclude that every HCN emission peak is related to a different kind of structural water elimination, such as free water in K
+ cavities, water coordinated to the Mn ion replacing the [Fe(CN)
6] vacancy, and water forming from the OH groups. Deng et al. [
3] reported no HCN emission below 300
oC for the defect-free K
2Mn[Fe(CN)
6], therefore confirming that the presence of structural water downshifts the onset decomposition temperatures. Additional evidence for different water units was presented by IR-spectroscopy: the signals of δ(H–O–H) at 1617 cm
−1 and ν(O–H) at 3600–3000 cm
−1 were detected (
Figure S1), and the intensity of these bands in ad-KMFCN was significantly higher than in vd-KMFCN. In the 3500-3580 cm
−1 region, bands related to the ν(O–H) vibrations of structural hydroxyls could be found in both samples [
7,
23]. The ν(CN) vibration bands of equal intensities were also found in both IR spectra at 2059 cm
−1, which corresponds to the Fe
II-CN-Mn
II mode, and had no additional signs of the M
III ion presence in both positions [
24].
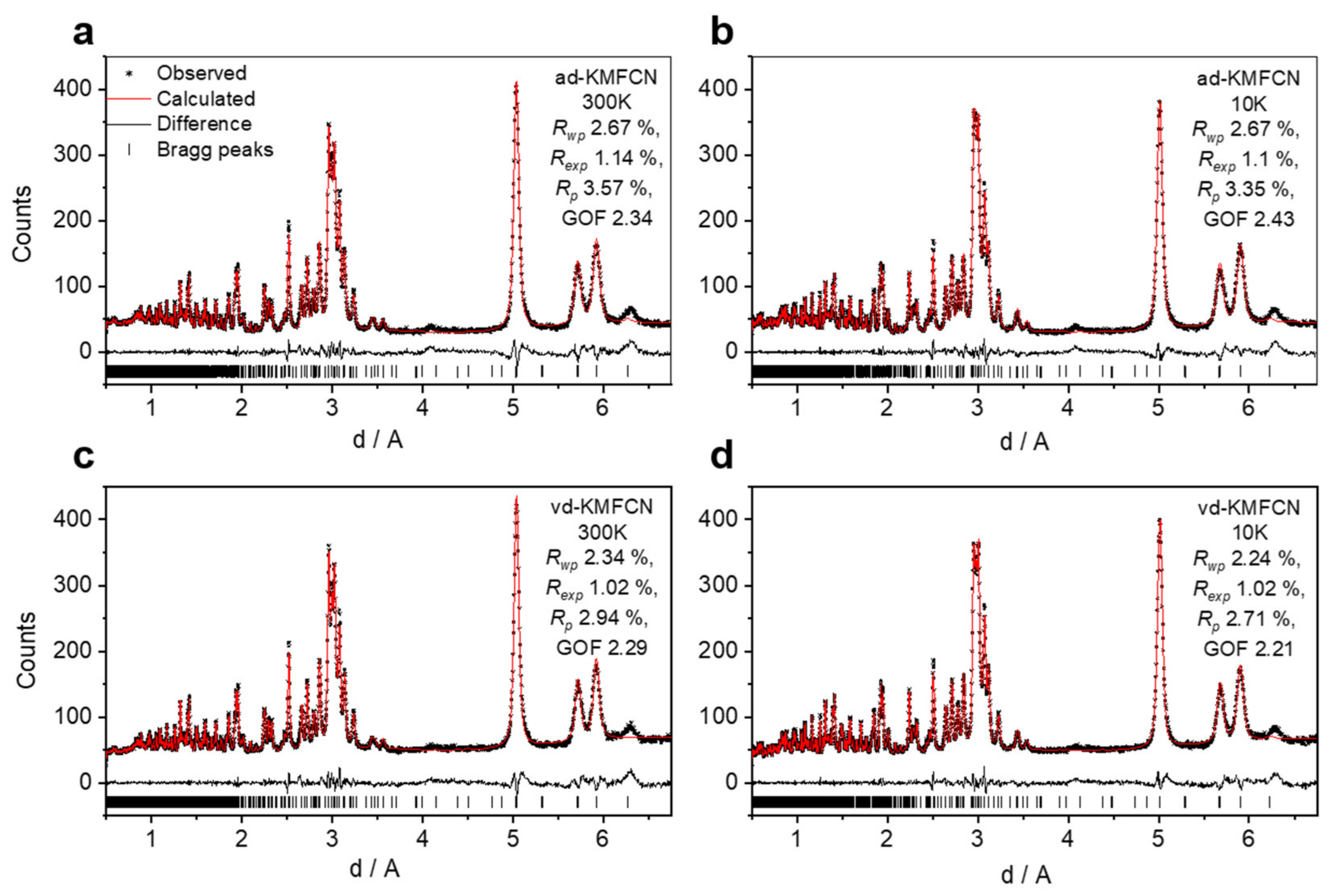
To further study a crystal structure and to locate water molecules, we collected TOF-NPD data at room temperature and 10 K for ad- and vd-samples (
Figure 2). In contrast to XRD analysis, the NPD method allows us to resolve light atoms such as carbon, nitrogen, oxygen and even hydrogen [
25]. In X-ray diffraction analysis, the atomic scattering factors of carbon, nitrogen and oxygen are very close and locating H
+ is even more problematic. To the best of our knowledge, no neutron diffraction investigation has been performed to analyze the light atoms in KMFCN, but some PBAs, as a class of electrode materials, have been investigated [
17,
26,
27,
28].
The detailed analysis of the NPD data at 10 K revealed no magnetic ordering. KMFCN is paramagnetic within a wide temperature range due to low-spin Fe
II (t
2g)
6 (S = 0) and high-spin Mn
II (t
2g)
3(e
g)
2 (S = 5), which is in accordance with Xue et al. [
29]. We did not observe any phase transitions when lowering the temperature; the unit cell parameters gradually decreased without anomalies. At the same time, due to the electrostatic effect of additional water and its interaction between oxygen and cations in ad-KMFCN, its unit cell volume was smaller than that of vd-KMFCN (
Table 1).
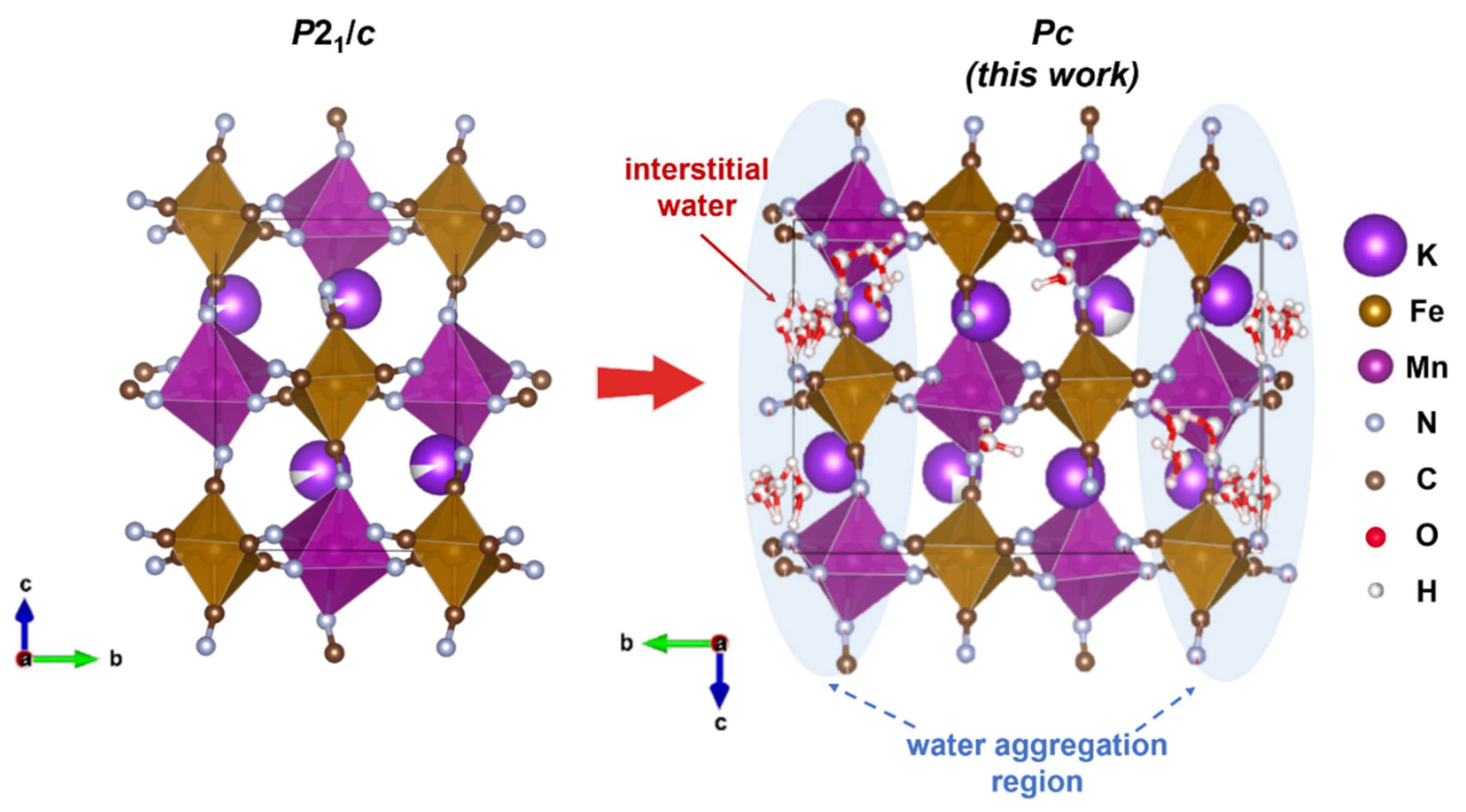
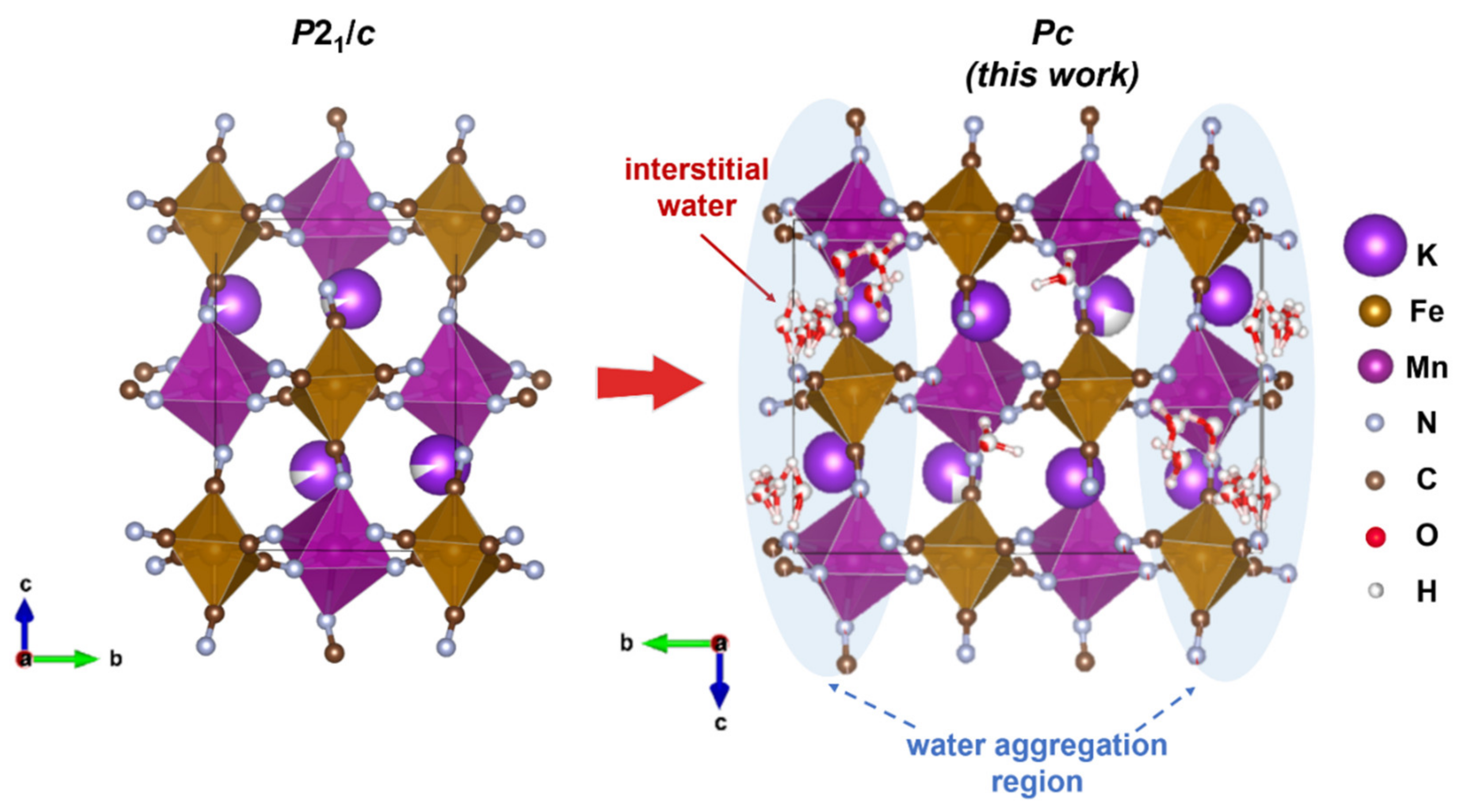
Both samples were indexed with the monoclinic symmetry. Attempting to apply the previously published
P2
1/
c space group to the NPD profile resulted in an unfitted peak at ~6.3Å and an insufficient peak intensity match of several other peaks. Therefore, we tested multiple space groups with assistance from the ISODISTORT tool and found that
Pc with a twice larger unit cell along the
b axis allows fitting the peak at ~6.3Å (indexed as
in
Pc), in contrast to the earlier proposed
P2
1/
c. Statistical indices also validate the choice of the lower symmetry space group resulting in
Rwp = 2.71% (GOF 2.30) vs. 3.68% (GOF 3.08) for
Pc and
P2
1/
c, respectively (as in the case of ad-KMFCN at 300 K). Additionally, the MnN
6 and FeC
6 octahedra revealed fewer distortions with the
Pc space group. It was found that all the Mn sites are fully occupied; however, half of the Fe sites have deficiencies clearly indicating vacancies. Later, in the refinement, we fixed all non-deficient positions to 1. Considering that the formation of such a large void must require a significant stabilization energy [
7], we suggested that each missed [Fe(CN)
6]
4− anion is replaced by six hydroxyl groups in such a manner that oxygen is connected to nearby manganese. To model this, we introduced mixed C/H and O/N occupancies. Some of these hydroxyls may actually be water molecules; however, sufficient discrimination between water and hydroxyl could not be achieved in this experiment; therefore, we kept them as hydroxyls only. To define the interstitial water molecule positions, we randomly placed four and eight water molecules into the unit cell corresponding to four potassium sites and doubled the amount to model a potential disorder. Then, we performed a simulated annealing with only water molecules to be freely refined. Water molecules were described as rigid bodies to simplify calculations, and the occupancy for each was set to 0.5. A simulated annealing resulted in a structure with a better fit than the original. After that, we refined the new water positions and their amounts considering the possibility of disorder and partial occupancies. The refinement factor
Rwp for the case of eight water molecules per unit cell reaches 2.47%, while for four water molecules, an
Rwp of 2.57% was achieved. Therefore, the eight water molecules model was considered more correct in the proposed model. It was found that all the water molecules are distributed along the potassium-accommodating cavities; though a large portion of water aggregated in the layers perpendicular to the
b axis (
Figure 3). Although we report certain water molecules’ positions, these should be considered as a merged extended region with a higher probability to locate water (marked in blue). The distribution of these water regions is apparently the origin of the doubling of the original
P2
1/
c unit cell and loss of centering bringing to the
Pc unit cell. Finally, taking into account both the EDX analysis and Rietveld refinement, the compositions were calculated as ad-K
1.90(5)Mn[(OH)
6]
0.015(3)[Fe(CN)
6]
0.985(3)∙1.14(4)H
2O and vd-K
1.90(5)Mn[(OH)
6]
0.015(3)[Fe(CN)
6]
0.985(3)∙1.03(4)H
2O. The slight electrical disbalance in these formulas can be attributed to the presence of the entire water molecules instead of hydroxyls, as well as a slight oxidation of transition metals during synthesis.
The unrefined peak at 4.1 Å in d-space did not move after the temperature increased from 10 K to 300 K compared to the others in both samples (
Figure S2); thus, it might correspond to an impurity, possibly coming from the neutron diffraction equipment.
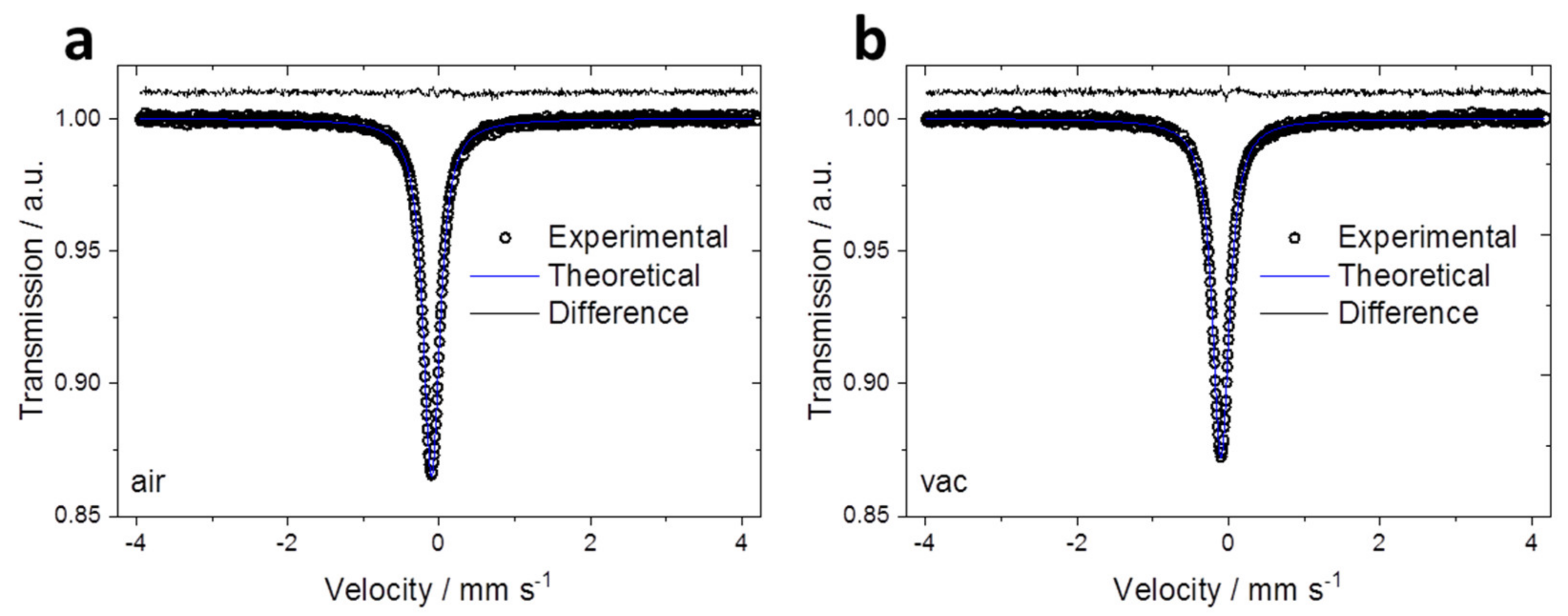
The oxidation state and spin state for Fe in ad- and vd-KMFCN were confirmed by
57Fe Mössbauer spectroscopy. The spectra of both samples display similar singlets (
Figure 4). Despite LS-Fe
II and LS-Fe
III having almost the same isomeric shift, due to the symmetric S = 0 configuration, the quadrupole splitting of LS-Fe
II in the octahedral coordination is small or equals 0 mm∙s
−1 [
30], as the experiment shows (
Table 2). Therefore, all the Fe in these samples can be reasonably assigned to the LS-Fe
II electronic state; thus, the drying conditions have no impact on the iron oxidation state change.
The further upscaling and industrial battery manufacturing requirements, taking both the electrochemical properties and time and money costs for the drying procedure into account, which is a significant part of the production process, incur about 18.2% of the costs [
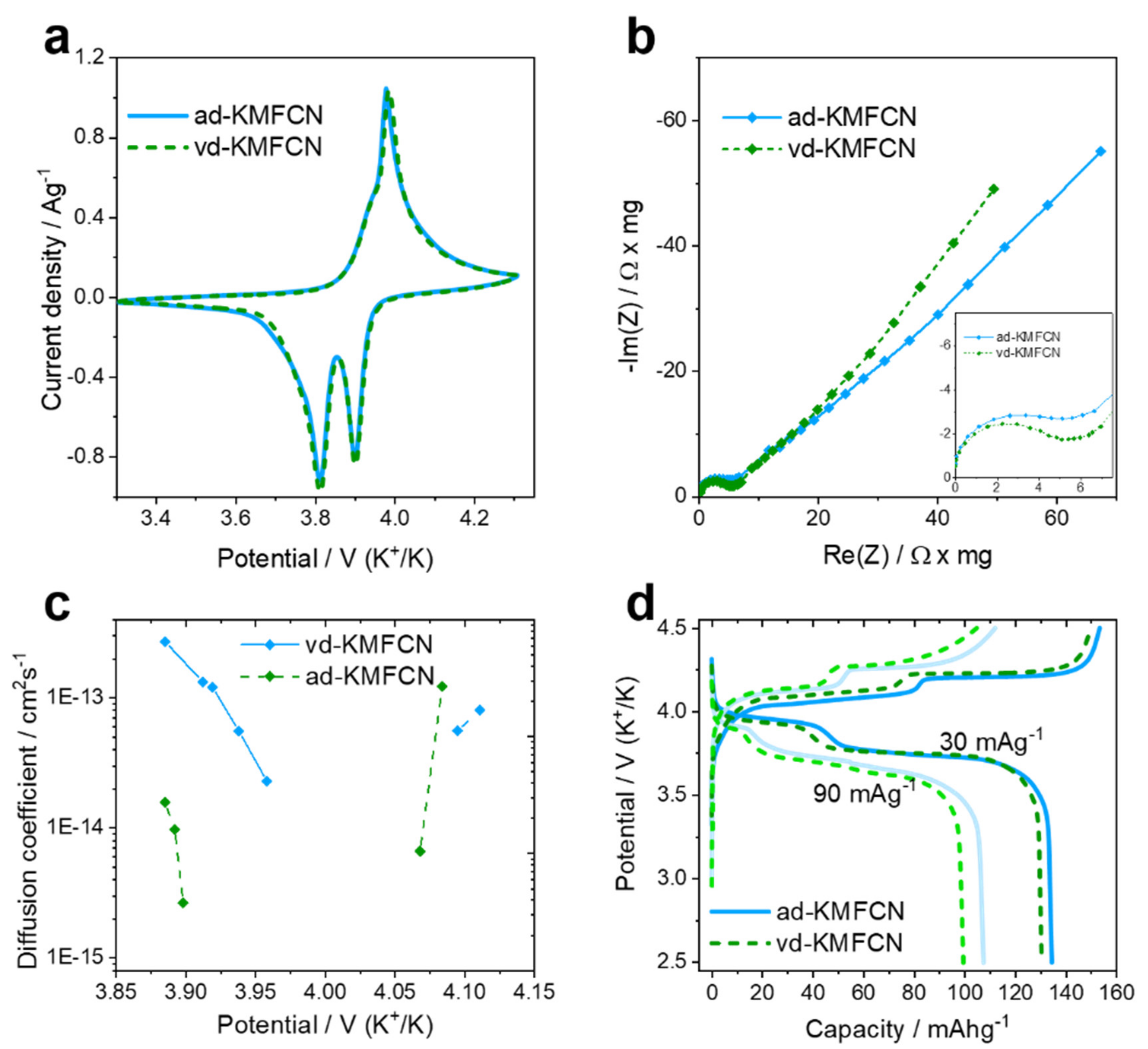
31], with the vacuum drying of prepared electrodes incurring about a quarter of this value. The electrochemical behavior changes were studied using a few methods, and CVs registered in three-electrode cells show two pairs of peaks between 3.6 and 4.1 V vs. K
+/K, corresponding to phase transitions after half and full potassium extraction in ad- and vd-KMFCN (
Figure 5a). The first redox process of vd-KMFCN is at 3.940/3.834 V and associates with the Fe
3+/2+ transition, while the second one at 4.006/3.922 V corresponds to the Mn
3+/2+ one [
15]. The redox processes in ad-KMFCN occur at 3.961/3.834 V and 3.998/3.920 V for Fe and Mn, respectively. The similarity of the CVs of ad- and vd-KMFCN indicates that no difference in phase transition potentials with higher and lower interstitial water levels was observed. The EIS spectra of the electrodes registered in the single-phase region (at 3.892 V) show a high-frequency semicircle, associated with the resistance of surface layers (
Figure 5b). The charge transfer-related semicircle cannot be resolved at low states of charge, yet the close values of the diameters of the high-frequency semicircles and the similarity of the shapes of the lower-frequency parts of the spectra indicate that the interfacial K
+ transfer kinetics are not affected by the drying method.
The apparent diffusion coefficients (D) of alkali cations in PBA structures were estimated many times for different sodium- and potassium-based PBA electrodes. Depending on the data analysis method, the reported D values varied from 10
−9 to 10
−14 cm
2 s
−1 [
4,
32]. In our work, we used PITT, and the estimated D values were in the range 10
−15–10
−13 cm
2 s
−1 in the narrow single-phase regions (
Figure 5c). Since D values typically decrease by orders of magnitude in the vicinity of the phase-transformation potentials [
22], and the single-phase regions are extremely narrow for the studied materials, it is more appropriate to compare the maximal D values for the samples, which amount to 10
−13 cm
2 s
−1 after full extraction, and differ by one order of magnitude before K
+ extraction (
Figure 5c). Based on these data, we conclude that the drying procedure had no critical influence on the ionic diffusivities in the PBA structures.
Galvanostatic measurements were performed for the KMFCN samples with small particles about 10–20 nm in size (
Figure S5) since the charge/discharge capacities of PBAs are severely affected by the particle’s size [
33]. The charge–discharge profiles of the nanosized ad- and vd-KMFCN samples reveal two plateau-like regions corresponding to two phase transitions. The discharge capacity (C
dis) of ad-KMFCN is slightly higher and equals 134.5 mAh g
−1 at 30 mA g
−1 (C/5) and 107.5 mAh g
−1 at 90 mA g
−1 (C/2), while the C
dis of vd-KMFCN amounts to 130.5 mAh g
−1 and 99.5 mAh g
−1, respectively (
Figure 5d). At the same time, the charge capacities are higher than the theoretical ones for this composition (C
theor ≈ 141 mAh g
−1), due to electrolyte decomposition and SEI formation in the first cycles [
15]. The electrochemical performance of the samples with large particles clearly correlated with that of the nanosized ones albeit being characterized by significantly lower capacities related to the poor K
+ diffusivities in the studied structures. The best cycling performance was achieved after drying under air at 75 °C (
Figure S6). Therefore, the vacuum drying procedure at higher temperatures (≥100 °C) is not a necessary step for PBA electrode material preparation and does not positively affect neither the electrochemical properties nor the water content of KMFCN.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}