
Isolation, Identification, Spectral Studies and X-ray Crystal Structures of Two Compounds from Bixa orellana, DFT Calculations and DNA Binding Studies

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Apparatus
2.2. Plant Material
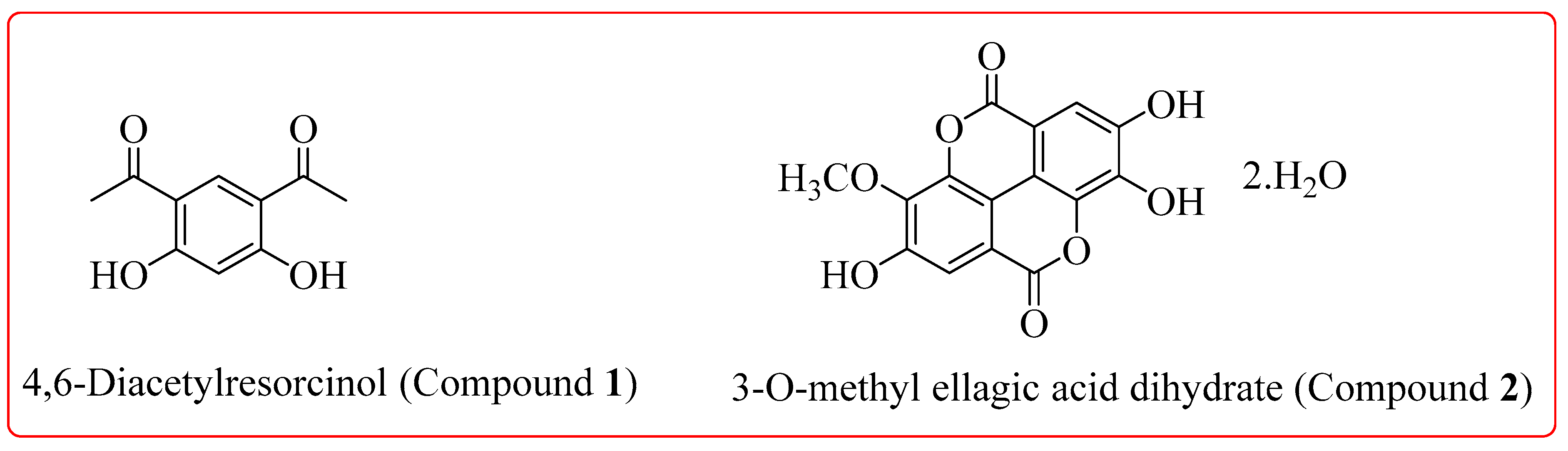
2.3. Extraction and Isolation
2.4. Spectral Analysis of Isolated Compounds
2.5. Crystallographic Analysis
2.6. Computational Procedure
2.7. ct-DNA Binding Studies
2.7.1. Sample Preparation for DNA Binding Experiments
2.7.2. UV-Visible Absorption Spectroscopy
2.7.3. Steady State Fluorescence
2.7.4. Circular Dichroism (CD)
2.7.5. Docking Studies
2.7.6. Prediction of Drug-Likeness (Lipinski’s Rule of Five)
2.7.7. Synthetic Accessibility and Bioavailability Score Prediction
3. Results and Discussion
3.1. Crystal Structure, Molecular Geometry and IR Spectral Analysis of 1 and 2
3.2. Frontier Molecular Orbital and UV-Visible Spectral Analysis
3.3. NMR Spectral Analysis
3.4. Molecular Electrostatic Potential (MEP)
3.5. UV-Visible Absorption Spectroscopy
3.6. Steady-State Fluorescence
3.7. Circular Dichroism (CD)
3.8. Docking
3.9. Prediction of DRUG-likeness (Lipinski’s Rule of Five)
3.10. Bioavailability Score and Synthetic Accessibility
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Silva, S.N.S.; Amaral, C.L.F.; Reboucas, T.N.H. Adoption of conservation practices on farm and selection of varieties by producers of annatto in the city of Vitoria da Conquista-BA. Rev. Bras. Agroecologica 2010, 5, 106. [Google Scholar]
- Correa, M.P. Dicionario das Plantas ´Uteis do Brasil e das´ Exoticas Cultivadas; Ministerio da Agricultura/IBDF: Rio ´de Janeiro, Brasil, 1978; Volume 4.
- Villar, R.; Calleja, J.M.; Morales, C.; Caceres, A. Screening of 17 Guatemalan medicinal plants for platelet antiaggregant activity. Phytother Res. 1997, 11, 441. [Google Scholar] [CrossRef]
- Aher, A.A.; Bairagi, S.M. Formulation and evaluation of herbal lipstick from colour pigments of Bixa Orellana (Bixaceae) seeds. Int. J. Pharm. Biol. Sci. 2012, 4, 357. [Google Scholar]
- Kang, E.J.; Campbell, R.E.; Bastian, E.; Drake, M.A. Invited review: Annatto usage and bleaching in dairy foods. J. Dairy Sci. 2010, 93, 3891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venugopalan, A.; Giridhar, P.; Ravishankar, G.A. Food, ethanobotanical and diversified applications of Bixa orellana L.: A scope for its improvement through biotechnological mediation. Ind. J. Fundament. Appl. Life Sci. 2011, 1, 9. [Google Scholar]
- Scotter, M. The chemistry and analysis of annatto food colouring: A review. Food Addit. Contam. 2009, 26, 1123. [Google Scholar] [CrossRef]
- Mercadante, A.Z.; Steck, A.; Pfander, H. Three minor carotenoids from annatto (Bixa orellana) seeds. Phytochemistry 1999, 52, 135–159. [Google Scholar] [CrossRef]
- Pino, J.A.; Correa, M.T. Chemical Composition of the Essential Oil from Annatto (Bixa orellana L.) Seeds. J. Essent. Oil Res. 2003, 15, 66. [Google Scholar] [CrossRef]
- Yong, Y.K.; Zakaria, Z.A.; Kadir, A.A.; Somchit, M.N.; Lian, G.E.C.; Ahmad, Z. Chemical constituents and antihistamine activity of Bixa orellana leaf extract. BMC Complement. Altern. Med. 2013, 13, 32. [Google Scholar] [CrossRef] [Green Version]
- Parveen, M.; Malla, A.M.; Ali, A.; Nami, S.A.A.; Silva, P.S.P.; Silva, M.R. Isolation, Characterization, Bioassay and X-ray Crystallographic Study of Phytoconstituents from Bixa orellana Leaves. Chem. Nat. Compd. 2015, 51, 62. [Google Scholar] [CrossRef]
- Khan, M.S.Y.; Sharma, S.; Husain, A. Synthesis and antibacterial evaluation of new flavonoid derivatives from 4,6-diacetyl resorcinol. Sci. Pharm. 2002, 70, 287. [Google Scholar] [CrossRef] [Green Version]
- SADABS. Area-Detector Absorption Correction; Siemens Industrial Automation, Inc.: Madison, WI, USA, 1996. [Google Scholar]
- Sheldrick, G.M. SHELXL-97: Program for Crystal Structure Refinement; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Platon, S.A. An Integrated Tool for the Analysis of the Results of a Single Crystal Structure Determination. Acta Crystallogr. Sect. A 1990, 46, C34. [Google Scholar]
- Platon, S.A. A Multipurpose Crystallographic Tool; Utrecht University: Utrecht, The Netherlands, 1998. [Google Scholar]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Cryst. 2012, 45, 849. [Google Scholar] [CrossRef]
- Grime, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03, Revision E.01; Gaussian Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Schmidt, J.R.; Polik, W.F. WebMO Enterprise, 18.1.001; WebMO LLC: Holland, MI, USA, 2016. [Google Scholar]
- Dennington, R.; Keith, T.; Millam, J. Gauss View Version 5; Semichem Inc.: Shawnee Mission, AR, USA, 2009. [Google Scholar]
- Chemcraft-Graphical Software for Visualization of Quantum Chemistry Computations. Available online: https://www.chemcraftprog.com (accessed on 1 November 2021).
- O’boyle, N.M.; Tenderholt, A.L. cclib: A library for package-independent computational chemistry algorithms. J. Comput. Chem. 2008, 29, 839. [Google Scholar] [CrossRef] [PubMed]
- Jayaram, B.; Singh, T.; Mukherjee, G.; Mathur, A.; Shekhar, S.; Shekhar, V. Sanjeevini: A freely accessible webserver for target directed lead molecule discovery. BMC Bioinform. 2012, 13, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Kokila, M.K.; Nirmala, K.A.; Shamala, P.N. Structure of 4,6-di acetylresorcinol. Acta. Cryst. 1992, C48, 1133. [Google Scholar]
- Rossi, M.; Erlebacher, J.; Zacharias, D.E.; Carrell, H.L.; Iannucci, B. The crystal and molecular structure of ellagic acid dihydrate: A dietary anti-cancer agent. Carcinogenesis 1991, 12, 2227. [Google Scholar] [CrossRef]
- Socrates, G. Infrared Characteristic Group Frequencies, 3rd ed.; Wiley Interscience Publications: New York, NY, USA, 1980. [Google Scholar]
- Kosar, B.; Albayrak, C. Spectroscopic investigations and quantum chemical computational study of (E)-4-methoxy-2-[(p-tolylimino)methyl]phenol. Spectrochim. Acta A 2011, 78, 160. [Google Scholar] [CrossRef]
- Alam, M.J.; Ahmad, S. Quantum chemical and spectroscopic investigations of 3-methyladenine. Spectrochim. Acta A 2014, 128, 653. [Google Scholar] [CrossRef] [PubMed]
- Tabatchnik, A.; Blot, V.; Pipelier, M.; Dubreuil, D.; Renault, E.; Le Questel, J.-Y. Theoretical Study of the Structures and Hydrogen-Bond Properties of New Alternated Heterocyclic Compounds. J. Phys. Chem. A. 2010, 114, 6413. [Google Scholar] [CrossRef] [PubMed]
- Afrin, S.; Rahman, Y.; Sarwar, T.; Husain, M.A.; Ali, A.; Tabish, M. Molecular spectroscopic and thermodynamic studies on the interaction of anti-platelet drug ticlopidine with calf thymus DNA. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2017, 186, 66. [Google Scholar] [CrossRef] [PubMed]
- Parveen, M.; Aslam, A.; Siddiqui, S.; Tabish, M.; Alam, M. Structure elucidation, DNA binding and molecular docking studies of natural compounds isolated from Crateva religiosa leaves. J. Mol. Struct. 2021, 1251, 131976. [Google Scholar] [CrossRef]
- Sirajuddin, M.; Ali, S.; Badshah, A. Drug-DNA interactions and their study by UV-Visible, fluorescence spectroscopies and cyclic voltametry. J. Photochem. Photobiol. B Biol. 2013, 124, 1–19. [Google Scholar] [CrossRef]
- Hussain, I.; Fatima, S.; Siddiqui, S.; Ahmed, S.; Tabish, M. Exploring the binding mechanism of β-resorcylic acid with calf thymus DNA: Insights from multi-spectroscopic, thermodynamic and bioinformatics approaches. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2021, 260, 119952. [Google Scholar] [CrossRef]
- Siddiqui, S.; Ameen, F.; Jahan, I.; Nayeem, S.M.; Tabish, M. A comprehensive spectroscopic and computational investigation on the binding of the anti-asthmatic drug triamcinolone with serum albumin. New J. Chem. 2019, 43, 4137. [Google Scholar] [CrossRef]
- Siddiqui, S.; Mujeeb, A.; Ameen, F.; Ishqi, H.M.; Rehman, S.U.; Tabish, M. Investigating the mechanism of binding of nalidixic acid with deoxyribonucleic acid and serum albumin: A biophysical and molecular docking approaches. J. Biomol. Struct. Dyn. 2021, 39, 570. [Google Scholar] [CrossRef]
- Siddiqui, S.; Ameen, F.; ur Rehman, S.; Sarwar, T.; Tabish, M. Studying the interaction of drug/ligand with serum albumin. J. Mol. Liq. 2021, 336, 116200. [Google Scholar] [CrossRef]
- Ameen, F.; Siddiqui, S.; Jahan, I.; Nayeem, S.M.; ur Rehman, S.; Tabish, M. A detailed insight into the interaction of memantine with bovine serum albumin: A spectroscopic and computational approach. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2022, 265, 120391. [Google Scholar] [CrossRef]
- Enmozhi, S.K.; Raja, K.; Sebastine, I.; Joseph, J. Andrographolide as a potential inhibitor of SARS-CoV-2 main protease: An in silico approach. J. Biomol. Struct. Dyn. 2021, 39, 3092. [Google Scholar] [CrossRef] [PubMed]
- Benet, L.Z.; Hosey, C.M.; Ursu, O.; Oprea, T.I. BDDCS, the Rule of 5 and drugability. Adv. Drug Deliv. Rev. 2016, 101, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathak, K.; Raghuvanshi, S. Oral bioavailability: Issues and solutions via nanoformulations. Clin. Pharmacokinet. 2015, 54, 325. [Google Scholar] [CrossRef] [PubMed]
- Zothantluanga, J.H.; Gogoi, N.; Shakya, A.; Chetia, D.; Lalthanzara, H. Computational guided identification of potential leads from Acacia pennata (L.) Willd. as inhibitors for cellular entry and viral replication of SARS-CoV-2. Future J. Pharm. Sci. 2021, 7, 201. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bonds (Ǻ) | Expt. | DFT | Angles (°) | Expt. | DFT | Dihedral Angles (°) | Expt. | DFT |
|---|---|---|---|---|---|---|---|---|
| 4,6-Diacetylresorcinol (1) | ||||||||
| C1-C2 | 1.38 | 1.39 | C2-C1-C6 | 121.0 | 120.2 | C6-C1-C2-H15 | −175.2 | −179.9 |
| C1-C6 | 1.41 | 1.43 | C2-C1-O14 | 117.9 | 118.1 | O14-C1-C2-C3 | 179.0 | 180.0 |
| C1-O14 | 1.34 | 1.33 | C6-C1-O14 | 121.0 | 121.6 | O14-C1-C6-C5 | −179.5 | −180.0 |
| C2-C3 | 1.37 | 1.39 | C1-C2-C3 | 119.8 | 120.8 | O14-C1-C6-C7 | −0.916 | −0.004 |
| C2-H15 | 0.99 | 1.08 | C1-C2-H15 | 116.9 | 119.5 | C1-C2-C3-O13 | −178.7 | −179.9 |
| C3-C4 | 1.42 | 1.43 | C3-C2-H15 | 122.8 | 119.5 | C15-C2-C3-O13 | −5.6 | −0.0143 |
| C3-O13 | 1.34 | 1.33 | C3-C4-C4 | 117.9 | 120.2 | C2-C3-O13-H23 | 174.6 | 179.9 |
| C4-C5 | 1.38 | 1.39 | C2-C3-O13 | 118.1 | 118.1 | C4-C3-O13-H23 | −5.0 | −0.0051 |
| C4-C10 | 1.46 | 1.46 | C4-C3-O13 | 120.9 | 121.6 | C3-C4-C10-O12 | 1.3 | 0.00076 |
| C5-C6 | 1.39 | 1.39 | C3-C4-C10 | 120.0 | 119.7 | C5-C4-C10-O12 | −178.2 | 179.9 |
| C5-H16 | 0.99 | 1.08 | C5-C6-C7 | 121.7 | 122.4 | C5-C6-C7-O9 | 176.9 | 179.9 |
| C6-C7 | 1.46 | 1.46 | C6-C7-C8 | 120.9 | 120.0 | O9-C7-C8-H17 | 10.5 | 0.0172 |
| C7-C8 | 1.48 | 1.51 | C6-C7-O9 | 119.8 | 121.0 | O9-C7-C8-H18 | −106.3 | −120.0 |
| C7-O9 | 1.23 | 1.23 | C8-C7-O9 | 119.1 | 118.8 | O9-C7-C8-H19 | 135.7 | 120.0 |
| O14-H24 | 1.05 | 0.99 | C1-C14-O24 | 101.6 | 106.9 | O12-C10-C11-H20 | −125.3 | −119.9 |
| 3-O-Methylellagic acid (2) | ||||||||
| O1-C12 | 1.35 | 1.34 | C12-O1-C28 | 115.9 | 120.4 | C28-O1-C12-C13 | 115.1 | 146.0 |
| O1-C28 | 1.44 | 1.43 | H3-O2-C13 | 109.5 | 108.9 | C28-O1-C12-C25 | −68.7 | −36.5 |
| O2-H3 | 0.82 | 0.96 | C17-O5-C18 | 122.1 | 122.7 | C12-O1-C28-H29 | −53.0 | −42.5 |
| O2-C13 | 1.34 | 1.35 | H7-O6-C19 | 109.4 | 108.0 | H3-O2-C13-C12 | 172.8 | 179.2 |
| O4-C17 | 1.20 | 1.19 | C24-O11-C25 | 122.4 | 123.6 | H3-O2-C13-C14 | −7.3 | −1.1 |
| O5-C17 | 1.37 | 1.39 | O1-C12-C13 | 118.7 | 116.0 | C18-O5-C17-O4 | −178.0 | 179.5 |
| O5-C18 | 1.38 | 1.36 | O1-C12-C25 | 122.0 | 126.0 | C18-O5-C17-C16 | 1.9 | −0.4 |
| O6-H7 | 0.81 | 0.96 | C13-C12-C25 | 119.0 | 117.7 | C17-O5-C18-C19 | 178.1 | −179.3 |
| O6-C19 | 1.33 | 1.34 | O2-C13-C12 | 114.8 | 115.7 | H7-O6-C19-C18 | −170.3 | 179.8 |
| C18-C27 | 1.38 | 1.39 | O2-C13-C14 | 124.3 | 122.9 | H7-O6-C19-C20 | 10.2 | −0.1 |
| C23-C24 | 1.45 | 1.46 | C13-C14-H15 | 120.4 | 120.8 | O10-C24-O11-C25 | 178.9 | −178.8 |
| C19-C20 | 1.40 | 1.41 | C13-C14-C16 | 119.1 | 120.0 | O10-C24-C23-C21 | 0.9 | −0.4 |
| C28-H31 | 0.95 | 1.09 | O8-C20-C19 | 113.9 | 113.6 | H15-C14-C16-C17 | −0.04 | 0.2 |
| C21-H22 | 0.93 | 1.08 | C19-C18-O5 | 118.3 | 118.4 | H15-C14-C16-C26 | 179.4 | −179.6 |
| C16-C25 | 1.39 | 1.40 | C18-C27-C26 | 118.7 | 118.5 | C18-C27-C23-C24 | 178.5 | 179.6 |
| FTIR | ωharmonic | Assignments | FTIR | ωharmonic | Assignments | ||
|---|---|---|---|---|---|---|---|
| 4,6-Diacetylresorcinol (1) | 3427 | 3295 | νOH | 3-O-Methylellagic acid dihydrate (2) | 3433 | 3848–3778 | νOH |
| 3291 | νasyOH | 3159 | 3184–3179 | νCH (ar) | |||
| 3082 | 3212 | νCH (ar) | 2927 | 3146, 3119, 3035 | νasyCH, νCH | ||
| 3207 | νCH (ar) | 1723 | 1820, 1813 | νC=O | |||
| 3147 | νCH (ar) | 1600 | 1659 | νCC, βHOC | |||
| 3146 | νCH (ar) | 1650, 1634, 1612, 1560, 1549, 1526 | νCC, νasyCC γHCC | ||||
| 2925 2851 | 3097 | νCH | 1518 | 1504 | βCOC, βHCH, τHCOC | ||
| 3036 | νCH | 1422 | 1489 | βHCH,τHCOC | |||
| 1652 | 1696 | νasyC=O | 1450 | νCC, βHOC | |||
| 1694 | νC=O | 1361 | 1382 | νO-C, νasyC-O | |||
| 1661 | νasyC=O, Ar-ring | 1340 | 1372, 1322 | νasyCC, βHOC | |||
| 1588 | 1627 | νCC, νasyCC, βHOC | 1293 | 1319 | νasyO-C, βHCC | ||
| 1527 | βHOC | 1297 | νO-C, γHOC, βHCC | ||||
| 1486 | 1482 | βHCH, τHCCC | 1190 | 1221 | βHOC, βHCH, τHCOC | ||
| 1472 | βHCH, τHCCC | 1175 | βHOC, βHCC | ||||
| 1425 | 1398 | βHCH | 1060 | 1167 | βHCH, τHCOC | ||
| 1290 | νO-Cand βHCC (ar) | 966 | 1053, 1000 | νO-C | |||
| 1266 | νCC, βHOC, βCCC | 912 867 | 921 | γCCC | |||
| 1214 | νO-C, βHCC (ar) | 861 | τHCCC | ||||
| 1092 | βCCC, τHCCC | 859 | outCCCC | ||||
| 964 | γ HCH, τHCCC | 800 | 807 | mix vibration | |||
| 914 | τHCCC | 759 | outOCOC | ||||
| 762 | βCCC | 699 | βCOC, outOCCC | ||||
| 725 | outCCCC | 640 | βCOC, βOCC, | ||||
| 674 | βCCC | 575 | 614 | βCOC, βOCC | |||
| 556 | βCCC, βCCO | 539 | νCC, βCCC, |
| λexp | λtheo | E(eV) | f | Composition (%) | λexp | λtheo | E(eV) | f | Composition (%) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 4,6-Diacetylresorcinol (1) | 273.6 | 268.5 | 4.6167 | 0.1915 | H→L + 1 (99) | 3-O-Methylellagic acid dihydrate (2) | 255.6 | 259.7 | 4.77 | 0.0498 | H-3→L(30) |
| 251.9 | 4.9212 | 0.7777 | H-1→L + 1 (69), H→L (25) | 252.9 | 4.90 | 0.0366 | H-5→L (25), H-4→L (50) | ||||
| 323.7 | 302.6 | 4.0968 | 0.1209 | H-1→L + 1 (26), H→L (73) | 365.3 | 351.7 | 3.52 | 0.2290 | H→L(94) | ||
| 300.1 | 4.1314 | 0.0366 | H-3→L + 1 (19), H-2→L (79) | 313.6 | 3.95 | 0.0269 | H-1→L (51), H→L + 1 (4) |
| System | Ksv (M−1) | Kq (M−1 s−1) | n | Kb (M−1) |
|---|---|---|---|---|
| Compound 2 | 1.9 × 104 | 1.9 × 1012 | 1.0 | 3.8 × 105 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parveen, M.; Azeem, M.; Aslam, A.; Azam, M.; Siddiqui, S.; Tabish, M.; Malla, A.M.; Min, K.; Rodrigues, V.H.; Al-Resayes, S.I.; et al. Isolation, Identification, Spectral Studies and X-ray Crystal Structures of Two Compounds from Bixa orellana, DFT Calculations and DNA Binding Studies. Crystals 2022, 12, 380. https://doi.org/10.3390/cryst12030380
Parveen M, Azeem M, Aslam A, Azam M, Siddiqui S, Tabish M, Malla AM, Min K, Rodrigues VH, Al-Resayes SI, et al. Isolation, Identification, Spectral Studies and X-ray Crystal Structures of Two Compounds from Bixa orellana, DFT Calculations and DNA Binding Studies. Crystals. 2022; 12(3):380. https://doi.org/10.3390/cryst12030380
Chicago/Turabian StyleParveen, Mehtab, Mohammad Azeem, Afroz Aslam, Mohammad Azam, Sharmin Siddiqui, Mohammad Tabish, Ali Mohammad Malla, Kim Min, Vitor Hugo Rodrigues, Saud I. Al-Resayes, and et al. 2022. "Isolation, Identification, Spectral Studies and X-ray Crystal Structures of Two Compounds from Bixa orellana, DFT Calculations and DNA Binding Studies" Crystals 12, no. 3: 380. https://doi.org/10.3390/cryst12030380
APA StyleParveen, M., Azeem, M., Aslam, A., Azam, M., Siddiqui, S., Tabish, M., Malla, A. M., Min, K., Rodrigues, V. H., Al-Resayes, S. I., & Alam, M. (2022). Isolation, Identification, Spectral Studies and X-ray Crystal Structures of Two Compounds from Bixa orellana, DFT Calculations and DNA Binding Studies. Crystals, 12(3), 380. https://doi.org/10.3390/cryst12030380